DOI: 10.1534/genetics.107.078998
Joint Estimation of Migration Rate and Effective
Population Size Using the Island Model
Garrick T. Skalski
1Department of Ecology and Evolutionary Biology, University of Kansas, Lawrence, Kansas 66045
Manuscript received July 17, 2007 Accepted for publication August 13, 2007
ABSTRACT
Using the island model of population demography, I report that the demographic parameters migra-tion rate and effective populamigra-tion size can be jointly estimated with equilibrium probabilities of identity in state calculated using a sample of genotypes collected at a single point in time from a single generation. The method, which uses moment-type estimators, applies to dioecious populations in which females and males have identical demography and monoecious populations with no selfing and requires that offspring genotypes are sampled following reproduction and prior to migration. I illustrate the estimation pro-cedure using the infinite-island model with no mutation and the finite-island model with three kinds of mutation models. In the infinite-island model with no mutation, the estimators can be expressed as simple functions of estimates of theF-statistic parametersFITandFST. In the finite-island model with mutation amongkalleles, mutation rate, migration rate, and effective population size can be simultaneously esti-mated. The estimates of migration rate and effective population size are somewhat robust to violations in assumptions that may arise in empirical applications such as different kinds of mutation models and deviations from temporal equilibrium.
P
OPULATION geneticists recognize that the demo-graphic characteristics of populations, such as mi-gration rates and population sizes, affect population genetic structure (Wright 1951). Accordingly, manypopulation genetic studies have investigated how demo-graphic properties might be inferred from genetic mea-surements in populations (e.g., Slatkin 1985; Waples
1989; Pudovkin et al. 1996; Beerli and Felsenstein
2001; Vitalisand Couvet2001; Wangand Whitlock
2003; Robledo-Arnuncio et al.2006). In parallel, the
cultivation of genomic resources in species that are amenable to field study has facilitated the application of genetic methodologies to estimate demographic rates in natural populations.
The island model of Wright(1951) is an important
model in population genetics. Under a simple version of this model, an infinite number of demes, each having population sizeN, exchange migrants at rate munder the assumption that migrants into a deme come from any of the other demes with equal probability. In the absence of mutation and other evolutionary forces, genetic polymorphism is maintained within demes via a balance between genetic drift and migration. An im-portant feature of the infinite-island model is that, at temporal equilibrium, the magnitude of genetic differ-entiation among demes,FST, is approximated by
FST
1 114mN;
where the approximation is intended to apply for small values of the migration rate,m(Wright1951). An
im-portant statistical consequence of the above result is that the product parametermN, the number of individuals migrating and reproducing per generation, may be estimated using data on FST (Slatkin 1985), but the parametersmandNcannot be estimated individually in this way. Although the indiscriminate application of the infinite-island model to interpret genetic data in terms of demographic rates has been discouraged (Whitlock
and Mccauley 1999), the island model continues to
support a variety of theoretical and empirical investiga-tions (e.g., Vitalisand Couvet 2001; Balloux et al.
2003; Ha¨ nflingand Weetman2006).
There is continuing interest in statistical approaches that estimate both migration rate,m, and effective pop-ulation size, N, from genetic data, including methods that are applicable to a sample taken from a single gener-ation at a single point in time (Beerliand Felsenstein
2001; Vitalisand Couvet2001; Wangand Whitlock
2003). Here, I report results that show how the island model of dioecious or monoecious populations can be used to simultaneously estimate migration rate,m, and effective population size,N, using a sample of selectively neutral markers taken from a single generation at a single point in time. In particular, at temporal equilib-rium under the infinite-island model with no mutation, the demographic parametersmandNcan be estimated
1Corresponding author:1637 Merion Pl., Lawrence, KS 66047.
E-mail: [email protected]
using data onFITandFST(Wright1951). At temporal
equilibrium under the finite-island model with ak-allele mutation scheme, the demographic parametersmand N, as well as the mutation rate, u, can be jointly esti-mated using data on probabilities of identity in state.
THE INFINITE-ISLAND MODEL WITH NO MUTATION
I first describe key results for the infinite-island model of a selectively neutral locus with no mutation; theoret-ical details are in theappendix. Under the infinite-island
model with no mutation, an infinite number of demes, each having effective population sizeN, exchange migrants at ratem. The results that follow apply to monoecious populations with no selfing (N adults) and dioecious populations when males and females have identical demography (N adults composed of N/2 females and N/2 males). This model is appropriate for highly fecund organisms with localized mating, including species of invertebrates, amphibians, fishes, and plants.
Population genetic structure can be characterized using probabilities of gene identity (e.g., Maruyama 1970;
MaynardSmith1970; Neiand Feldman1972; Crow
and Aoki1984; Epperson1999; Rousset2001; Vitalis
2002). Accordingly, letQ1(t),Q2(t), andQ3(t)be the prob-abilities (summed overkalleles at one locus) that genes within individuals, between individuals within a deme, and between individuals between demes are the same allele at timet, respectively. Hence,Q1(t),Q2(t), andQ3(t) are probabilities of identity in allelic state. A key idea in the following theory is that the interpretation of the probabilities of identity can depend on the timing of the sampling of genotypes within the sequence of demo-graphic events that defines the life cycle (Vitalis2002).
I first assume that the sampling of genotypes follows a premigration census in the sense that genotypes are sam-pled from offspring immediately following reproduc-tion and prior to migrareproduc-tion. This kind of sampling is appropriate for highly fecund organisms with localized mating in which many offspring may be available follow-ing reproduction for genotypfollow-ing. Under the premigra-tion census in the infinite-island model with no mutapremigra-tion, the probabilities of identity at temporal equilibrium satisfy
Q1ðt11Þ¼Q1ðtÞ¼ ð1mÞ2Q2ðtÞ1½1 ð1mÞ2Q3ðtÞ
Q2ðt11Þ¼Q2ðtÞ¼
1
2Nð11Q1ðtÞÞ1ð1mÞ
2 1 1
N
Q2ðtÞ
1½1 ð1mÞ2 11
N
Q3ðtÞ
Q3ðt11Þ¼Q3ðtÞ¼Q3ðtÞ:
ð1Þ
Equation 1 is the same as Equation A1.4 of Vitalis(2002)
when assuming an infinite number of demes with no mu-tation and no sex-specific dispersal in the latter. Although recursions similar to Equation 1 have been presented
and analyzed (MaynardSmith1970; Vitalisand Couvet
2001; Vitalis2002; Ballouxet al.2003; see theappendix
for details), previous work seems to have overlooked the idea that Equation 1 can be used to jointly estimatemand N. Indeed, at temporal equilibrium, the parametersFIT andFSTare distinct and are given by
FIT¼
Q1Q3
1Q3
¼ ð1mÞ
2
½1 ð1mÞ22N1ð1mÞ2
ð12mÞ
ð12mÞ14mN
FST ¼
Q2Q3
1Q3
¼ 1
½1 ð1mÞ22N1ð1mÞ2
1
ð12mÞ14mN;
where the approximation omits terms proportional to m2½the approximation is given here solely to connect these
findings to Wright’s (1951) classic result that FST
1=ð114mNÞ. Equation 14 in Vitalis(2002) assuming
no mutation, an infinite number of demes, and no sex-specific dispersal is the same as the equation for FST given above, but Vitalis(2002) does not report an
ex-pression forFIT. Thus, migration rate,m, and effective population size,N, can be expressed in terms ofFITand FST, without approximation, via
m¼1
ffiffiffiffiffiffiffi FIT FST r ¼1 ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
Q1Q3
Q2Q3
s
N ¼ 1FIT
2ðFSTFITÞ
¼ 1Q1
2ðQ2Q1Þ
:
Hence, the above expressions can be used to estimatem andN via the moment-based estimators
ˆ
m¼1
ffiffiffiffiffiffiffiffi ˆ FIT ˆ FST s ¼1 ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi ˆ Q1Qˆ3
ˆ Q2Qˆ3
s
ˆ
N¼ 1FˆIT
2ðFˆSTFˆITÞ¼
1Qˆ1
2ðQˆ2Qˆ1Þ; ð2Þ
whereFˆIT;FˆST;Qˆ1ðtÞ;Qˆ2ðtÞ;andQˆ3ðtÞdenote estimates
ofFIT,FST,Q1(t),Q2(t), andQ3(t), respectively. Methods for estimatingFIT,FST,Q1(t),Q2(t), andQ3(t)are discussed by Rousset(2001). Equation 17 in Vitalis(2002) gives
an estimator for sex-specific dispersal rates similar to the estimator ofmin Equation 2, but, importantly, the for-mer requires estimates ofFSTfrom a sequence of sam-ples taken pre- and postmigration, rather than estimates of FITandFSTfrom a single sample as in Equation 2. Fontanillaset al. (2004) also give estimators for
sex-specific dispersal based on the idea of Vitalis (2002)
that require estimates ofFSTfrom samples taken pre- and postmigration. Vitalis (2002) and Fontanillas et al.
(2004) do not report estimators of effective population size. Estimates ofmandNcan be calculated from multiple loci by calculating Qˆ1ðtÞ; Qˆ2ðtÞ;andQˆ3ðtÞ over loci (or,
offspring genotypes are sampled following migration using a postmigration census scheme (Vitalis2002),
then the recursions for the probabilities of identity are different from those for the premigration census with the consequence thatFIT¼FST. Hence, the parameters mandNcannot be jointly estimated in this way using a postmigration census.
To verify the recursions in Equation 1, and thus that the estimators in Equation 2 work as intended, I sim-ulated genotype data under the infinite-island model with no mutation for dioecious and monoecious (with no selfing) populations at temporal equilibrium over a range of migration rates (0.02, 0.05, 0.10, and 0.20) and effective population sizes (10, 20, and 50). In the sim-ulations, individual genotypes were tracked forward in time using a Monte Carlo implementation of the prob-ability model defined by the life cycle using a premig-ration census. For each replicate simulation, diploid genotypes were initialized using random pairs of alleles, the life cycle was iterated until the system reached tem-poral equilibrium, and offspring genotypes were sampled prior to migration. I numerically solved the analytical recursions in Equation 1 to identify, in advance of the stochastic simulations, a sufficient number of genera-tions required for the system of probabilities of identity to reach equilibrium to a precision of 104(equilibrium
to four decimal places; 1000 generations is sufficient for all parameter combinations under the infinite-island model). Means of the probabilities of identity calculated over replicate simulations are in close agreement with those calculated from the analytical recursions. The simulations were carried out using 20 independent eight-allele loci (with equally frequent eight-alleles) at which 50 offspring were genotyped from each of 20 demes. Sim-ulated data were combined over loci to calculateQˆ1ðtÞ;
ˆ
Q2ðtÞ;andQˆ3ðtÞ:Negative estimates ofN(equivalent to
infinite-valued estimates ofN) andmwere set equal to 1000 and zero, respectively.
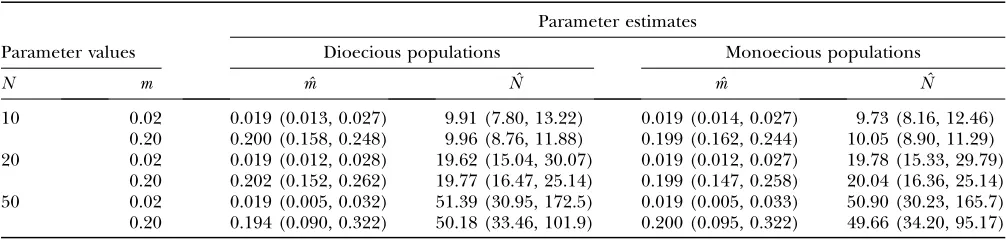
The simulations of the infinite-island model show, given sufficient data collected using a premigration census, that estimates of migration rate and effective population size using Equation 2 are close to their true values for both dioecious (Figure 1A; Table 1) and monoecious populations (Figure 1B; Table 1). The precision of the estimates ofNdecreases with increasingN, and the pre-cision of the estimates of mdecreases with increasing mandN. Additional simulation results are in supplemen-tal Table S1 at http://www.genetics.org/supplemensupplemen-tal/.
THE FINITE-ISLAND MODEL WITH
k-ALLELE MUTATION
I now describe key results for the finite-island model with mutation following the k-allele mutation model; theoretical details are in theappendix. Under the
finite-island model,sdemes, each having effective population sizeN, exchange migrants at ratem, and genes can
mu-tate into other alleles after gamete production accord-ing to thek-allele mutation model. The results that follow again apply to monoecious populations with no selfing and dioecious populations when males and females exhibit identical demography (including identical rates of mutation).
Under the premigration census in the finite-island model withk-allele mutation, the probabilities of identity at tem-poral equilibrium satisfy
Q1ðt11Þ¼Q1ðtÞ¼U21ðU1U2Þ½M1Q2ðtÞ1ð1M1ÞQ3ðtÞ Q2ðt11Þ¼Q2ðtÞ¼
U1 2N1 1
1 2N
U21 1
2NðU1U2ÞQ1ðtÞ
1M1 1 1
N
ðU1U2ÞQ2ðtÞ
1ð1M1Þ 1 1
N
ðU1U2ÞQ3ðtÞ Q3ðt11Þ¼Q3ðtÞ¼U21ðU1U2Þ½M2Q2ðtÞ1ð1M2ÞQ3ðtÞ;
ð3Þ Figure1.—Infinite-island model: estimates of migration rate and effective population size. (A) Estimates of migration rate,
where
M1¼ ð1mÞ21
m2
s1
M2¼
1M1
s1
U1¼ ð1uÞ21
u2
k1
U2¼
1U1
k1:
Equation 3 is the same as Equation A1.4 of Vitalis
(2002) when the latter is modified to havek-allele mu-tation (rather than infinite-allele mumu-tation) and no sex-specific dispersal. Although recursions similar to Equation 3 have been presented and analyzed (MaynardSmith
1970; Vitalisand Couvet2001; Vitalis2002; Balloux
et al.2003; see appendix for details), the subsequent
estimation ofu, m, and Nbased on Equation 3 seems not to have been recognized in previous work. Indeed, Equation 3 can be used to jointly estimate the mutation rate,u, migration rate,m, and effective population size, N, by solving the system of equations
ˆ
Q1ðtÞ¼U21ðU1U2Þ½M1Qˆ2ðtÞ1ð1M1ÞQˆ3ðtÞ
ˆ Q2ðtÞ¼
U1
2N 1 1 1 2N
U21
1
2NðU1U2ÞQˆ1ðtÞ
1M1 1
1 N
ðU1U2ÞQˆ2ðtÞ
1ð1M1Þ 1
1 N
ðU1U2ÞQˆ3ðtÞ
ˆ
Q3ðtÞ¼U21ðU1U2Þ½M2Qˆ2ðtÞ1ð1M2ÞQˆ3ðtÞ ð4Þ
foru, m, and N. The values of u,m, andN that satisfy Equation 4, denoted byuˆ;mˆ;andNˆ;respectively, are the respective moment-based estimators of u, m, and N. The estimatesuˆ;mˆ;andNˆare calculated assuming that the number of demes, s, and the number of possible alleles,k, are known. Estimates can be calculated from
multiple loci by summing the left- and right-hand sides of Equation 4 over loci (kmay be locus specific in theU1 andU2terms in the right-hand side of Equation 4) and calculating the parameter values that solve the moment equations obtained by setting the left-hand sum over loci equal to the right-hand sum over loci.
To verify the recursions in Equation 3, and thus that the estimators uˆ; mˆ; andNˆ based on Equation 4 work as intended, I simulated genotype data under the finite-island model with k-allele mutation for dioecious and monoecious (with no selfing) populations at temporal equilibrium over a range of migration rates (0.02, 0.05, 0.10, and 0.20) and effective population sizes (10, 20, and 50) at three different mutation rates (0.001, 0.0005, and 0.0001). These values for the mutation rate are con-sistent with values used in similar simulation studies (e.g., Vitalisand Couvet2001; Wangand Whitlock2003;
Excoffier et al. 2005) and estimates from empirical
data (Estoupet al.2001; Laiand Sun2003; Excoffier
et al.2005). Simulations were executed as described above for the infinite-island model: individual genotypes were tracked forward in time using a Monte Carlo implemen-tation of the probability model defined by the life cycle using a premigration census forsdemes. I numerically solved the analytical recursions in Equation 3 to identify a sufficient number of generations for the system to reach equilibrium to a precision of 104(5000 generations for
u¼0.001, 6000 generations foru¼0.0005, and 15,000 generations foru¼0.0001). Models with smaller values ofuandmand larger values ofN require more gener-ations to reach equilibrium. Means of the probabilities of identity calculated over replicate simulations are in close agreement with those calculated from the analyt-ical recursions. The simulations were carried out using 30 independent eight-allele loci (with equally frequent alleles; hence,k¼8) at which 100 offspring were geno-typed from each of 20 demes. Simulated data were com-bined over loci to calculateQˆ1ðtÞ;Qˆ2ðtÞ;andQˆ3ðtÞ;and
the estimatorsuˆ;mˆ;andNˆdefined by Equation 4 were calculated numerically using a nonlinear least-squares
TABLE 1
Medians (5th, 95th percentiles) of the estimates of migration rate and effective population size using genotype data simulated under the infinite-island model at different values of migration rate,m, and effective population size,
N, based on 400 replicate simulations for each pair of model migration rate and effective population size parameters for dioecious and monoecious model populations
Parameter estimates
Parameter values Dioecious populations Monoecious populations
N m mˆ Nˆ mˆ Nˆ
10 0.02 0.019 (0.013, 0.027) 9.91 (7.80, 13.22) 0.019 (0.014, 0.027) 9.73 (8.16, 12.46)
0.20 0.200 (0.158, 0.248) 9.96 (8.76, 11.88) 0.199 (0.162, 0.244) 10.05 (8.90, 11.29)
20 0.02 0.019 (0.012, 0.028) 19.62 (15.04, 30.07) 0.019 (0.012, 0.027) 19.78 (15.33, 29.79)
0.20 0.202 (0.152, 0.262) 19.77 (16.47, 25.14) 0.199 (0.147, 0.258) 20.04 (16.36, 25.14)
50 0.02 0.019 (0.005, 0.032) 51.39 (30.95, 172.5) 0.019 (0.005, 0.033) 50.90 (30.23, 165.7)
procedure in the software application MATLAB (The Mathworks). Estimates ofu,m, andNwere constrained according to 0#uˆ#1, 0#mˆ #1, and 2#Nˆ#1000, respectively, to obtain realistic estimates and to account for the possibility of infinite-valued estimates ofN (cf. Waples 1989; Williamson and Slatkin 1999; Wang
and Whitlock2003).
The simulations of the finite-island model show, given sufficient data collected using a premigration census, that estimates of migration rate and effective population size using Equation 4 are close to their true values for both dioecious (Figure 2, A–C; Table 2) and monoecious populations (Figure 2D; Table 2). The precision of the estimates ofNdecreases with increasing values ofN, and the precision of the estimates of m decreases with in-creasing values ofmand N. The precision of the esti-mates ofmandNdecreases with lower mutation rates. Estimates of mutation rate, given sufficient data, are similarly close to their true values (Table 2). The preci-sion of the estimates ofudecreases with decreasing values ofuand increasing values of N. Additional simulation results are in supplemental Table S2 at http://www. genetics.org/supplemental/.
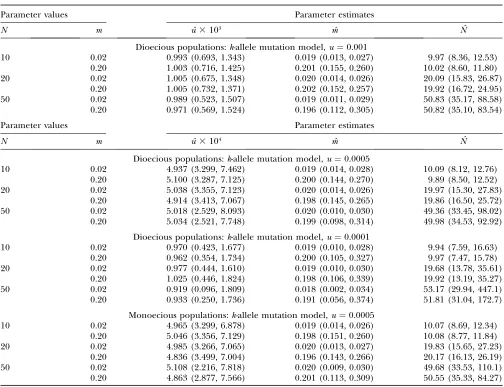
Examples of the sampling distributions of the param-eter estimates are shown in Figure 3 for dioecious pop-ulations with u ¼ 0.0005, m ¼ 0.05, and N ¼ 20 for samples of 100 genotypes from 20 demes genotyped at 30 eight-allele loci. The distributions of uˆ and mˆ are approximately symmetrical (Figure 3, A and B), and the distribution ofNˆ is positively skewed (Figure 3C). The estimates ofmandNare strongly negatively correlated and fall along the line defined bym ˆˆN¼mN(Figure 3D). The estimates ofuandmare positively correlated, and the estimates ofuandNare negatively correlated (the correlation betweenuˆandNˆis strong, but not as strong as that exhibited bymˆandNˆ). These results suggest that the product parametermNis well estimated, but that the
individual estimates ofu,m, andNare more difficult to identify precisely from data.
In empirical situations,k, the number of possible alleles at a locus, might be expected to vary across loci. In this case, estimates ofu,m, andNmay be constructed from the moment equations defined by summing the left-and right-hleft-and sides of Equation 4 over loci left-and setting the left-hand sum equal to the right-hand sum. Simula-tions of dioecious populaSimula-tions in the finite-island model (100 individuals genotyped from each of 20 demes) with k-allele mutation (u¼0.0005) for 30 loci having a ran-dom mixture of 4, 8, or 12 possible alleles show, given sufficient data collected using a premigration census, that estimates of mutation rate, migration rate, and ef-fective population size are close to their true values and have similar properties to those calculated when all loci have the same number of possible allelic states (simu-lation results are in supplemental Table S3 at http:// www.genetics.org/supplemental/).
ROBUSTNESS OF THE ESTIMATION PROCEDURES
The extent to which a statistical procedure yields use-ful parameter estimates typically depends on the ap-propriateness of the assumptions to a given data set. Accordingly, I assessed the properties of the parameter estimates when some of the assumptions of the models are violated.
I used thek-allele mutation model to describe the mu-tation process in the estimation procedure described above. The infinite-allele model (Kimura and Crow
1964) can be employed using Equation 4 by settingkto a large (and effectively infinite) value. An alternative mu-tation model that may be appropriate for some markers such as microsatellite sequences is the stepwise muta-tion model (Ohtaand Kimura1973). The stepwise
and posits that mutation occurs between allelic states that are adjacent in the ordering. Accordingly, I applied the estimation procedure to data generated using the simulation approach described above for the finite-island model except that I implemented mutation according to two simple kinds of stepwise mutation models: a step-wise mutation model with no bounds on allele length (unbounded stepwise mutation model; Ohtaand Kimura
1973) and a stepwise mutation model with lower and upper bounds on allele length (bounded stepwise mu-tation model; constrained to eight allelic states). I simu-lated genotype data under the finite-island model with dioecious populations over a range of migration rates (0.02, 0.05, 0.10, and 0.20) and effective population sizes (10, 20, and 50) at a mutation rate ofu¼0.0005. Hence, each generation every gene mutates to an adja-cent allele with probabilityu, mutating to either
neigh-boring allele with equal probability. In the bounded stepwise mutation model, genes at the lower bound mutate toward the upper bound and genes at the upper bound mutate toward the lower bound. The simulations were carried out using 30 independent eight-allele loci (with equally frequent alleles) at which 100 offspring were genotyped from each of 20 demes after 6000 gen-erations. Simulated data were combined over loci to calculateQˆ1ðtÞ;Qˆ2ðtÞ;andQˆ3ðtÞ:I setk¼10,000 to mimic
the infinite-allele mutation model for parameter estima-tion using data generated from the unbounded stepwise mutation model, and I setk¼8 for parameter estima-tion using data generated from the bounded stepwise mutation model.
The accuracy (when comparing the medians of rep-licate estimates to their parametric values) of estimates of migration rate and effective population size for data
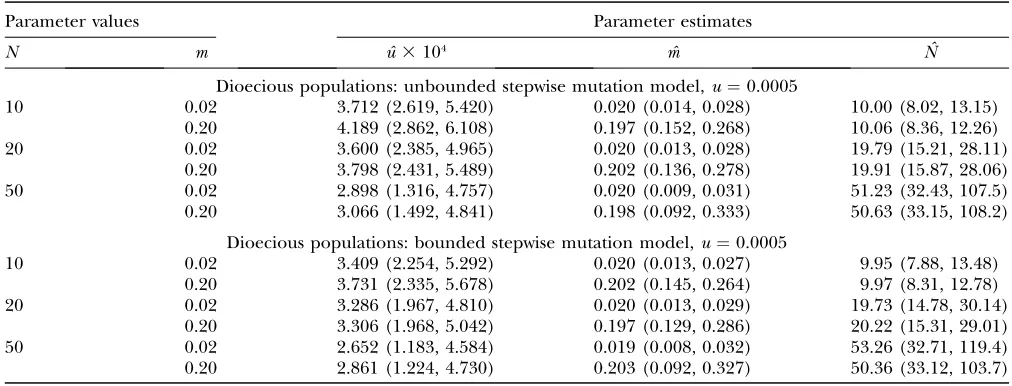
TABLE 2
Medians (5th, 95th percentiles) of the estimates of mutation rate, migration rate, and effective population size using genotype data simulated under the finite-island model with thek-allele mutation model at different values of mutation
rate,u, migration rate,m, and effective population size,N, based on 400 replicate simulations for each pair of model migration rate and effective population size parameters for dioecious (u¼0.001,
u¼0.0005, andu¼0.0001) and monoecious (u¼0.0005) model populations
Parameter values Parameter estimates
N m uˆ 3103 mˆ Nˆ
Dioecious populations:k-allele mutation model,u¼0.001
10 0.02 0.993 (0.693, 1.343) 0.019 (0.013, 0.027) 9.97 (8.36, 12.53)
0.20 1.003 (0.716, 1.425) 0.201 (0.155, 0.260) 10.02 (8.60, 11.80)
20 0.02 1.005 (0.675, 1.348) 0.020 (0.014, 0.026) 20.09 (15.83, 26.87)
0.20 1.005 (0.732, 1.371) 0.202 (0.152, 0.257) 19.92 (16.72, 24.95)
50 0.02 0.989 (0.523, 1.507) 0.019 (0.011, 0.029) 50.83 (35.17, 88.58)
0.20 0.971 (0.569, 1.524) 0.196 (0.112, 0.305) 50.82 (35.10, 83.54)
Parameter values Parameter estimates
N m uˆ 3104 mˆ Nˆ
Dioecious populations:k-allele mutation model,u¼0.0005
10 0.02 4.937 (3.299, 7.462) 0.019 (0.014, 0.028) 10.09 (8.12, 12.76)
0.20 5.100 (3.287, 7.125) 0.200 (0.144, 0.270) 9.89 (8.50, 12.52)
20 0.02 5.038 (3.355, 7.123) 0.020 (0.014, 0.026) 19.97 (15.30, 27.83)
0.20 4.914 (3.413, 7.067) 0.198 (0.145, 0.265) 19.86 (16.50, 25.72)
50 0.02 5.018 (2.529, 8.093) 0.020 (0.010, 0.030) 49.36 (33.45, 98.02)
0.20 5.034 (2.521, 7.748) 0.199 (0.098, 0.314) 49.98 (34.53, 92.92)
Dioecious populations:k-allele mutation model,u¼0.0001
10 0.02 0.970 (0.423, 1.677) 0.019 (0.010, 0.028) 9.94 (7.59, 16.63)
0.20 0.962 (0.354, 1.734) 0.200 (0.105, 0.327) 9.97 (7.47, 15.78)
20 0.02 0.977 (0.444, 1.610) 0.019 (0.010, 0.030) 19.68 (13.78, 35.61)
0.20 1.025 (0.446, 1.824) 0.198 (0.106, 0.339) 19.92 (13.19, 35.27)
50 0.02 0.919 (0.096, 1.809) 0.018 (0.002, 0.034) 53.17 (29.94, 447.1)
0.20 0.933 (0.250, 1.736) 0.191 (0.056, 0.374) 51.81 (31.04, 172.7)
Monoecious populations:k-allele mutation model,u¼0.0005
10 0.02 4.965 (3.299, 6.878) 0.019 (0.014, 0.026) 10.07 (8.69, 12.34)
0.20 5.046 (3.356, 7.129) 0.198 (0.151, 0.260) 10.08 (8.77, 11.84)
20 0.02 4.985 (3.266, 7.065) 0.020 (0.013, 0.027) 19.83 (15.65, 27.23)
0.20 4.836 (3.499, 7.004) 0.196 (0.143, 0.266) 20.17 (16.13, 26.19)
50 0.02 5.108 (2.216, 7.818) 0.020 (0.009, 0.030) 49.68 (33.53, 110.1)
generated under the stepwise mutation models is sim-ilar to that observed under thek-allele mutation model (Figure 4; Table 3), despite the fact that the k-allele mutation model is the assumed mutation process in Equation 4. In particular, the medians of the estimates ofmandNare close to their respective parametric values. The estimates ofmandNfor data generated under the stepwise mutation models (Figure 4; Table 3) exhibit levels of precision similar to, but slightly lower than, the estimates of those parameters for data generated under the equivalentk-allele mutation model (Figure 2B; Table 2). Hence, the estimates ofmandNbased on Equation 4 are somewhat robust to violations in the assumptions of the mutation model. In contrast, the estimates of mu-tation rate, when summarized using their medians, are negatively biased (Table 3), suggesting that the mutation rate estimates are sensitive to violations in the assump-tions of the mutation model in the estimation procedure. Additional simulation results are in supplemental Table S4 at http://www.genetics.org/supplemental/.
The assumption of temporal equilibrium in the prob-ability of identity recursion equations is used to estimate mutation rate, migration rate, and effective population size. To assess this assumption, I applied the estimation procedure to genotype data simulated under nonequi-librium conditions using the infinite-island model with no mutation and the finite-island model with mutation for dioecious populations with parameter values m ¼ 0.05 andN¼20. For the finite-island model, I simulated data under thek-allele mutation model, the unbounded stepwise mutation model, and the bounded stepwise mutation model using a mutation rate ofu¼0.0001 (the value of the mutation rate requiring the most genera-tions to reach temporal equilibrium). At these param-eter values, the infinite-island model is very close to temporal equilibrium after 50 generations, whereas the
finite-island model withk-allele mutation requires10,000 generations to reach equilibrium to four decimal places. Diploid genotypes were initialized using random pairs of alleles, the life cycle was iterated for 10, 20, 50, 100, or 200 generations, and genotypes were then sampled prior to migration (100 offspring genotyped at 30 eight-allele loci in each of 20 demes).
The accuracy of the estimates of mutation rate, mig-ration rate, and effective population size, measured using the median of replicate estimates relative to their parametric values, increases as the number of gener-ations increases from 10 to 200 for both the infinite- and the finite-island models (Figure 5). The estimates of u under the finite-island models are strongly positively biased at 10–200 generations (Figure 5A). In contrast, the estimates ofmunder the infinite-island model and the finite-island model with unbounded stepwise muta-tion are positively biased at 10 generamuta-tions, but are nearly unbiased after $20 generations (Figure 5B). The estimates ofmunder the finite-island models with k-allele and bounded stepwise mutation models are neg-atively biased, but the bias is relneg-atively small, especially after$100 generations (Figure 5B). The estimates ofN exhibit the least bias among the estimated parameters, indicating essentially no bias in the infinite-island model and the finite island model with unbounded stepwise mutation at$10 generations and a small negative bias in the finite-island models with k-allele and bounded stepwise mutation models after 10 and 20 generations and very little bias for$50 generations (Figure 5C). The precision (measured via 5th and 95th percentiles in Figure 5) of the estimates ofmandNis high relative to estimates of those parameters under equilibrium condi-tions (see supplemental Table S2 at http://www.genetics. org/supplemental/: u ¼ 0.0001, m ¼ 0.05, N ¼ 20). Hence, the estimation procedures do not require Figure3.—Sampling distributions of parameter estimates. (A) Sampling distribution of estimates of mutation rate,u. (B) Sampling distribution of estimates of migration rate,m. (C) Sampling dis-tribution of estimates of effective population size,
N. (D) Estimates of effective population size,N, graphed against estimates of migration rate, m. Estimates are from data simulated under the fi-nite-island model for dioecious populations with
k-allele mutation using parameters u ¼ 0.0005,
equilibrium conditions to provide reasonable estimates of migration rate and effective population size, and the nonequilibrium conditions explored here actually in-crease the precision of these estimates. In contrast, the nonequilibrium conditions examined here result in inaccurate estimates of the mutation rate. Simulations suggest that at least 4000 generations under the finite-island model with k-allele mutation are required to obtain accurate estimates of the mutation rate whenu¼ 0.0001,m¼0.05, andN¼20.
The estimation of parameters using Equation 4 assumes thatk, the number of possible allelic states at a locus, is known. In practice,kmight be estimated using the total number of alleles observed in all of the data for each locus, and there would be uncertainty in its value. Ac-cordingly, using data generated under the finite-island models (100 offspring genotyped at 30 eight-allele loci in each of 20 demes after 6000 generations; hencek¼8) withk-allele mutation, unbounded stepwise mutation,
and bounded stepwise mutation (u¼0.0005,m¼0.05, N ¼20), I calculated estimates of mutation rate, mig-ration rate, and effective population size using values for kin Equation 4 equal to 2, 4, 8, 16, and 100.
The medians of the estimates of migration rate and effective population size are close to their parametric values over the range of assumed values ofk(2, 4, 8, 16, and 100), but the medians of the estimates of mutation rate deviate from the parametric values for most of the as-sumed values ofk, with most cases exhibiting negative bias. The precision (measured via 5th and 95th per-centiles) of the estimates ofmandNis similar over the range of assumed values of kwith the exception that estimates ofmare slightly less precise fork¼2 under the k-allele mutation model. Estimates ofu for k¼2 are quite variable and some are very near zero; otherwise the precision of the estimates ofuis similar across the different assumed values ofk. Hence, estimates ofmand N are robust to uncertainty in the value of k, and, in contrast, estimates ofuare more sensitive to deviations from the parametric value ofk.
DISCUSSION
Population geneticists have actively studied the idea that demographic parameters such as migration rate and effective population size might be estimable from genetic data (e.g., Slatkin1985; Waples1989; Pudovkin
et al.1996; Beerliand Felsenstein2001; Vitalisand
Couvet 2001; Wang and Whitlock 2003; Robledo
-Arnuncio et al.2006). Using the classic island model
(Wright1951), I report that migration rate and
effec-tive population size can be jointly estimated from prob-abilities of identity using neutral markers in dioecious or monoecious populations when offspring genotypes are collected prior to migration from a single genera-tion at a single point in time. The life cycle and sampling model are appropriate for highly fecund organisms with localized mating, including species of invertebrates, amphibians, fishes, and plants; hence the method has the potential for broad taxonomic utility.
The estimation procedure works because assuming a dioecious population—or monoecious populations with no selfing—in which offspring genotypes are sampled prior to migration has the consequence thatQ1(t)6¼Q2(t) and hence provides additional information that is not available for other mating systems and sampling schemes that result inQ1(t)¼Q2(t)(e.g., random selfing resulting in random pairing of gametes from all adults during mating, including pairing of gametes from the same adult; Maruyama 1970; Nei and Feldman 1972; Nagylaki
1983; Crow and Aoki 1984; Epperson 1999) orQ1(t)
very nearly equal toQ2(t)(e.g., the finite-island model un-der a postmigration census). Previous studies in pop-ulation ecology (e.g., Caswell2001) and genetics (e.g.,
Nagylaki 1983; Waples 1989; Vitalis 2002) have
recognized that the mating system and/or timing of Figure4.—Finite-island model: stepwise mutation models.
sampling can affect the interpretation of demographic quantities, but the application of these ideas to the present scenario of joint estimation of migration rate and effective population size using a sample from a single generation collected at a single point in time seems not to have been analyzed in prior investigations. Several studies have examined recursions for probabil-ities of identity in state that are similar to those used here, but these studies do not identify the estimation procedures developed here. In theappendix, I outline
how various recursions for probabilities of identity that have been studied (Maruyama1970; MaynardSmith
1970; Nei and Feldman 1972; Nagylaki 1983; Crow
and Aoki 1984; Epperson 1999; Vitalisand Couvet
2001; Vitalis2002; Ballouxet al.2003) can be derived
under the pre- and postmigration census schemes, thus helping to explain the different forms of these equa-tions that occur in the literature. Indeed, Vitalisand
Couvet(2001) estimatemandNusing probabilities of
identity, and their Equation 5 with no selfing is equi-valent to the infinite-island model withk-allele mutation under a premigration census as defined here; but Vitalis
and Couvet(2001) assume an infinite-island model with
mutation among an infinite number of alleles, and they use the approximationFST1=ð114mNÞalong with a two-locus identity measure (a fourth-moment quantity) rather than the single-locus quantities FITand FSTor Q1(t),Q2(t), andQ3(t)(all second-moment quantities).
Like other genetic methods for estimating demographic parameters (e.g., Waples 1989; Pudovkin et al.1996;
Williamsonand Slatkin1999; Beerliand Felsenstein
2001; Vitalisand Couvet2001; Wangand Whitlock
2003), the procedure described here will typically
re-quire considerable data to recover accurate and precise estimates. The accuracy and precision achieved will, in general, depend on several factors, including the values of the parametersu,m, andN, as well as the number of demes, number of individuals genotyped, number of loci used, and the appropriateness of the model to the empirical system under investigation. Previous studies (Waples1989; Pudovkinet al.1996; Williamsonand
Slatkin 1999; Vitalisand Couvet 2001; Wangand
Whitlock2003) have shown that effective population
size is more difficult to estimate asNincreases, and my simulation results are consistent with the findings in these earlier studies. Indeed, my results suggest thatQ1(t)and Q2(t) become increasingly similar as N increases, with the consequence that estimates ofN approach infinity asQˆ1ðtÞ andQˆ2ðtÞ become equal. Further, negative
esti-mates ofNare possible with Equation 2 ifQˆ1ðtÞ.Qˆ2ðtÞ:
Because effective population size influences genetic quantities via the function 1/Nin standard models, it is not surprising that larger population sizes are more difficult to estimate with genetic data because this in-volves estimating an effect of magnitude1/N—a small number for even moderately sizedN. Hence, genetic-based methods for estimating effective population size work best for small populations, and the numbers of loci and individuals genotyped must increase with increas-ing N to maintain a given level of precision (Waples
1989; Pudovkin et al.1996; Williamsonand Slatkin
1999; Vitalisand Couvet2001; Wangand Whitlock
2003). Accordingly, the method presented here is most likely to be useful for populations with a metapopulation structure defined by many small demes. Detailed guid-ance on the accuracy and precision of the estimators for
TABLE 3
Medians (5th, 95th percentiles) of the estimates of mutation rate, migration rate, and effective population size using genotype data simulated under the finite-island model with stepwise mutation models (employing either unbounded or bounded
ranges in allele lengths;u¼0.0005) at different values of migration rate,m, and effective population size,N, based on 400 replicate simulations for each pair of model migration rate and effective population size parameters for
dioecious model populations
Parameter values Parameter estimates
N m uˆ3104 mˆ Nˆ
Dioecious populations: unbounded stepwise mutation model,u¼0.0005
10 0.02 3.712 (2.619, 5.420) 0.020 (0.014, 0.028) 10.00 (8.02, 13.15)
0.20 4.189 (2.862, 6.108) 0.197 (0.152, 0.268) 10.06 (8.36, 12.26)
20 0.02 3.600 (2.385, 4.965) 0.020 (0.013, 0.028) 19.79 (15.21, 28.11)
0.20 3.798 (2.431, 5.489) 0.202 (0.136, 0.278) 19.91 (15.87, 28.06)
50 0.02 2.898 (1.316, 4.757) 0.020 (0.009, 0.031) 51.23 (32.43, 107.5)
0.20 3.066 (1.492, 4.841) 0.198 (0.092, 0.333) 50.63 (33.15, 108.2)
Dioecious populations: bounded stepwise mutation model,u¼0.0005
10 0.02 3.409 (2.254, 5.292) 0.020 (0.013, 0.027) 9.95 (7.88, 13.48)
0.20 3.731 (2.335, 5.678) 0.202 (0.145, 0.264) 9.97 (8.31, 12.78)
20 0.02 3.286 (1.967, 4.810) 0.020 (0.013, 0.029) 19.73 (14.78, 30.14)
0.20 3.306 (1.968, 5.042) 0.197 (0.129, 0.286) 20.22 (15.31, 29.01)
50 0.02 2.652 (1.183, 4.584) 0.019 (0.008, 0.032) 53.26 (32.71, 119.4)
specific empirical scenarios can be obtained using simulations. Source code used to simulate data under the infinite-island model with no mutation and the finite-island model withk-allele mutation is available at http://www.genetics.org/supplemental/.
The simulation results suggest that the estimates of migration rate and effective population size are some-what robust to violations of the model assumptions. Reasonable estimates ofmand N can be obtained for loci exhibiting stepwise mutation (Ohta and Kimura
1973), under nonequilibrium conditions, or if the number of possible allelic states is not precisely known.
In contrast, estimates of the mutation rate,u, are sen-sitive to violations in the assumptions and can be quite biased in these settings. The precision of the estimates ofmandNis higher for data simulated with a high mu-tation rate and for data simulated under nonequilibrium conditions, suggesting that the procedure works better at higher levels of genetic diversity. The estimation procedure under the finite-island model requires that the number of demes,s, be known, but it does not re-quire that all demes be sampled because all demes are identical in island models. In non-island models, the set of demes that is exchanging migrants generally must be known to estimate migration rates among the demes (Beerliand Felsenstein2001; Wangand Whitlock
2003; Slatkin2005).
Many studies investigate the estimation of demographic parameters from genetic data (Slatkin 1985; Waples
1989; Pudovkinet al.1996; Wangand Whitlock2003;
Robledo-Arnuncioet al.2006); however, few methods
exist for jointly estimating parameters like migration rate and effective population size from genetic data col-lected from a sample taken from a single generation at a single point in time (Beerliand Felsenstein 1999,
2001; Vitalis and Couvet 2001). For example, the
product parameter mN can be estimated from single-generation data onFSTunder the infinite-island model (Slatkin1985), and effective population size alone can
be estimated from multiple samples on allele frequen-cies from two or more generations (Waples1989) or from
a single sample of offspring assuming unrelated parents using heterozygote excess (Pudovkinet al.1996). Vitalis
(2002) and Fontanillaset al.(2004) use two samples
(both pre- and postmigration samples) to estimate mig-ration rate alone using F-statistics under the infinite-island model with sex-specific dispersal. Extending the idea in Waples(1989), if allele frequency data are
avail-able from multiple samples from multiple generations from two or more demes, then migration rate and ef-fective population size can be jointly estimated (Wang
and Whitlock2003). Using data from a single
gener-ation, the method of Beerliand Felsenstein(2001)
estimates the deme-specific product parameters 4uN andm/uforsdemes under a general migration scheme under the assumption that effective population size is sufficiently large so that the coalescent model of genetic drift is appropriate and that m and u are sufficiently small so that the quantitiesmNanduNremain finite as N goes to infinity. In a two-deme version of their coa-lescent procedure, Beerliand Felsenstein(1999)
ini-tially estimate 4uNand m/u using moment estimators based on the probability-of-identity equations of Nei
and Feldman(1972), which can be derived for randomly
mating monoecious populations under a postmigration census scheme. The method of Vitalisand Couvet
(2001) uses one- and two-locus probabilities of identity to estimatemandNunder the infinite-island model with infinite-allele mutation and random selfing, assuming Figure5.—Estimation under nonequilibrium conditions.
(A) Estimates of mutation rate,u, (on the log scale) as a func-tion of number of generafunc-tions using data generated under finite-island models with different mutation models for dioe-cious populations. (B) Estimates of migration rate, m, as a function of number of generations using data generated un-der the infinite-island model with no mutation and finite-is-land models with different mutation models for dioecious populations. (C) Estimates of effective population size,N, as a function of number of generations using data generated under the inisland model with no mutation and finite-island models with different mutation models for dioecious populations. The medians (symbols) and 5th and 95th per-centiles (error bars) of estimates from 400 replicate simula-tions are plotted for each model at generasimula-tions 10, 20, 50, 100, and 200. Dashed lines denote the parameter values
that u ¼0 and m is sufficiently small so that the ap-proximation FST1=ð114mNÞ might be valid. In a somewhat different demographic scenario that tackles the same issues, if migration occurs via the dispersal of male gametes and genotype data are available from off-spring and their mothers (e.g., pollen dispersal with genotype data from seeds and their mother plant), then the gamete dispersal curve can be estimated indepen-dently of effective population density by making use of probabilities of identity, and an approximate estimate of effective population density can also be calculated (Robledo-Arnuncioet al.2006). The method of Robledo
-Arnuncioet al.(2006) is nonequilibrium in the sense
that it does not model mutation and it estimates dis-persal in the most recent generation assuming that par-ents are unrelated. Under a demographic model of admixture of previously separated demes (vs.demes ex-hibiting continuous migration and drift), computation-ally intensive Bayesian procedures based on coalescent models have been used to estimate demographic param-eters (e.g., the admixture proportion) that are consistent with a set of observed summary statistics, including es-timates ofFST(Estoupet al.2001; Excoffieret al.2005). Under the standard coalescent model, only the product parameter uN is estimable unless additional infor-mation onu(orN) is available (Estoupet al.2001) or
the sampling scheme and demography mimic samples taken from the same deme over different generations (cf. Waples1989; Excoffieret al.2005). The results from
these studies illustrate the challenges of estimating de-mographic parameters from genetic data.
The method I describe here requires only a sample from a single generation at a single point in time; it can jointly estimate mutation rate, migration rate, and ef-fective population size; it is relatively simple computa-tionally and, given the parametric model, need not make assumptions concerning the values of parameters that might be estimated; but, at present, it has not been de-veloped to accommodate more general demographic situations. However, it may be possible to extend the method to include other demographic and genetic sce-narios, such as a time series of samples (Wang and
Whitlock2003), stepping-stone dispersal, more general
migration models (e.g., Beerliand Felsenstein2001),
deme-specific effective population sizes, and other mu-tation models (e.g., Laiand Sun 2003). More general
forms of the model can lead to additional (but still linear) recursions for the probabilities of identity in state, but if the probability of identity within individuals remains different from the probability of identity among indi-viduals within demes in these more general settings, then information may be available to jointly estimate migration rates and effective population sizes in more detailed models.
I thank Mark Holder, Steve Hudman, John Kelly, Rasmus Nielsen, Bruce Weir, and two anonymous reviewers for assistance, conversa-tions, and/or comments concerning this research. I acknowledge
funding from the University of Kansas and the National Science Foundation (DEB 06-09722).
LITERATURE CITED
Balloux, F., L. Lehmannand T.deMeeuˆ s, 2003 The population genetics of clonal and partially clonal diploids. Genetics 164: 1635–1644.
Beerli, P., and J. Felsenstein, 1999 Maximum-likelihood estima-tion of migraestima-tion rates and effective populaestima-tion numbers in two populations using a coalescent approach. Genetics 152: 763–773.
Beerli, P., and J. Felsenstein, 2001 Maximum likelihood estima-tion of a migraestima-tion matrix and effective populaestima-tion size inn sub-populations by using a coalescent approach. Proc. Natl. Acad. Sci. USA98:4563–4568.
Caswell, H., 2001 Matrix Population Models: Construction, Analysis,
Interpretation.Sinauer Associates, Sutherland, MA.
Crow, J. F., and K. Aoki, 1984 Group selection for a polygenic be-havioral trait: estimating the degree of population subdivision. Proc. Natl. Acad. Sci. USA81:6073–6077.
Epperson, B. K ., 1999 Gene genealogies in geographically struc-tured populations. Genetics152:797–806.
Estoup, A., I. J. Wilson, C. Sullivan, J. Cornuetand C. Moritz, 2001 Inferring population history from microsatellite and en-zyme data in serially introduced cane toads,Bufo marinus. Genet-ics159:1671–1687.
Excoffier, L., A. Estoupand J. Cornuet, 2005 Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics169:1727–1738.
Fontanillas, P., E. Petitand N. Perrin, 2004 Estimating sex-specific dispersal rates with autosomal markers in hierarchically struc-tured populations. Evolution58:886–894.
Ha¨ nfling, B., and D. Weetman, 2006 Concordant genetic estima-tors of migration reveal anthropogenically enhanced source-sink population structure in the river sculpin,Cottus gobio.Genetics 173:1487–1501.
Kimura, M., and J. Crow, 1964 The number of alleles that can be maintained in a finite population. Genetics49:725–738. Lai, Y., and F. Sun, 2003 The relationship between microsatellite
slippage mutation rate and the number of repeat units. Mol. Biol. Evol.20:2123–2131.
Maruyama, T., 1970 Effective number of alleles in a subdivided pop-ulation. Theor. Popul. Biol.1:273–306.
MaynardSmith, J., 1970 Population size, polymorphism, and the rate of non-Darwinian evolution. Am. Nat.104:231–237. Nagylaki, T., 1983 The robustness of neutral models of geographic
variation. Theor. Popul. Biol.24:268–294.
Nei, M., and M. W. Feldman, 1972 Identity of genes by descent within and between populations under mutation and migration pressures. Theor. Popul. Biol.3:460–465.
Ohta, T., and M. Kimura, 1973 A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet. Res.22:201–204.
Pudovkin, A. I., D. V. Zaykinand D. Hedgecock, 1996 On the po-tential for estimating the effective number of breeders from het-erozygote-excess in progeny. Genetics144:383–387.
Robledo-Arnuncio, J. J., F. Austerlitzand P. E. Smouse, 2006 A new method of estimating the pollen dispersal curve indepen-dently of effective density. Genetics173:1033–1045.
Rousset, F., 2001 Inferences from spatial population genetics, pp. 239–269 in Handbook of Statistical Genetics, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, New York.
Slatkin, M., 1985 Gene flow in natural populations. Annu. Rev. Ecol. Syst.16:393–430.
Slatkin, M., 2005 Seeing ghosts: the effect of unsampled popula-tions on migration rates estimated for sampled populapopula-tions. Mol. Ecol.14:67–73.
Vitalis, R., 2002 Sex-specific genetic differentiation and coales-cence times: estimating sex-biased dispersal rates. Mol. Ecol. 11:125–138.
Wang, J., and M. C. Whitlock, 2003 Estimating effective popula-tion size and migrapopula-tion rates from genetic samples over space and time. Genetics163:429–446.
Waples, R. S., 1989 A generalized approach for estimating effective population size from temporal changes in allele frequency. Ge-netics121:379–391.
Whitlock, M. C., and D. E. McCauley, 1999 Indirect measures of gene flow and migration:FST6¼1/(4Nm11). Heredity82:117–125.
Williamson, E. G., and M. Slatkin, 1999 Using maximum likeli-hood to estimate population size from temporal changes in allele frequencies. Genetics152:755–761.
Wright, S., 1951 The genetical structure of populations. Ann. Eu-gen.15:323–354.
Communicating editor: R. Nielsen
APPENDIX
I consider parametric expressions involvingQ1(t),Q2(t), andQ3(t), the probabilities of identity in allelic state within one selectively neutral locus at timet, in ans-deme finite-island model with genetic drift, migration, and mutation. I adopt part of the derivation strategy of Nagylaki(1983) and outline the life cycle for the models that I consider.
Starting withNdiploid, monoecious adults in each ofsdemes, reproduction begins with each adult producing a large (i.e., infinite) number of haploid gametes. The allele in each gamete may then mutate into a different allele according to a general mutation model. The reproduction phase is completed by the random pairing of gametes from different adults within demes; no offspring are produced using two gametes from the same adult (i.e., no selfing occurs). Offspring then migrate among demes so that, following migration, a fractionmof the individuals in a deme are migrants and a fraction 1mof the individuals are residents. Population regulation completes the life cycle withN offspring chosen at random within each deme to compose the adults that will produce the next generation. The life cycle just described is the diploid dispersion life cycle considered by Nagylaki(1983). The equilibrium results that
follow also apply to dioecious populations when males and females are equal in number (the populations within demes are regulated toN/2 females andN/2 males so that the total adult effective population size isN), migrate at the same rate, and experience the same mutation model.
Because demographic and genetic measures can depend on the timing of sampling within the life cycle (e.g., Waples 1989; Caswell 2001; Vitalis 2002), I consider two census schemes, a premigration census and a
postmigration census. Assuming pre- and postmigration census schemes for dioecious populations with sex-specific dispersal following the infinite-allele mutation model in a finite number of demes, Vitalis(2002) gives recursions for
probabilities of identity by descent (premigration, Equation A1.4; postmigration, Equation A1.1; Vitalis2002) that
can be readily modified to obtain the results that follow. Note that the second and third columns in the matrixA following Equation A1.1 in Vitalis(2002) should have terms like (12/N) consistent with Equation 4 in that article,
rather than terms like (11/N).
First, I consider the premigration census under the infinite-island model. Under the premigration census, the sampling of offspring occurs immediately following reproduction and prior to migration. LetQ1(t),Q2(t), andQ3(t)be the probabilities (summed overkalleles at one locus) that genes within individuals, between individuals within a deme, and between individuals between demes are the same allele at timet, respectively. Under the premigration census scheme, Equation A1.4 of Vitalis(2002) modified for an infinite number of demes with no mutation and no
sex-specific dispersal yields, at temporal equilibrium, the recursions
Q1ðt11Þ¼Q1ðtÞ¼ ð1mÞ2Q2ðtÞ1½1 ð1mÞ2Q3ðtÞ
Q2ðt11Þ¼Q2ðtÞ¼
1
2Nð11Q1ðtÞÞ1ð1mÞ
2 1 1
N
Q2ðtÞ1½1 ð1mÞ2 1
1 N
Q3ðtÞ
Q3ðt11Þ¼Q3ðtÞ¼Q3ðtÞ:
Solving forQ1(t)andQ2(t)yields
Q1ðtÞ¼
ð1mÞ2
1 ð1mÞ2ð1 ð1=2
NÞÞ 1 2N
1 ½1 ð1mÞ
2
1 ð1mÞ2ð1 ð1=2
NÞÞQ3ðtÞ
Q2ðtÞ¼
1
1 ð1mÞ2ð1 ð1=2NÞÞ
1 2N
1 ½1 ð1mÞ
2
1 ð1mÞ2ð1 ð1=2NÞÞ 1
1 2N
Accordingly, the parametersFITandFST(Wright1951) are given by
FIT¼
Q1Q3
1Q3
¼ ð1mÞ
2
½1 ð1mÞ22N1ð1mÞ2
ð12mÞ
ð12mÞ14mN
FST¼
Q2Q3
1Q3
¼ 1
½1 ð1mÞ22
N1ð1mÞ2
1
ð12mÞ14mN;
where the approximation omits terms proportional tom2. Equation 14 in Vitalis(2002) assuming no mutation, an
infinite number of demes, and no sex-specific dispersal is the same as the equation forFSTgiven above, but Vitalis (2002) does not report an expression for FIT. Thus, migration rate, m, and effective population size, N, can be expressed exactly in terms ofFITandFSTvia
m¼1
ffiffiffiffiffiffiffi
FIT
FST
r
¼1
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
Q1Q3
Q2Q3
s
N ¼ 1FIT
2ðFSTFITÞ
¼ 1Q1
2ðQ2Q1Þ
:
Equation 16 in Vitalis(2002) gives an expression for sex-specific dispersal rates similar to the expression formgiven
here, but, importantly, the former is a function ofFSTfrom a sequence of samples taken pre- and postmigration, rather than a function ofFITandFSTfor a single sample as given here. Vitalis(2002) does not report an expression forN.
Under the postmigration census scheme, Equation A1.1 of Vitalis(2002) modified for an infinite number of
demes with no mutation and no sex-specific dispersal yields, at temporal equilibrium, the recursions
Q1ðt11Þ¼Q1ðtÞ¼Q2ðtÞ
Q2ðt11Þ¼Q2ðtÞ¼ ð1mÞ2
1
2Nð11Q1ðtÞÞ1ð1mÞ
2 11
N
Q2ðtÞ1½1 ð1mÞ2Q3ðtÞ
Q3ðt11Þ¼Q3ðtÞ¼Q3ðtÞ:
Solving forQ1(t)andQ2(t)gives
Q1ðtÞ¼Q2ðtÞ¼
ð1mÞ2
1 ð1mÞ2ð1 ð1=2NÞÞ
1 2N
1 ½1 ð1mÞ
2
1 ð1mÞ2ð1 ð1=2NÞÞQ3ðtÞ:
In this case, becauseQ1(t)¼Q2(t), the parametersFITandFSTare given by
FIT¼
Q1Q3
1Q3
¼FST
FST¼
Q2Q3
1Q3
¼ ð1mÞ
2
½1 ð1mÞ22
N1ð1mÞ2
ð12mÞ
ð12mÞ14mN;
where the approximation omits terms proportional tom2. Equation 12 in Vitalis(2002) assuming no mutation and
an infinite number of demes is the same as the equation forFSTgiven above, but Vitalis(2002) does not report an expression forFIT. Hence, unlike the premigration census, the parametersmandNcannot be uniquely determined fromFITandFSTunder a postmigration census.
Under the finite-island model with ak-allele mutation scheme, the number of demes,s, is finite, and each gene occupies one ofkallelic states, mutates with probabilityuper generation, and, given a mutation event, mutates to each of the otherk1 alleles with equal probability. Under a premigration census, modifying Equation A1.4 of Vitalis
(2002) to assume the finite-island model with ak-allele mutation scheme with no sex-specific dispersal yields, at temporal equilibrium, the recursions
Q1ðt11Þ¼Q1ðtÞ¼U21ðU1U2Þ½M1Q2ðtÞ1ð1M1ÞQ3ðtÞ
Q2ðt11Þ¼Q2ðtÞ¼
U1
2N 1 1 1 2N
U21
1
2NðU1U2ÞQ1ðtÞ
1M1 1
1 N
ðU1U2ÞQ2ðtÞ1ð1M1Þ 1
1 N
ðU1U2ÞQ3ðtÞ
where
M1 ¼ ð1mÞ21
m2
s1
M2 ¼
1M1
s1
U1 ¼ ð1uÞ21
u2
k1
U2 ¼
1U1
k1:
The above equations, being linear, can be solved explicitly for the equilibrium values ofQ1(t),Q2(t), andQ3(t). However, because I could not identify a simple form for the resulting expressions, I do not list them here. Under a postmigration census, modifying Equation A1.1 of Vitalis(2002) to assume the finite-island model with ak-allele mutation scheme
with no sex-specific dispersal yields, at temporal equilibrium, the recursions
Q1ðt11Þ¼Q1ðtÞ¼U21ðU1U2ÞQ2ðtÞ
Q2ðt11Þ¼Q2ðtÞ¼M1
U1
2N 1 1 1 2N
U2
1M1
2NðU1U2ÞQ1ðtÞ
1M1 1
1 N
ðU1U2ÞQ2ðtÞ1ð1M1ÞðU1U2ÞQ3ðtÞ
Q3ðt11Þ¼Q3ðtÞ¼M2
U1
2N 1 1 1 2N
U2
1M2
2NðU1U2ÞQ1ðtÞ
1M2 1
1 N
ðU1U2ÞQ2ðtÞ1ð1M2ÞðU1U2ÞQ3ðtÞ:
Hence, at temporal equilibrium under a postmigration census,Q1(t)is nearly equal toQ2(t)because the mutation rate, u, is typically a very small number. Thus, the estimation ofu,m, andNunder a postmigration census should be difficult. In the case of dioecious populations (assuming the locus is not sex-linked), probabilities of identity within and between individuals must be specified for male and female pairs of genes so thatQF
1ðtÞandQ1MðtÞare the probabilities
that genes are identical in state within female and male individuals;QFF
2ðtÞ;Q2MMðtÞ;andQ2FMðtÞare the probabilities that
genes are identical in state between two females, between two males, and between a female and a male within a deme; andQFF
3ðtÞ;Q3MMðtÞ;andQ3FMðtÞare the probabilities that genes are identical in state between two females, between two males,
and between a female and a male for individuals in different demes (e.g., Vitalis2002). Under a premigration census,
modifying Equation A1.4 of Vitalis(2002) to have thek-allele mutation model with no sex-specific dispersal yields, at
temporal equilibrium, the dioecious population recursions
Q1Fðt11Þ¼Q1Mðt11Þ¼U21M1ðU1U2ÞQ2FMðtÞ1ð1M1ÞðU1U2ÞQFM3ðtÞ
QFF2ðt11Þ¼QMM2ðt11Þ¼QFM2ðt11Þ¼
U1
2N 1 1 1 2N
U21
1
2NðU1U2Þ 1 2 ðQ
FF
1ðtÞ1Q1MMðtÞÞ
11
2½M1ðU1U2ÞQ
FM
2ðtÞ1ð1M1ÞðU1U2ÞQ3FMðtÞ
1M1
1
4
1 2N
ðU1U2ÞðQ2FFðtÞ1Q2MMðtÞÞ
1ð1M1Þ
1
4
1 2N
ðU1U2ÞðQFF3ðtÞ1QMM3ðtÞÞ
QFF3ðt11Þ¼QMM3ðt11Þ¼QFM3ðt11Þ¼U21M2
1
4 ðU1U2ÞðQ
FF
2ðtÞ1QMM2ðtÞ12QFM2ðtÞÞ
1ð1M2Þ
1
4 ðU1U2ÞðQ
FF
3ðtÞ1Q3MMðtÞ12QFM3ðtÞÞ:
Hence, at temporal equilibrium the probabilities of identity for dioecious populations are identical to those in monoecious populations (with no selfing) when males and females have identical demography.
and Equation 1 of Neiand Feldman(1972) can be derived in the present context by assuming a finite-island model,
mutation among an infinite number of alleles, and monoecious populations with random mating½including random selfing; hence,Q1(t)¼Q2(t)under a postmigration census. Equation 78 of Nagylaki(1983) with zero selfing can be derived by assuming a finite-island model, mutation among an infinite number of alleles, and monoecious populations with no selfing under a postmigration census. Equation 5 of Crowand Aoki(1984) can be derived by
assuming a finite-island model, mutation among k alleles, and monoecious populations with random mating ½including random selfing; hence,Q1(t)¼Q2(t)under a postmigration census. Equation 2 of Epperson(1999) can be derived by assuming a finite-island model with general between-deme migration rates, no mutation, and monoecious populations with random mating under a postmigration census. Recursions forQ1(t) andQ3(t)½a one-generation recursion forQ2(t)is not presentedin MaynardSmith(1970) can be derived by assuming a finite-island model, mutation among an infinite number of alleles, and monoecious populations with no selfing under a premigration census. Equation 5 of Vitalisand Couvet(2001) with zero selfing can be derived by assuming an infinite-island
model, mutation among k alleles, and monoecious populations with no selfing under a premigration census. Equations A1.1 and A1.4 of Vitalis(2002), assuming pre- and postmigration census schemes, respectively, can be
derived assuming dioecious populations with sex-specific dispersal following the infinite-allele mutation model in a finite number of demes. Finally, the juvenile life stage recursions of Ballouxet al.(2003) with no selfing and no clonal