Antisense suppression of the nonsense mediated decay factor Upf3b as a potential treatment for diseases caused by nonsense mutations
Full text
Figure
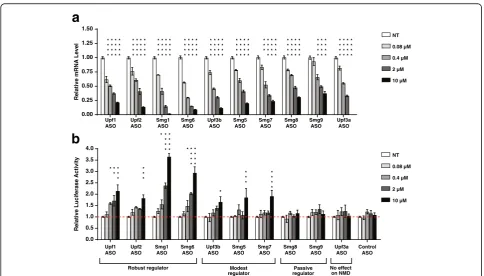
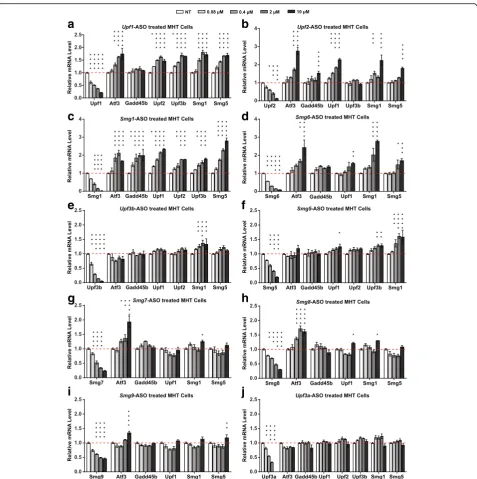
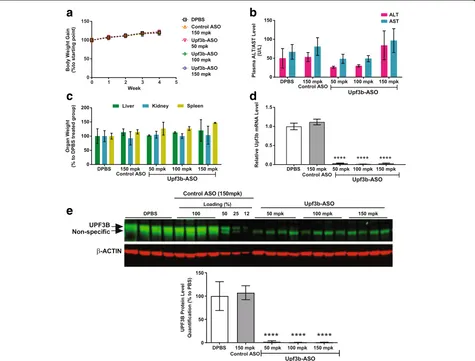
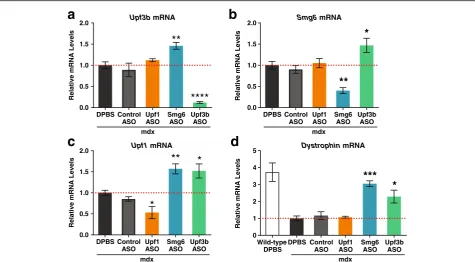

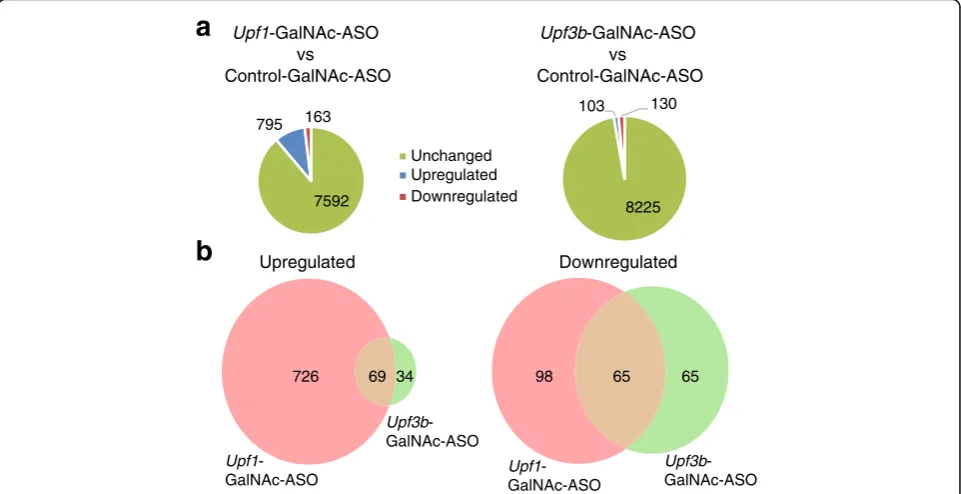
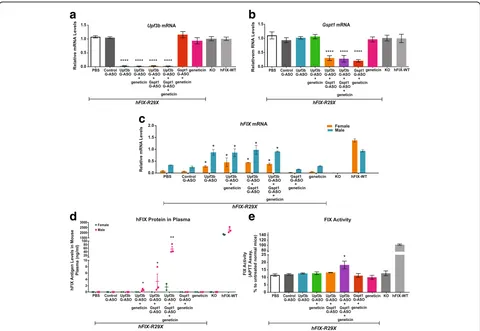
Related documents
Solar light mediated Photocatalytic degradation of organic dyes such as brilliant yellow, brilliant blue, rhodamine b and proclor red Mx- 5B dye, two mixed dye (rhodamine b
A., and Hagin, R.: Specific reading disability: teaching through stimulation of. deficit
An introduction to the Buddhist history of Buryatia serves as the backdrop for Tenzin Chodron ’ s autobiographical materials, which discuss her early life, education, professional
Some Millerites and early Sabbatarian Adventists employed several terms to express the concept, such as the typical Day of Atonement, breastplate of judgment,
Where you have refinanced your loan, any amount that has been paid to you under a Critical Illness claim or Cancer claim under the terms of your previous loan protection
• Personal cover with no (direct) employment link, p/h pays premiums from net income?. • Various GIO’s and life-event
