Ames Laboratory Publications
Ames Laboratory
3-2006
Fundamental-Measure Density Functional Theory
Study of the Crystal-Melt Interface of the Hard
Sphere System
Vadim B. Warshavsky
Iowa State University
Xueyu Song
Iowa State University, [email protected]
Follow this and additional works at:
http://lib.dr.iastate.edu/ameslab_pubs
Part of the
Chemistry Commons
The complete bibliographic information for this item can be found at
http://lib.dr.iastate.edu/
ameslab_pubs/369
. For information on how to cite this item, please visit
http://lib.dr.iastate.edu/
howtocite.html
.
Fundamental-Measure Density Functional Theory Study of the
Crystal-Melt Interface of the Hard Sphere System
Abstract
Two versions of the fundamental measure density functionals together with a new interfacial density profile
parametrization were used to study the hard-sphere crystal-melt interface in the framework of the
fundamental measure density functional theory. The equilibrium interfacial density profiles and interfacial
free energies were found as a result of minimization of grand canonical potential of system with respect to
parameters of density profile. We found that the average interfacial free energy is about 0.78, which is in
reasonable agreement with simulation results.
Disciplines
Chemistry
Comments
This article is from
Physical Review E
73 (2006): 031110, doi:
10.1103/PhysRevE.73.031110
. Posted with
permission.
Fundamental-measure density functional theory study of the crystal-melt interface of the hard
sphere system
Vadim B. Warshavsky and Xueyu Song
Ames Laboratory and Department of Chemistry, Iowa State University, Ames, Iowa 50011, USA 共Received 16 December 2005; published 15 March 2006兲
Two versions of the fundamental measure density functionals together with a new interfacial density profile parametrization were used to study the hard-sphere crystal-melt interface in the framework of the fundamental measure density functional theory. The equilibrium interfacial density profiles and interfacial free energies were found as a result of minimization of grand canonical potential of system with respect to parameters of density profile. We found that the average interfacial free energy is about 0.78, which is in reasonable agree-ment with simulation results.
DOI:10.1103/PhysRevE.73.031110 PACS number共s兲: 64.10.⫹h
I. INTRODUCTION
Investigations of the interfaces between a crystal and its melt play an important role for the better understanding of the processes of crystal growth and melting. One of the key quantities of the crystal-melt interface is the interfacial free energy. Experimentally, this quantity can be extracted from nucleation data关1兴 or from experimentally measured Wulff shape of the crystal-melt system 关2兴. The former is based upon the classical nucleation theory and only the overall av-erage of interfacial free energy can be obtained. The latter can yield anisotropy of the interfacial free energies, but so far only a few systems have been studied关3兴. Thus efficient and accurate theoretical-computational methods will be valu-able alternatives to provide relivalu-able information on the crystal-melt interfaces.
The computational methods to study the thermodynamics and structures of the interface which accounts for the micro-scopic structure are molecular simulations, either Monte Carlo共MC兲or molecular dynamics共MD兲simulations. There are two main strategies to extract the interfacial free energies from molecular simulations. The first one is the cleaving potential method, which is based on the basic thermody-namic statement that the interfacial free energy is equal to the reversible work to create that interface关4–6兴. The second one is the capillary wave method, which is based on the measurements of interfacial fluctuations, which is related to the interfacial stiffness that in turn yields the interfacial free energy关7–10兴. These methods have been applied to various model systems, but the computational cost is still quite de-manding. For the hard sphere system, it was found that the anisotropy in the interfacial free energies related to the dif-ferent crystal surface orientations is weak and the average value is⬃0.6关5,10兴.
For the past two decades a number of density functional theories were developed for the investigation of the proper-ties of the hard sphere system including the interfaces. From these theories, phase diagram, the equilibrium density profile of the interface, and the interfacial free energies have been obtained 共see Ref. 关11兴兲. Furthermore, the thermodynamic properties of systems with soft interaction can be computed with the help of the hard sphere system together with the
appropriate perturbative theories 共see, for example, Refs.
关12,13兴兲.
For the density functional theory共DFT兲 study of interfa-cial properties the choice of interfainterfa-cial density parametriza-tion is crucial in addiparametriza-tion to the choice of various versions of density functionals. The commonly used interfacial density parametrization is the two-parameter one proposed by Curtin
关14兴 共which is based on a earlier form by Haymetet al.关15兴兲. As it turns out the Curtin parametrization could become un-physical in some parameter region共see Sec. II兲. Another pa-rametrization which includes more than 105independent
pa-rameters was proposed 关16兴, but the minimization for the interface free energy of such a large parameter space is com-putational very demanding.
As for the density functionals used, all the studies are based on earlier versions of DFT involving either the trun-cated expansion of the free energy关15,17,18兴or more elabo-rated nonperturbation types of DFT such as weighted density approximation 共WDA兲, modified weighted density approxi-mation共MWDA兲, generalized effective liquid approximation
共GELA兲 and their modifications 关14,16,19–22兴. The results of these works are not consistent with each other where the interfacial free energy falls in the range of 0.25− 4.0 共see brief review Ref.关23兴and Table II兲.
Based on some geometrical properties of the hard sphere system, a fundamentally different form of density functional, called fundamental measure 共FM兲 DFT, was proposed关24兴. With some modifications this functional is very successful to describe the different properties of hard sphere共HS兲liquid, solid phases and their coexistence共for the recent review see Ref.关25兴兲, although up to now the HS solid/liquid interface properties have not been explored yet.
The present work was undertaken to study the interfacial properties of the hard sphere system using the fundamental density functional. We have proposed a more physical pa-rametrization of the interfacial density profile to overcome the drawbacks of previous methods. The resulting interfacial free energy is reasonably in agreement with simulation re-sults.
The article is organized as the following. In Sec. II the fundamental measure DFT is briefly presented to make the paper self-contained. In Sec. III the coexistence properties of
PHYSICAL REVIEW E73, 031110共2006兲
the bulk solid and liquid phases are calculated to pave the way for our presentation of interfacial free energy calcula-tions. In Sec. IV the parametrization of the interfacial density profile are described. The results for the equilibrium density profiles and the interfacial free energies at various crystal orientations are obtained in Sec. IV.
II. FUNDAMENTAL MEASURE DENSITY FUNCTIONAL THEORY
In the framework of DFT the free energy FHS of a HS
system can be written as a functional of number density pro-file共rជ兲. It consists of two parts: the ideal gas contribution
Fid关兴 and the excess free energy Fex关兴 over the ideal-gas part
FHS关共rជ兲兴=Fid关共ជr兲兴+Fex关共rជ兲兴, 共1兲 where
Fid关共rជ兲兴=kBT
冕
drជ共ជr兲兵ln关共rជ兲⌳3兴− 1其, 共2兲andkB is the Boltzmann constant,T temperature, and ⌳ de
Broglie wavelength.
From the above functional the corresponding grand ca-nonical potential functional⍀HSmay also be constructed
⍀HS关共rជ兲兴=FHS关共rជ兲兴−
冕
drជ共rជ兲+冕
drជVext共rជ兲共rជ兲, 共3兲 whereis the chemical potential of the system and Vext共rជ兲the potential of the external field acting on the system. The variational principle
冏
␦⍀HS关共rជ兲兴␦共rជ兲
冏
,T= 0 共4兲 yields the equation for the equilibrium density profile in an external potential.
In the framework of Rosenfeld Fundamental Measure DFT the excess part of the free energyFexis expressed as a
volume integration on the Rosenfeld functional⌽关24–28兴
Fex=
冕
drជ⌽兵n␣共rជ兲其. 共5兲The scalarn2,n3, vectornជv2, and tensornˆweighted densities
in this expression are given by the integral convolutions of the number density共rជ兲with corresponding weight functions
n␣共rជ兲=
冕
drជ⬘
共rជ⬘
兲␣共rជ−rជ⬘
兲. 共6兲 The weight functions␣in these expression were defined as2共y兲=␦
冉
2 −y
冊
, 3共y兲=⌰冉
2 −y
冊
, 共7兲ជv2共yជ兲=eជy␦
冉
2 −y
冊
, ˆjk共yជ兲=eជjeជk␦冉
2 −y
冊
. 共8兲 Here eជy is a unit vector along the yជ direction and is thediameter of the hard sphere, ␦共r兲 the Dirac delta-function,
⌰共r兲the Heaviside step function, ជv2 andˆjkare the vector
and tensor weight functions, respectively.
Among different approximations for the Rosenfeld func-tional⌽, we chose the two which successfully describe the liquid, solid phases, and their coexistence. The first version
关26兴 共denoted as V2PY version兲is independent of the tensor weighted densities
⌽共V2PY兲= − n2
2 ln共1 −n3兲+
n22−nv22
2共1 −n3兲
+共
n22−nv
2
2兲3
24n23
1
共1 −n3兲2 共9兲
and in the homogeneous density limit reduces to the Percus-Yevick共PY兲free energy expression. Another version共T2CS兲
关27,28兴is also a functional of tensor densities
⌽共T2CS兲= − n2
2ln共1 −n3兲+
n22−nv
2
2
2共1 −n3兲+
1 8n32
冉
n3
共1 −n3兲2
+ ln共1 −n3兲
冊
兵nជv2nˆnជv2−n2nជv22
− tr共nˆ3兲+n2tr共nˆ2兲其, 共10兲 which in the homogeneous density limit gives the Carnahan-Starling共CS兲free energy. It is known that the CS expression is more accurate to describe the HS liquid free energy as compared to the Percus-Yevick one, whereas the PY-based FM DFT functionals are more accurate to describe the HS solid properties共see discussion in Ref.关28兴兲.
III. HS SOLID AND LIQUID PHASES COEXISTENCE
To study the interfacial properties, we first need to outline the coexistence conditions under the functionals used. Below we briefly describe the necessary ingredients for the calcula-tion of the coexistence condicalcula-tions using the funccalcula-tionals given by Eqs. 共9兲 and 共10兲 共more details of the calculations are available in Refs.关13,26–28兴兲.
The fcc solid phase lattice parameter isa=共4 /兲1/3, where
is the number density in the bulk solid
关
=1V兰Vs共rជ兲dV兴
.The density profiles共rជ兲 in the bulk solid can be accurately
parametrized by a sum of Gaussian density distributions around the solid lattice sites兵Rជ其
s共rជ兲=
兺
i⌬共rជ−Rជi兲=
冉
␣
冊
3/2
兺
i e−␣共rជ−Rជi兲2, 共11兲where ␣ is the width of the Gaussian distribution. The weighted densities can be written as a sum of the correspond-ing contributions from different lattice sites
VADIM B. WARSHAVSKY AND XUEYU SONG PHYSICAL REVIEW E73, 031110共2006兲
n␣共rជ兲=
兺
i
n⌬共␣兲共rជ−Rជi兲. 共12兲
All the weighted densitiesn⌬共␣兲共rជ兲can be found in analytical forms关13,30兴. For a given bulk density the value of free energyF关␣;兴can be calculated as a function of the Gauss-ian parameter␣. The minimum ofF关␣;兴with respect to␣ gives the equilibrium value of free energyF关兴and equilib-rium value of parameter␣. The dependence of the FM DFT solid free energy on the bulk density was found to be in a good agreement with the result of MC simulations. The knowledge of the functional dependence of free energyFon the bulk density in the solid and liquid phases allows to compute the coexisting conditions between the solid and the liquid phases. The results are given in Table I. It is shown that both versions of FM free energy functional agree well with simulation results, except that T2CS version provides slightly better Lindemann ratio共together with the Gaussian parameter␣兲at coexistence.
IV. HARD SPHERE CRYSTAL/MELT INTERFACE
Now let us consider the interface between the coexisting HS bulk solid and liquid. In the interfacial region the prop-erties of bulk solid are smoothly transformed to the proper-ties of the bulk liquid. The interfacial free energy is defined as
␥=⍀共,T兲−⍀bulk共,T兲
A =
⍀+PV
A , 共13兲
where⍀is the grand canonical potential in the system with the interface,⍀bulk=⍀solid=⍀liquidis the grand canonical
po-tential of the equilibrium solid and liquid phases共in the bulk
⍀= −PV,Pis the coexistence pressure,Vthe volume of the system兲,Athe area of the interface. To calculate the interfa-cial grand canonical potential ⍀ an appropriate interfacial density parametrization is needed.
In the literature, a widely used method to parameterize the density profile of the interface is关14兴
共rជ兲=l+共s−l兲f0共z兲+
兺
G⫽0GfG共z兲eiG
ជ·rជ
, 共14兲
where
fG共z兲=
冦
1, 兩z兩艋z0,
1
2
冋
1 + cos冉
共z−z0兲⌬zG
冊
册
, z0⬍兩z兩艋zG,
0, zG⬍兩z兩,
共15兲
⌬zG=兩zG−z0兩=
冉
G1G
冊
⌬z共⌬z0=⌬z兲, 共16兲
0艋艋1,G艌G1. 共17兲
Herezis the coordinate in the perpendicular direction to the interface,Gជ is a reciprocal lattice vector, G its magni-tude, G1 the magnitude of the smallest nonzero reciprocal
lattice vector. The parameter⌬zis the width of the interface and the parameter controls the rate of broadening of the solid density peaks. The interface is located in the region
关z0;z0+⌬z兴; forz⬍z0the density profile reduces to the
den-sity profile in the bulk solid s共rជ兲=s+兺G⫽0GeiG
ជ·rជ
written with reciprocal lattice vectors; forz⬎zg=z0+⌬zit gives the
homogeneous density in the bulk liquidl. To calculate the
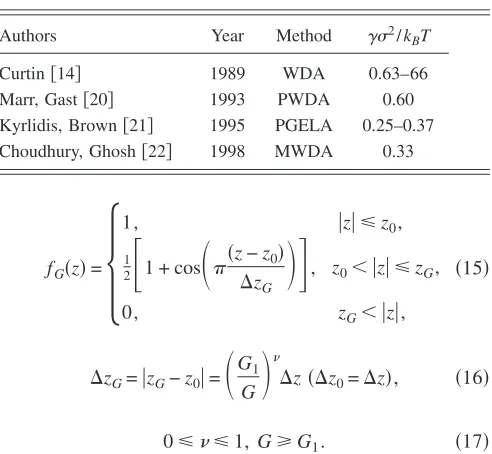
interfacial free energy some authors used this parametriza-tion together with the different types of DFT. Table II sum-marizes these results. It is seen that the results for the inter-facial free energy differ from each other for various types of DFT used.
We also started with this interfacial density profile param-etrization together with the fundamental measure DFT. All the corresponding weighted densities in the interfacen2,n3,
nជv2, andnˆwere computed analytically共except for the regions
关z0−/ 2 ;z0+/ 2兴 and 关zg−/ 2 ;zg+/ 2兴 where the
weighted densities were reduced to the one dimensional in-tegrals兲. For various and ⌬z we did not find the global minimum for the interfacial free energy␥. For example, for every given⌬zwith increasingfrom= 0 to= 1 the␥just gradually decreases even to some negative value for big.
There is also another problem with the density parametri-zation of Eqs.共14兲–共17兲. It is known that an important con-dition for the HS system is the non-overlapping of hard spheres关16,31–33兴. In the integral form this condition can be written as
冕
d3r⬘
⌰冉
[image:5.612.315.561.89.316.2]2 −兩rជ−rជ
⬘
兩冊
共rជ⬘
兲艋1, 共18兲 which gives restrictions on the values of the weighted den-sityn3 关Eqs.共6兲and共7兲兴TABLE I. HS fcc solid-liquid coexisting parameters;land s are the liquid and solid densities,L=共3 /␣兲1/2/athe Lindemann
ra-tio,Ppressure at coexistence. Results of MC simulation are taken from Ref.关29兴.
l s L P
T2CS 0.934 1.023 0.133 11.29
V2PY 0.937 1.020 0.113 12.26
MC 0.943 1.041 0.129 11.7
TABLE II. The density functional theory results for the hard-sphere solid-liquid interfacial free energy with the interfacial den-sity profile parametrized by Eqs.共14兲–共17兲.
Authors Year Method ␥2/k
BT
Curtin关14兴 1989 WDA 0.63–66
Marr, Gast关20兴 1993 PWDA 0.60
Kyrlidis, Brown关21兴 1995 PGELA 0.25–0.37 Choudhury, Ghosh关22兴 1998 MWDA 0.33
FUNDAMENTAL-MEASURE DENSITY FUNCTIONAL¼ PHYSICAL REVIEW E73, 031110共2006兲
n3共r兲艋1. 共19兲 For previous DFT approximations共such as WDA, MWDA, GELA兲the condition of Eq.共18兲is not intrinsic to the func-tional, although an attempt to adopt constrained MWDA that satisfies this condition was undertaken 关31兴. In the frame-work of the fundamental measure DFT, which is based on some geometrical properties of hard spheres关34兴, the condi-tion of the nonoverlapping of hard spheres is automatically accounted for by the special form of the free energy func-tional⌽, where the conditionn3艌1 leads to divergent
func-tional ⌽ in Eqs. 共9兲 and 共10兲. We have observed that for some small values of⌬zand big values of at some points of the interface the local packing fractionn3is larger than 1,
which indicates the unphysical nature of this density profile parametrization. This unphysical behavior can also been seen from the following argument. For = 0 the density profile can be shown to be
共rជ兲=l关1 −f0共z兲兴+f0共z兲s共rជ兲. 共20兲
It can be easily seen that the density profile is a mixing of a bulk liquid densityland a solid density profiles共rជ兲, which
under certain conditions will leads to overlapping hard spheres, i.e.,n3艌1.
To avoid the abovementioned difficulties with the param-etrization Eqs. 共14兲–共17兲 we introduced a new parametriza-tion of the density profile. The main feature of this param-etrization is that the fcc lattice structure now is spread all over the system and the density profile of the whole system may be written in a similar way to the one in the bulk solid, but with different widths
共rជ兲=
兺
Rជ
冉
␣共Rជ兲
冊
3/2
e−␣共Rជ兲共rជ−Rជ兲2, 共21兲 now the Gaussian parameter␣ depends on the site position
Rជ. Let the interface be in perpendicular to thezdirection. For
the lattice sites at the bulk solid共z⬍z0兲the Gaussian param-eter␣=␣共Rជ兲equals to␣s, the equilibrium Gaussian width of
the coexistence solid phase. For the ones in the bulk liquid
共z⬎z0+⌬z兲the parameter␣should be small enough to pro-vide the sum of the widely overlapping Gaussians to be equal to the uniform density distribution 共we chose␣l= 1兲.
For the interfacial region between the bulk solid and liquid
共z0⬍z⬍z0+⌬z兲 there are N layers of lattice sites whose
equilibrium values of the Gaussian parameters兵␣i其i=1 N
can be found by minimizing the interfacial free energy. The Gauss-ian parameters␣iwithin the same layer are the same and the
parameters change from layer to layer in the interfacial re-gion with the obvious restriction ␣s艌␣1艌␣2艌¯艌␣N
艌␣l. Thus, the values of␣iand␣共Rជ兲are related by
␣共Rជ兲=
冦
␣s, Rz,i艋z0
␣i, z0⬍Rz,i⬍z0+⌬z,
␣l= 1, Rz,i⬍z0+⌬z,
whereRz,iis projection of the lattice vector of theishell on
thezaxis.
In addition, the fcc solid lattice spacingasin the plane of
the interface共i.e., along thexandyaxis兲is fixed to be
ax,i=ay,i=as=共4/s兲1/3, 共22兲
whereas in the perpendicular direction to the interface共along thez-axis direction兲it increases fromas=共4 /s兲1/3atz⬍z0to
al= 4 /共las 2兲
at z⬎z0+⌬z. Such choice of al provides the
value of the homogeneous density at z⬎z0+⌬z to be equal
to the coexisting liquid density l. The lattice parameter in
the transition area is chosen to be equal to
az,i共Rជ兲= 4/关sm共Rជ兲as 2兴
, 共23兲 wheresm共z兲 is some smoothed density profile in the
inter-face region which may be parametrized by an expression with hyperbolic tangent 共other reasonable function forms give the same results兲
sm共z兲=l+共s−l兲
冦
1, 兩z兩艋z0, 1
2
冋
1 − tanh冉
6共z−z0−⌬z/2兲
⌬z
冊
册
, z0⬍兩z兩艋z0+⌬z.0, z0+⌬z⬍z.
共24兲
In the bulk fcc crystal lattice the distancedbetween layers along different crystal orientations are d110=a/ 2
冑
2, d100=a/ 2,d111=a/
冑
3 共d110⬍d100⬍d111兲. In this lattice 共110兲 ismore densely packed, but the most densely packed plane is
共111兲. Extending this to the whole system 共bulk solid +interfacial region+bulk liquid兲 we can write the interlayer spacingdialong the normal direction to the interface共along
thezaxis兲as
di共100兲= az,i
2 , di
共110兲= az,i
2
冑
2,di共111兲=az,i
冑
3. 共25兲For a given crystal direction the distance between layers,
⌬Rz,i=di, increases with the increasingzfrom the bulk solid 共z⬍z0兲 to bulk liquid 共z⬎z0+⌬z兲. The width of the
transi-tion region⌬z is
VADIM B. WARSHAVSKY AND XUEYU SONG PHYSICAL REVIEW E73, 031110共2006兲
⌬z=
兺
i=1 N
di. 共26兲
Finally, to find the values of lattice parameter az,i in the
transition region a set of Eqs. 共23兲–共26兲 are solved by the iterations.
With the above interfacial density parametrization, all weighted densities can be written conveniently a sums of the contributions from different lattice sites. Moreover every contribution can be given by an analytical expression.
To find the grand canonical potential⍀关Eq.共3兲兴 incorpo-rated into the expression for the interfacial free energy␥关Eq.
共13兲兴 the three-dimensional volume integral can be calcu-lated numerically. To reduce the volume of integration a sim-plex of the integration volume can be found by taking into account the symmetry of the system. This simplex is chosen to be a prism with its rib along thez direction and its base being a triangle in thex-yplane. The equation of this triangle is for 共100兲 orientation 0艋x艋as
4, y⬍x and as
4艋x艋 as
2, y
⬍as
2−x, and its area A= as2
16; for 共110兲 orientation: 0艋x
艋 as
2冑2 and 0艋y艋 as
2, A= as2
4冑2; for 共111兲 orientation: 0艋x
艋 as
2冑2 and − x
冑3艋y艋冑x3,A=
as2
8冑3.
Now for a given crystal orientation, the number of varia-tional parametersNand every set of Gaussian parameters in transition region兵␣i其i=1
N
,␥共␣1, . . . ,␣N兲can be calculated
nu-merically. To minimize␥ with respect to兵␣i其s we used the
numerical downhill simplex method in multidimensions关35兴. We performed the minimization of functional␥ with dif-ferent numbers of layers N艌2. It was found that with the increasing of the numberNthe equilibrium␥decreases until it reaches a limiting value forNⲏ17. The obtained results of the interfacial free energy for T2CS and V2PY versions of FM free energy functional are compared in Table III with the results of MD关5兴and MC关10兴simulations and also DFT of Ref.关16兴. Reference关16兴used the WDA functional but with the interfacial density profile parametrized by almost 1 mil-lion independent parameters and at the same time, the con-dition Eq.共18兲was taken into account. It can be seen that the average interfacial energy ␥ obtained in Ref. 关16兴 is two times smaller than the one obtained from simulations, whereas the␥obtained at the present study overestimates by 37%共T2CS version兲or 26%共V2PY version兲.
It should be noted that␥100overestimates the simulation
results by only 10% 共V2PY version兲. The reason that the V2PY version of FM DFT provides better agreement with the simulation results than the T2CS version may be ex-plained as the following. From the discussion in Ref.关28兴it may be concluded that the versions of FM DFT based on the PY approximation provides a better description of the HS crystal, whereas the ones based on CS approximation are more accurate for the HS fluid. In our model solid/liquid interfacial system, all the system is considered to be the sol-idlike, with the atoms located at the sites of model solid lattice. The density profile in such solidlike system has the form of a sum of Gaussian distributions around these solid lattice sites, with the Gaussian parameter ␣ decreases and lattice parametera increases toward the bulk liquid side. It seems that according to the discussions in Ref. 关28兴 a PY version will be indeed a better choice than the CS one for such system. In Ref. 关28兴 the tensor version of Rosenfeld functional T2PY is provided, which gives rather crude coex-isting densitiess= 0.985 andl= 0.892. So it seems that the
vector version V2PY of Ref. 关24兴 provides the best choice for the calculation of the interfacial free energy in the frame-work of the present solidlike model.
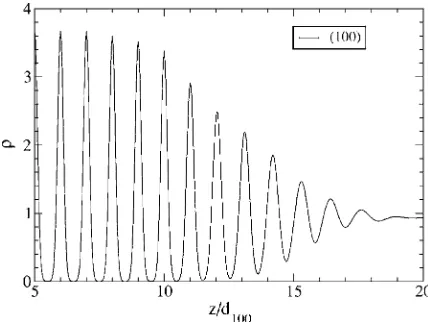
The result for the equilibrium interfacial planar averaged density profile 共z兲=A1兰兰dxdy共rជ兲 are shown in Fig. 1 for the 100 crystal orientation. The obtained width of interface may be estimated as approximately 7 − 9 interfacial layers for all the orientations, which is in line with the results of MC
关21兴, MD simulations关36,37兴, although the WDA DFT关16兴 provides slightly narrower interface. The other crystal orien-tations gives similar agreements between our calculation and the simulations.
V. CONCLUSIONS
[image:7.612.329.543.56.217.2]We applied the fundamental measure density functional theory to study the interface between coexisting hard sphere
TABLE III. The results for the HS solid-liquid surface tension 共in the units ofkBT/2兲 for the different crystal interface
orienta-tions obtained by MD共Davidchak and Laird关5兴兲, MC共Mu, Houk and Song关10兴兲simulations, WDA DFT共Ohnesorge, Lowen, Wag-ner关16兴兲and also the results of the present work with T2CS and V2PY versions of FM free energy density functional.
MD MC DFT T2CS V2PY
␥100 0.62± 0.01 0.64± 0.02 0.35 0.79 0.68
␥110 0.64± 0.01 0.62± 0.02 0.30 0.89 0.84
␥111 0.58± 0.01 0.61± 0.02 0.26 0.87 0.82
␥ 0.61± 0.01 0.62± 0.02 0.30 0.85 0.78
FIG. 1. The interfacial density profile 共z兲 as a function of z/d100for the共100兲direction. As a small change in the initial part
of the interfacial density profile will be accumulated over the whole profile, thus a direct comparison with simulation results is not illu-minating. We did compare the overall features of our density profile with simulations关37兴and they are consistent with each other for the three directions computed here.
FUNDAMENTAL-MEASURE DENSITY FUNCTIONAL¼ PHYSICAL REVIEW E73, 031110共2006兲
liquid and its fcc solid. The minimization of the interfacial free energy using earlier interfacial parametrization of the density profile关14兴was found to give nonphysical configu-rations of the interfacial density.
As a result a more physical parametrization of the density profile was introduced and the minimization of the interfacial free energy provides good equilibrium density profile in the interfacial region. The resulting interfacial free energy has an average value of ␥= 0.78 共V2PY version兲. The interfacial free energies along different crystal directions are also calcu-lated. We found 26% difference between the interfacial free energy obtained in the present study and the one obtained by the MC and MD simulations关5,10兴; it seems that the aniso-tropy could not be resolved reliably from the current density function formulation.
The fact that the obtained values for the interfacial free energy␥ overestimate the simulation ones means that there is enough “room” to continue the minimization. Indeed it seems that for more flexible parametrization or for larger number of the minimization parameters the minimization of the grand canonical functional may decrease the result for␥ further. The increasing the number of minimization param-eters may be achieved with introducing the tensor values of parameter 兵␣其s in the interface rather the scalar ones.
An-other possibility is to utilize as the minimization parameters the values of the interlayer interfacial distances兵az其s rather
than using the parametrization 共23兲 and 共24兲. However, it should be noted here that the increasing of the numbers of minimization parameters makes the numerical computation more extensive.
Applications of the present strategy to various model sys-tems where simulation results are known 关9,38兴 should be interesting. From such calculations we may have a better error estimate of the density functional approach to interfa-cial free energies. The power of the present approach may lie in the applications to the interfaces of multicomponent sys-tems, where computer simulations are much more computa-tional intensive.
ACKNOWLEDGMENTS
This research was sponsored in part by the Division of Materials Sciences and Engineering, Office of Basic Energy Sciences, U. S. Department of Energy, under Contract No. W-7405-ENG-82 with Iowa State University 共V.B.W. and X.S.兲and by a NSF Grant No. CHE0303758共X.S.兲.
关1兴M. E. Glicksman and C. Vold, Acta Metall. 17, 1共1969兲. 关2兴R. E. Napolitano, S. Liu, and R. Trivedi, Interface Sci.10, 217
共2002兲.
关3兴J. J. Hoyt, M. Asta, T. Haxhimali, A. Karma, R. E. Napolitano, R. Trivedi, B. Laird, and J. R. Morris, MRS Bull. 29, 935 共2004兲.
关4兴J. Q. Broughton and G. H. Gilmer, J. Chem. Phys. 84, 5759 共1986兲.
关5兴R. L. Davidchack and B. B. Laird, Phys. Rev. Lett. 85, 4751 共2000兲.
关6兴Y. Mu and X. Song, J. Chem. Phys. 124, 034712共2006兲. 关7兴J. J. Hoyt, M. Asta, and A. Karma, Phys. Rev. Lett. 86, 5530
共2001兲.
关8兴J. R. Morris, Z. Y. Lu, Y. Y. Ye, and K. M. Ho, Interface Sci. 10, 143共2002兲.
关9兴J. R. Morris and X. Song, J. Chem. Phys. 119, 3920共2003兲. 关10兴Y. Mu, A. Houk, and X. Song, J. Phys. Chem. B 109, 6500
共2005兲.
关11兴R. Evans, inFundamentals of Inhomogeneous Fluid, edited by D. Henderson共Wiley, New York, 1992兲.
关12兴J. D. Weeks, D. Chandler, and H. C. Andersen, J. Chem. Phys. 54, 5237共1971兲.
关13兴V. B. Warshavsky and X. Song, Phys. Rev. E 69, 061113 共2004兲.
关14兴W. A. Curtin, Phys. Rev. B 39, 6775共1989兲.
关15兴A. D. J. Haymet and D. W. Oxtoby, J. Chem. Phys. 74, 2559 共1981兲.
关16兴R. Ohnesorge, H. Lowen, and H. Wagner, Phys. Rev. E 50, 4801共1994兲.
关17兴S. M. Moore and H. J. Raveche, J. Chem. Phys. 85, 6039 共1986兲.
关18兴W. E. McMullen and D. W. Oxtoby, J. Chem. Phys. 88, 1967 共1988兲.
关19兴W. A. Curtin, Phys. Rev. Lett. 59, 1228共1987兲.
关20兴D. W. Marr and A. P. Gast, Phys. Rev. E 47, 1212共1993兲. 关21兴A. Kyrlidis and R. A. Brown, Phys. Rev. E 51, 5832共1995兲. 关22兴N. Choudhury and S. K. Ghosh, Phys. Rev. E 57, 1939共1998兲. 关23兴D. W. Marr, J. Chem. Phys. 102, 8283共1995兲.
关24兴Y. Rosenfeld, Phys. Rev. Lett. 63, 980共1989兲.
关25兴H. Löwen, J. Phys.: Condens. Matter 14, 11897共2002兲. 关26兴Y. Rosenfeld, M. Schmidt, H. Lowen, and P. Tarazona, Phys.
Rev. E 55, 4245共1997兲.
关27兴R. Roth, R. Evans, A. Lang, and G. Kahl, J. Phys.: Condens. Matter 14, 12063共2002兲.
关28兴P. Tarazona, Physica A 306, 243共2002兲.
关29兴W. G. Hoover and F. H. Ree, J. Chem. Phys. 49, 3609共1968兲. 关30兴B. Groh and B. Mulder, Phys. Rev. E 61, 3811共2000兲. 关31兴R. Ohnesorge, H. Lowen, and H. Wagner, Phys. Rev. A 43,
2870共1991兲.
关32兴J. K. Percus, J. Stat. Phys. 52, 1157共1988兲.
关33兴W. A. Curtin and K. Runge, Phys. Rev. A 35, 4755共1987兲. 关34兴Y. Rosenfeld, J. Chem. Phys. 89, 4272共1988兲.
关35兴W. H. Press, B. P. Flannery, S. A. Teukovsky, and W. T. Vet-terling, Numerical Recipes (FORTRAN version) 共Cambridge University Press, Cambridge, 1989兲.
关36兴A. Mori, R. Manabe, and K. Nishioka, Phys. Rev. E 51, R3831 共1995兲.
关37兴R. L. Davidchack and B. B. Laird, J. Chem. Phys. 108, 9452 共1998兲.
关38兴R. L. Davidchack and B. B. Laird, Phys. Rev. Lett.94, 086102 共2005兲.
VADIM B. WARSHAVSKY AND XUEYU SONG PHYSICAL REVIEW E73, 031110共2006兲