Vol. 3, No. 1 (2015): 550-556 Research Article
Open Access
I
ISSSSNN::22332200--22224466
An Efficient Method for Extraction of Genomic DNA
from Insect Gut Bacteria - Culture dependent
Mani Kannan, Thangaiyan Suganya,
Vimalanathan Arunprasanna, Neelamegam
Rameshkumar
and Muthukalingan Krishnan*
Insect Molecular Biology Laboratory, Department of Environmental Biotechnology, Bharathidasan University, Tiruchirappalli – 620 024, Tamil Nadu, India
* Corresponding author:Muthukalingan Krishnan; email: [email protected]
ABSTRACT
Extraction of high quality genomic DNA from bacteria is the most important step for understanding their microbial physiology, diversity and evolution. In this methodology paper, we have extracted the genomic DNA from culture dependent bacteria using gut bacteria lysis (GBL) buffer in single step, cheap and non-toxic chemical lysis method followed by DNA precipitation. Electrophoresis of the extracted DNA revealed its high quality in sizes above 10 kb without contamination. UV spectrophotometry showed a maximum concentration of 6.2µg / mL (A260/A280 ratio) with purity range of 1.72 (A260/A230 ratio). Further, restriction digestion and
RAPD-PCR analysis confirmed that the DNA subjected for molecular analysis did not exhibit any obstruction. To compare the efficacy of our proposed method, DNA was extracted by a commercial kit (Genomic DNA Extraction kit, Real Genomics (RBC)). DNA isolated by commercial kit has disadvantaged in molecular biology application when compared with the results obtained by our present method. This confirmed that the DNA concentration and purity obtained by our new method was highly suitable for molecular biology applications. Apart from the quality of the DNA, our present method is rapid and uses less toxic chemicals with fewer steps, in consequence of which it should be suitable for microbiological applications.
Keywords:
Insects, Gut Bacteria, Genomic DNA extraction, RAPD-PCR, Restriction enzyme digestion.1.0 INTRODUCTION
The genomes of insects lack many of the catabolic enzymes necessary to digest food fully in order to extract energy. Thus, gut microbes play a pivotal role in the digestion of cellulose, fiber, lignin, starch, tannin etc. to provide energy for the host [1, 2]. Genomics study provides a new avenue for studying the gut microbial communities as it can reveal the relationship between the insect and its microbiome. The extraction of bacterial DNA has diverged into culture dependent and culture independent approaches. At present, there are several reports that describe the direct isolation of DNA from diverse sources like Soil [3], salivary gland [4], dental pulp [5] guts of insect [1, 2, 6, 7] and human [8, 9] for assessing the bacterial communities. At the same time recently there is no simple and rapid method available for the isolation of genomic DNA of gut microbes by a culture dependent method. The
polysaccharides, glycoprotein and lipopolysaccharides have been reported as interfering with the isolation process by co-precipitating with DNA [18]. To overcome this problem, numerous procedures have been developed and subjected to either physical, chemical and/or enzyme-based cell lysis followed by phenol-chloroform extraction, chaotropic-based fractionation or size exclusion column purification for the extraction of high quality genomic DNA [19, 20]. Now a days, several scientific companies offer commercial kits with a lot of advanced technology to accelerate the process of DNA isolation, as an alternative approach to the conventional methods. However, the kits are problematic in their use of toxic chemicals, a large number of steps and solution and buffer composition of which is not apparent to the researchers [21, 22]. Hence, there is a need for a simple and standard protocol for the extraction of genomic DNA. For molecular biology applications the extracted DNA needs to be of sufficient concentration and of high quality [23]. Any significant method for genomic DNA isolation to provide ample yields of pure DNA should be rapid, less laborious and eco-friendly. To emphasize these decisive factors, we have developed a simple method for the extraction of bacterial genomic DNA from insect gut. This could pave the way to identify and characterize novel microbes which may produce enzymes of industrial potential.
2.0 MATERIALS AND METHODS
2.1 Solutions and BuffersStock solutions were prepared as follows: 1 M Tris HCL, 4 M NaCl, 10% SDS, 0.5 % EDTA. 100 mL GBL working buffer solution contained 1 mL 1M Tris, 10 mL 4M NaCl, 0.4 mL 0.5 % EDTA, 6.8 mL 10% SDS and 80.2
mL DDH2O). TE Buffer contained 10 mM Tris HCL and
1 mM EDTA (pH 7.5).
2.2 Isolation of Insect Gut
The gut was isolated from V instar larvae of the
lepidopteran insect, Bombyx mori (i.e. during the active
feeding stage). Prior to dissection, the larvae were sterilized by wiping with 70% ethanol on the cuticle for 5 seconds [6, 7]. Dissection scissors were used to cut laterally behind the head capsule and the gut was removed and washed in 1X PBS to remove the leaf litter. Then, it was transferred into a 1.5 mL micro centrifuge tube for further processing.
2.3 Isolation of Gut Microbiota
The gut was sonicated in ultrasonic baths for 30s, vortexed for 1 min and then centrifuged at 10,000 rpm
for 5 minto separate bacterial cells from the gut wall.
The supernatant was serially diluted up to 10-7; each
dilution was plated on tryptic soybean agar plate (10 % TSA). The plates were incubated for 12-24 h at 37°C [24]. Colonies were picked on the basis of their morphological features and a pure culture was obtained through quadrant streaking. Each colony was sub-cultured twice to check the purity of the culture before DNA extraction. For the assessment of the proposed method, we choose both gram positive and
gram negative bacteria by gram staining [25]. A single colony was inoculated in 5mL of nutrient broth and incubated at 37°C for overnight in a rotating shaker (Scigenics Biotech, ORBITECH, India) in 220 rpm to reach 0.6 OD at 600nm.
2.4 Extraction of Genomic DNA
The bacterial pellet was collected from the overnight culture by centrifugation at 10,000 rpm for 3 min. The pellet was resuspended in 300 µl of GBL buffer by pipetting and vortexing. Immediately, 200 µl of chloroform and 150 µl of 6 M NaCl were added and the tubes were inverted ten times. The mixture was centrifuged at 10,000rpm for 5 min. The supernatant was transferred to a fresh 1.5 ml Eppendorf tube. An equal volume of absolute ethanol was added carefully down the wall of the tube. The mixture was centrifuged again at 10,000 rpm for 5 min and the supernatant discarded. Subsequently, the precipitated DNA was pipette out to a new Eppendorf tube and washed twice with 70 % ethanol by centrifugation at 5000 rpm for 5 minutes. The pellet was air dried at room temperature for 10 minute. Finally the pellet was dissolved in 50-100 µl of TE buffer (pH 8).
2.5 Yield, purity and integrity of DNA
The isolated DNA was subjected to spectrophotometric analysis (Ultrospec 2100, Amersham Bioscience, Hong Kong) to determine the quality and quantity. The DNA purity was determined from the ratios 260/280 nm (indicator of protein contamination) and 260/230nm (indicator of organic solvent residues). The size and intactness of the isolated DNA was checked by agarose gel electrophoresis. The isolated DNA was loaded on 1% agarose gel stained with ethidium bromide (1 µg/µl) and run for 30 min at 60 V. For image acquisition, a LAB India gel documentation system (Infinity, UK) was used. The isolated genomic DNA size was determined by using a 1kb DNA ladder (Biotool, Spain).
2.6 RAPD-PCR analysis
RAPD-PCR reaction was performed in a 50 μl reaction
volume, consisting of: 25 µl of 2X Premix Solution (Genet Bio, Germany) and 6 µl each of 0.5µM RAPD1-5'-GTTTCGCTCC-3' and RAPD2- 5'-GTAGACCCGT-3' primers (Synthesized from Eurofins, Bangalore, India ), 8 μl of water, and 5 µl (50 ng/µl) of genomic DNA. The amplification programme consisted of an initial denaturation at 94°C for 5 min, denaturation at 95°C for 1 min, annealing 28°C for 2 min, and extension at 72°C for 3 min, step 2 extended to 30 cycles and a final extension step of 10 min. The PCR products were resolved on a 1.5 % agarose gel stained with ethidium bromide (1 µg/µl) and run for 30 min at 60 V. Image acquisition, as before used a LAB India gel documentation system.
2.7 Digestion with restriction enzymes
The reaction was carried out in a total volume of 20 μl,
New England, incubated at 37ºC for 3 h. The digested DNA was subjected to 1.5 % of agarose gel stained with ethidium bromide (1 µg/µl), run for 30 min at 60 V and image acquisition was as above.
3.0 RESULTS AND DISCUSSION
Since a variety of commercial kits and modified protocols are reported for the extraction of bacterial
genomic DNA. Only a few of the methods achieve an optimum yield of DNA, but they are unsatisfactory in subsequent molecular biology applications [22]. Moreover, these methods are time consuming, costly and use significant amounts of toxic chemicals. Table 1, exhibited the comparison of commercial kit, standard protocol and proposed method.
Table 1. Comparative methodology for isolation of genomic DNA from insect gut microbes
Commercial Kit Conventional method GBL method
Bacterial Pellet was obtained (3ml culture) by centrifugation for 10 min at 5000 x g(7500 rpm).
Bacterial pellet was suspended in 180 µl of the appropriate enzyme solution (20 mg/ml lysozyme or 200 µg/ml lysostaphin; 20 mMTris·HCl, pH 8.0; 2 mM EDTA; 1.2% Triton).
It was incubated for at least 30 min at 37°C.
A 20 µl of proteinase K and 200 µl Buffer AL was added. It was mixed by vortexing.
It was incubated at 56°C for 30 min and then for a further 15 min at 95°C. Then centrifuged for a few seconds.
First 4 µl of RNase A was added (100 mg/ml), mixed by pulse-vortex for 15 s, and incubated for 2 min at room temperature. It was briefly centrifuged to remove drops from the lid before adding 200 µl Buffer AL to the sample. Again it was mixed by pulse-vortexing for 15 s, and incubated at 70°C for 10 min.
A 200 µl ethanol was added (96–100%) to the sample, and mixed by pulse-vortexing for 15s. After mixing, it was briefly centrifuged to remove drops from inside the lid.
The mixture was carefully applied from step 6 (including the precipitate) to the Commerical kit Mini spin column (in a 2 ml collection tube) without wetting the rim. The cap was closed, and centrifuged at 6000 x g (8000 rpm) for 1 min. The Commercial kit Mini spin column was placed in a clean 2 ml collection tube (provided), and filtrate was discarded.*
The Commercial kit Mini spin column was carefully opened and 500 µl of Buffer AW1 was added without wetting the rim. The cap was closed, and centrifuged at 6000 x g (8000 rpm) for 1 min. the Commercial kit Mini spin column was placed in a clean 2 ml collection tube (provided), and the filtrate was discarded.*
The Commercial kit Mini spin column was opened and 500 µl Buffer AW2 was added without wetting the rim. The cap was closed and centrifuged at full speed (20,000 x g ;
A 5 ml of the culture in a microcentrifuge was centrifuged for 2 min, or until a compact pellet forms. The supernatant was discarded.
The pellet was resuspended in 567 µl TE buffer by repeated pipetting.
A 30 µl of 10% SDS and 3 µl of 20 mg/ml proteinase K to give a final concentration of 100 µg/ml proteinase K in 0.5% SDS were added. It was mixed thoroughly and incubated for1 hr at 37°C.
A 100 µl of 5 M NaCl was added and mixed thoroughly.
A 80 µl of CTAB (1%)/NaCl (5M) solution was added. Mixed thoroughly and incubated 10 min at 65°C.
An approximately equal volume (0.7 to 0.8 ml) of chloroform/isoamyl alcohol was added, mixed thoroughly, and it was spin 4 to 5 min in a microcentrifuge.
Aqueous, viscous supernatant was transferred to a fresh microcentrifuge tube, by leaving the interface behind. An equal volume of phenol/chloroform/isoamyl alcohol was added, extracted thoroughly, and it was spin in a microcentrifuge for 5 min.
The supernatant was transferred to a fresh tube. A 0.6 vol of isopropanol was added to precipitate the nucleic acids. The tube was shaked back and forth until a stringy white DNA precipitate becomes clearly visible. At that point it was possible to transfer the pellet to a fresh tube containing 70% ethanol by hooking it onto the end of a micropipet that has been heat-sealed and bent in a Bunsen flame. Alternatively, the precipitate can be pelleted by spinning briefly at room temperature.
1 mL of bacterial culture was taken and centrifuged at 10,000 rpm at room temperature for 1 minute.
The pellet was resuspended in 300 µl of GBL by repeated pipetting or vortexing for 3 minute.
Then 150 µl of saturated 6 M NaCl and 300 µl of Chloroform was added and vortex for 1 minute, followed by centrifugation at 10,000 rpm for 10 minutes.
3.1 Influence of DNA Extraction Method on Quality and Yield
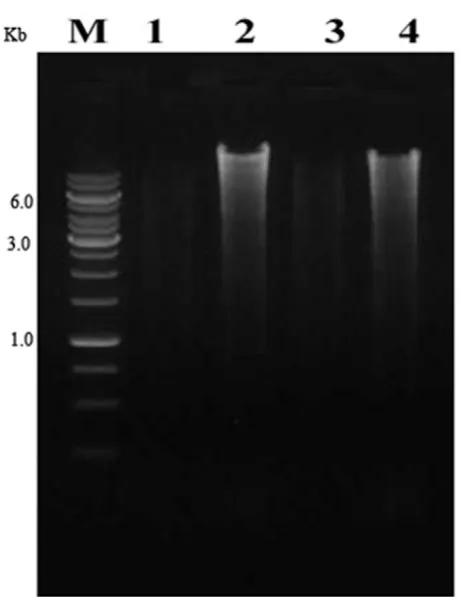
In the present study, we have isolated numerous bacterial colonies from insect gut. Among them, both a gram positive and a negative bacterium were chosen for the isolation of genomic DNA. This was to assess the effectiveness, reproducibility and efficiency of the method. The extracted genomic DNA from both types of bacteria was found to be intact by agarose gel electrophoresis. The size was found to be above 10 Kbp with no trace of smaller breakdown impurity when compared with a commercial kit (Figure 1). In the extraction process, a single step was used to lyse the bacterial cells with GBL buffer. Following lysis, there was no need of incubation for the removal of organic materials because it was precipitated by chloroform. In order to remove RNA, a minimum concentration RNase
(10mg/ml) was used instead of lithium chloride (LiCl) which was selectively used for RNA isolation [26, 27]. CTAB and β mercaptoethanol were used to eliminate the polysaccharide during DNA extraction [20]. However, few researchers used sodium acetate and isopropanol to get rid of the polysaccharide and secondary metabolites from the lysate [28, 29]. In few of the studies, high concentration of polyvinylpyrrolidone (PVP) was used to facilitate the precipitation of phenolic compounds through physical interaction and it was well reported that the PVP have the ability to protect the DNA from oxidizing agent of secondary metabolite and DNase in the lysed bacteria [29, 30]. This proposed methodology also subjected to DNA extraction and yielded sufficient concentration of DNA from other sources like cyanobacteria and other culturable bacteria (Data not shown).
Figure 1. Qualitative analysis of isolated genomic DNA from both Gram negative and Gram positive bacteria. M: DNA size
marker (1 kb), Lane 1: Gram negative culture DNA isolated by commercial kit, Lane 2: Gram negative culture DNA isolated by our GBL procedure, Lane 3: Gram positive culture DNA isolated by commercial kit, Lane 4: Gram positive culture DNA isolated
by our GBL procedure.
14,000 rpm) for 3 min.
The Commercial kit Mini spin column was placed in a new 2 ml collection tube (not provided) and the old collection tube was discarded with the filtrate. Centrifuged at full speed for 1 min.
The Commercial kit Mini spin column was placed in a clean 1.5 ml microcentrifuge tube (not provided), and the collection tube containing the filtrate was discarded. The Commercial kit Mini spin column was placed carefully and 200 µl Buffer AE or distilled water was added. Incubated at room temperature for 1 min, and then centrifuged at 6000 x g (8000 rpm) for 1 min.
Repeat the above step
The DNA was washed with 70% ethanol to remove residual CTAB and it was respin 5 min at room temperature to repellet it. The supernatant was removed carefully and the pellet was dried in a lyophilizer.
The pellet was redissolved in 100 µl TE buffer
Subsequently, the precipitated DNA was pipette out to a new Eppendorf tube and washed twice with 70 % ethanol by centrifugation at 5000 rpm for 5 minutes. The pellet was air dried at room temperature for 10 minute.
3.2 Comparative analysis of conventional and commercial kit with proposed methodology
Both the commercial kit and existing manual methods can recover purified nucleic acid for subsequent analysis. However, our method is rapid, requires fewer steps (seven) and notably reduces the amount of toxic chemical in the extraction process (Table 2). UV Spectrophotometric analysis showed the concentration of genomic DNA isolated from our proposed method
was 6.2 µg/ml (A260/A280 ratio) and the quality was
exhibited in the range of 1.72 at 260/230nm (Table 2). The good quality of DNA visualized by agarose gel electrophoresis remembers the quality of DNA
obtained by spectrophotometer analysis. Lower concentration of DNA from the commercial kit might be due to the greater number of column steps during purification, because the column may trap the DNA along with impurities. Moreover, only limited volume of sample can be purified in the commercial column. The toxic chemicals such as chloroform and SDS were used in the present methodology. However, the present methodology utilized less or reduced the usage of other toxic chemicals. The table.2 clearly states that the features such as quality, quantity, cost, steps, etc from the present methodology was highly accessible for all basic microbiology laboratories.
Table 2. Comparative table for features of different genomic DNA extraction methods
3.3 Molecular validation of the extracted DNA
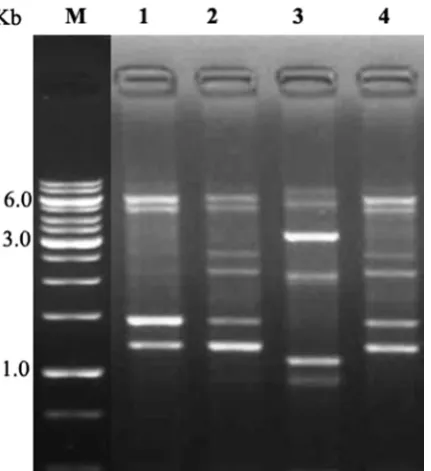
In order to check the efficiency and reliability of the genomic DNA isolated by our proposed method, DNA was subjected to restriction digestion and RAPD-PCR analysis. The electrophoresis results illustrated the
complete digestion of genomic DNA by Eco RI (Figure
2). It confirmed that the quality of DNA was good, as there was no inhibitory effect of chemicals used in the extraction of genomic DNA. Further, to evaluate the quality of genomic DNA it was counter checked by RAPD-PCR analysis. The electrophoresis results showed an intact banding pattern analysis ranging from 8000 to 1000 bp (Figure 3). The PCR result clearly suggests that it does not contain any inhibitors. It was very useful to study the genetic polymorphism with high resolution. Hence, the current protocol is cost effective and does not involve the usage of any toxic chemicals or allergen. Moreover, it is adaptable to large scale isolation of genomic DNA. The current method will be useful in the study of the insect gut microbial community, and its interaction with host. On the basis of DNA quality and quantity the GBL buffer method is novel, reliable and versatile method for large scale DNA extraction. This methodology also done in triplicate and the results were observed frequently (data not shown). In addition, an extracted DNA can be revalidated by next generation sequencing (NGS) and its accurate quality can be revalidated by Fluorimeter in near future; hence it will pay the base line to study the genome assembly, genetic diversity and quality of obtained DNA.
Figure 2. Restriction digestion analysis of isolated genomic
DNA from both Gram negative and positive bacterial genomic DNA. M: DNA size marker (1 kb), Lane 1: Gram negative culture DNA isolated by commercial kit, Lane 2: Gram negative culture DNA isolated by our GBL procedure, Lane 3: Gram positive culture DNA isolated by commercial kit, Lane 4: Gram positive culture DNA isolated by our GBL procedure.
Features Commercial DNA extraction kit Conventional DNA extraction GBL DNA extraction
Steps (No’s) 15 10 7
Volume of culture (ml) 1.5 5 1.0
Duration of DNA extraction
(Hours) 1.43 1.30 0.30
Quality (260/230nm) 1.76 1.68 1.72
Quality in agarose gel Good Good Good
Yield (µg/ml) 2.2 5.4 6.2
Toxic chemical Secrecy in solution Phenol, Isoamyl alcohol and
isopropanol,CTAB, Proteinase K No involvement of such a toxic chemical like conventional method
Figure 3. RAPD analysis of isolated genomic DNA from both negative and positive bacterial genomic DNA. M: DNA size marker (1 kb), Lane 1: Gram positive culture 1, Lane 2: Gram negative culture 1, Lane 3: Gram positive culture 2, Lane 4: Gram negative culture 2.
4.0 CONCLUSION
In summary, the overall results of our method provide a robust podium for the isolation of high quality genomic DNA from bacteria of any source. The time taken for the extraction is 30 minutes and the procedure also has been validated by inter-laboratory researchers. The purpose of this method is to establish an economical, simple and rapid method for extracting genomic DNA from culturable bacteria for industrial applications. In future, the present methodology will be useful to students for learning basic and advanced molecular microbiological techniques.
5.0 Acknowledgements
The authors are grateful to Prof. Linsay Sawyer, Emeritus Professor, University of Edinburg for his grammar revision and thoughtful comments. The authors are extremely thankful to the BRNS, DBT, UGC-Innovative programme, DST-FIST for a department infrastructure development facility.
6.0 REFERENCES
1. Adams, D and Douglas, AE. (1997). How symbiotic bacteria influence plant utilisation by the polyphagous aphid, Aphis fabae. Oecologia. 110: 528–532
2. Broderick, NA, Raffa, KF, Goodman, RM et al (2004). Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl Environ Microbiol. 70: 293-300
3. Plassart, P, Terrat, S, Thomson, B et al (2012). Evaluation of the ISO Standard 11063 DNA Extraction Procedure for Assessing Soil Microbial Abundance and Community Structure. PLoS One. 7: e44279
4. Lazarevic, V, Gaia, N, Girard, M et al (2013). Comparison of DNA Extraction Methods in Analysis of Salivary Bacterial Communities. PLoS One. 8:e67699
5. Tran-Hung, L, Tran-Thi, N, Aboudharam, G et al (2007). A New Method to Extract Dental Pulp DNA: Application to
6. Pandiarajan, J, Suganya, T, Krishnan, M. (2014). Gut resident microbes in Groundnut pest Amsacta albistriga (Red Hairy Caterpillar). Current Research in Microbiology and Biotechnology. 2: 340-346
7. Arun Prasanna, V, Kayalvizhi, N, Rameshkumar, N et al (2014). Characterization of amylase producing Bacillus megaterium from the gut microbiota of Silkworm Bombyx mori. Research Journal of Chemistry and Environment. 18(17): 38 - 45.
8. Carbonero, F, Nava, GM, Benefiel, AC et al (2011). Microbial DNA extraction from intestinal biopsies is improved by avoiding mechanical cell disruption. Journal of Microbiological Methods. 87:125-127
9. Yuan, S, Cohen, DB, Ravel, J et al (2012). Evaluation of Methods for the Extraction and
10. Purification of DNA from the Human Microbiome. PLoS One. 7: e33865
11. Kalia, A, Rattan, A, Chopra, PA. (1999). Method for Extraction of High Quality and High Quantity Genomic DNA Generally Applicable to Pathogenic Bacteria. Anal Biochem. 275:1-5 12. Aldous, WK, Pounder, JI, Cloud, JL et al (2005). Comparison
of Six Methods of Extracting Mycobacterium tuberculosis DNA from Processed Sputum for Testing by Quantitative Real-Time PCR. J Clin Microbial 43: 2471-2473
13. Ulrich, RL, Hughes, TA. (2001). A Rapid Procedure for Isolating Chromosomal DNA from Lactobacillus species and Other Gram Positive Bacteria. Lett Appl Microbiol. 32: 52-56 14. Omidi, Y, Barar, J, Akhtar, S. (2005). Toxicogenomics of
Cationic Lipid Based Vectors for Gene Therapy: Impact of Microarray Technology. Curr Drug Deliv. 2: 429-441
15. Walsh, PS, Metzger, DA, Higuchi, R. (1991). Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 10:506-1
16. Head, IM, Saunders, JR, Pickup, RW. (1998). Microbial evolution, diversity, and ecology: A decade of ribosomal RNA analysis of uncultivated microorganisms. Microbial Ecology. 35:1-21
17. Pace, NR. (1997). A molecular view of microbial diversity and the biosphere. Science. 276: 734–740
18. Chan, JW and Goodwin, PH. (1995). Extraction of Genomic DNA from Extracellular Polysaccharide Synthesizing Gram-Negative Bacteria. Bio Techniques. 18: 418-422
19. Hebron, HR, Yang, Y and Hang, J. (2009). Purification of Genomic DNA with Minimal Contamination of Proteins. J Biomol Tech. 20: 278-281
20. Ausubel, FM, Brent, R, Kingston, RE et al (1994). Current Protocols in Molecular Biology. John Wiley & Sons, NY. 21. Sambrook, J, Fritsch, EF, Maniatis, T. (1989). Molecular
Cloning: A Laboratory Manual. 2nd ed, CSH Laboratory Press, Cold Spring Harbor, NY.
22. Atashpaz, S, Barzegari, A, Azarbaijani, R. (2008). General DNA extraction kit. Iran Pat. 48024
23. Chen, H, Rangasamy, M, Tan, SY et al (2010). Evaluation of Five Methods for Total DNA Extraction from Western Corn Rootworm Beetles. PLoS One. 5: e11963
24. Li, JT, Yang, J, Chen, DC et al (2007). An optimized mini preparation method to obtain high quality genomic DNA from mature leaves of sunflower. Genet Mol Res. 6: 1064-1071
25. Vasanthakumar, A, Jr, ID, Handelsman, J et al (2006). Characterization of Gut-Associated Bacteria in Larvae and Adults of the Southern Pine Beetle, Dendroctonus frontalis Zimmermann. Environ Entomol. 35: 1710-1717
26. Gram, C. (1884). The differential staining of Schizomycetes in tissue sections and in dried preparations. General Microbiology. 2: 185-189
27. Hong, Y, Kim, S, Polne Fuller M et al (1995). DNA Extraction Conditions from Porphyra perforate Using LiCl. J Appl Phycol. 7: 101-107
28. Lev, Z. (1987). A Procedure for Large Scale Isolation of RNA Free Plasmid Phage DNA without the use of RNase. Anal Biochem. 160: 332-336
29. Ogram, A, Sayler, GS, Barkay, T. (1987). The Extraction and Purification of Microbial DNA from Sediments. J Microbiol Meth. 7:57-66
© 2015; AIZEON Publishers; All Rights Reserved
This is an Open Access article distributed under the terms of the Creative Commons Attribution License which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Chain Reaction Mediated Amplification. J Appl Bacteriol. 74: 78-85
31. Anderson, KL and Lebepe-Mazur, S. (2003). Comparison of Rapid Methods for the Extraction of Bacterial DNA from Colonic and Caecal Lumen Contents of the Pig. J Appl. Microbiol. 94: 988-993.