Ab Initio Prediction of Atomic Location of Third Elements in B2-Type TiNi
Sukeyoshi Yamamoto
1,2, Tomohito Yokomine
1, Kazunori Sato
1,*, Tomoyuki Terai
1, Takashi Fukuda
1and Tomoyuki Kakeshita
11Department of Materials Science and Engineering, Graduate School of Engineering, Osaka University, Suita 565–0871, Japan 2Nippon Steel & Sumitomo Metal Corporation, Futtsu 298–8511, Japan
Based on ab initio total energy calculations, we predict the atom location of the third element X (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni and Cu) in B2-type TiNi. The formation energy of Ti-substitutional X and Ni-substitutional X are estimated by using the Vienna Ab initio Simulation Package (VASP). It is found that Sc prefers the Ti-substitutional site and Cr, Mn, Fe and Co prefer the Ni-substitutional site. The location of V and Cu may depend on the composition of Ti and Ni. The chemical trend of the formation energy can be interpreted by the crys-tal orbicrys-tal Hamiltonian population (COHP) analysis. [doi:10.2320/matertrans.M2017333]
(Received November 2, 2017; Accepted December 13, 2017; Published February 2, 2018)
Keywords: ab initio calculation, density functional theory (DFT), titanium alloys, shape memory alloys (SMA), crystal orbital Hamiltonian population (COHP)
1. Introduction
Ti-Ni based shape memory alloys (SMAs) are now widely used in commercial products such as actuators and damp-ers1,2) because of their excellent shape memory and
super-elastic properties, which originate from the thermosuper-elastic martensitic transformations of the alloys. The parent phase of Ti-Ni based SMAs has the B2-type structure and the mar-tensite phase has the B19 -type, B19-type or a trigonal (R-phase) structures, depending on the composition and the heat treatment2). The shape memory and superelastic
proper-ties of Ti-Ni based alloys significantly depend on the struc-ture of the martensite phase. Since the crystal strucstruc-ture of the martensite phase and the transformation temperature sig-nificantly depend on the addition of third elements, a large number of studies have focused on the effect of third ele-ments on the transformation behavior3,4). In order to
com-prehensively understand the effect of third elements, their site occupancy should be clarified: whether a third element occupies the Ni-site or the Ti-site. The site occupancy of the third elements will help us construct model crystals for sim-ulating the transformation behavior of Ti-Ni based SMAs containing third elements. Several experimental and theoret-ical works have been made to clarify the site occupancy of the third elements in Ti-Ni based SMAs.
Nakata et al.5–8) and Tadaki et al.9) systematically
exam-ined the site preference of third elements in Ti-Ni SMAs us-ing the atom location by channelus-ing enhanced microanalysis (ALCHEMI) technique10). They quantitatively evaluated the
Ti- and Ni site occupancy of the third elements X (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Pd and Au) in Ti-Ni-X SMAs with various compositions. They revealed that the tendency of site preference of third elements can be classified into three types: (1) Fe, Co and Pd occupy the Ni-site regardless of the alloy compositions; (2) Sc occupies the Ti-site regard-less of the alloy compositions; (3) Cr, Mn, Cu, Au and V oc-cupy both the Ti- and Ni- sites and the occupancy depends on their compositions. They also tried to explain these
ex-perimental results on the basis of the Bragg-Williams ap-proximation. Although they considered only nearest neigh-bor pair potential as the internal energy of their model, a part of the experimental results are explained qualitatively. However, some discrepancy arose between the experimental results and their model in the occupancy of Ti- and Ni-sites. For instance, although their model predicts that the elements categorized in type (3) mentioned above occupy completely either Ti-site or Ni-site in the cases of Ti-poor or Ni-poor al-loys, respectively, the experimental results indicate that a part of the third elements occupy the other site. In addition, Cr tends to occupy Ni site a little more than Ti site despite that Cr is classified in (3).
Hosoda et al. made a thermodynamic approach for pre-dicting the site occupation of third elements in Ti-Ni based SMAs by employing a pseudo-ground state analysis11), and
summarized the site preference of the third elements in Ti-Ni-X alloys as follows12); (1) Au, Pd and Co substitute Ni
site. (2) Sc and V substitute Ti site. (3)Fe, Cr, Mn and Cu substitute deficient site of Ti or Ni. The site preference of V, Au and Fe disagree with the results of the ALCHEMI analy-sis by Nakata et al.7,8)
Bozzolo et al. performed Monte Calro simulations to pre-dict the site preference of Pd in Ti-Ni alloy by employing the Bozzolo-Ferrante-Smith (BFS) method13,14) composed of
1024 atoms. They concluded that Pd substitutes deficient site of Ti or Ni. This result disagrees with the ALCHEMI analysis made by Nakata et al.6), in which Pd substitutes Ni
site regardless of alloy composition. Bozzolo et al. also ap-plied the BFS method to Fe, Pt, Au, Cu, Zr and Hf as the third elements15). Their results of Au and Cu were in good
agreement with those obtained by ALCHEMI7,8), while
dis-crepancies were found in the other elements.
Sheng et al.16) investigated the substitution behavior of
al-loying elements in TiNi by using discrete variational (DV)-Xα cluster method. They calculated the bond order be-tween Ti or Ni and the third elements employing the Mulliken population analysis17–20). Their results were in
good agreement with Nakata s experimental analysis of Fe, Co, Sc, Cr, Mn, Cu and V. The only exception was Au, but
the reason for this was not clear.
Sun et al.21) employed a generalized atomic site
occupa-tion model with quasi chemical bond approximaoccupa-tion to pre-dict the site preference of Co in Ti-Ni alloy. They evaluated the change in free energy caused by the addition of Co to the TiNi alloys based on their model. In their model, they used a super lattice of Ni7Ti8Co as a Ni-poor alloy. They calculated
the total energy of their model using the CASTEP of the first principle calculation program software. They insist that Co substitutes Ni-site in their model, which agrees with the ALCHEMI analysis by Nakata et al.7,8) However,
calcula-tions for a Ti-poor alloy might be needed to discuss the site preference of Co.
Singh et al.22) used Vienna Ab initio Simulation Package
(VASP) to predict the site preference of third elements in the B2-type TiNi. They employed super lattice models of Ni15Ti16X and Ni16Ti15X (X = Mg, Al, Si, Sc, Ti, V, Cr, Mn,
Fe, Co, Ni, Cu, Zn, Ga, Ge, Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, In, Sn, Sb, Hf, Ta, W, Re, Os, Ir, Pt and Au). Their results were as follows; (1) Mg, Sc, Y, Ag, Cd, In, Sn and Hf substitute Ti site. (2) Cr, Mn, Fe, Co, Cu, Ga, Ge, Nb, Mo, Tc, Ru, Rh, W, Re, Os, Ir and Pt substitute Ni site. (3) V, Zn,Nb, Pd, Ta and Au substitute deficient site of Ti or Ni. (4) Zr substitutes excessive site of Ti or Ni. Among these ele-ments, Sc, Fe and Co agree with the results of ALCEMI analysis by Nakata et al.7,8) However, Cr, Mn, Cu and Pd
disagree with those.
As reviewed above, site preference of third additive ele-ments in B2 type TiNi alloys have been investigated experi-mentally using ALCHEMI method and theoretically by sev-eral methods. However, the agreement between experimental results and theoretical calculations is insufficient. The pur-pose of the present study is to explain the experimental re-sults reported by Nakata et al. by first-principles calcula-tions, especially focusing on the 3d transition metal elements. For this purpose, we perform ab initio total energy calculations and estimate formation energy of substitutional X in B2 type Ti-Ni-X (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, and Cu). Moreover, we calculate the crystal orbital Hamiltonian population (COHP)23) for bonding analysis in Ti-Ni-X to
discuss chemical trend of the site preference of the X elements.
2. Calculation Method
In order to predict the location of the third element X al-loyed into TiNi in B2 structure, the formation energy (Ef) of X located at Ti site (XTi) and Ef of X located at Ni site (XNi)
are calculated for X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni and Cu. For example, Ef(XTi) = [E(Ti1−xXxNi) + xE(Ti) − E(TiNi) − xE(X)]/x, and Ef(XNi) = [E(TiNi1−xXx) + xE(Ni) − E(TiNi) − xE(X)]/x, where x is concentration of X and E(A) is total energy per B2 unit cell of the system A. In order to calculate formation energy per one X atom, the energy dif-ference is divided by concentration x. In this paper, total en-ergy is calculated by using the projector augmented wave (PAW) method implemented in the Vienna Ab initio Simulation Package (VASP)24,25). For the exchange
correla-tion funccorrela-tional, we use the generalized gradient approxima-tion (GGA) parameterized by Perdew, Burke and Ernzerhof
(PBE)26). Calculated k-points are generated by the
Monkhorst-Pack method27). The cutoff energy is set to
520 eV.
In order to calculate total energy of TiNi with the third el-ement, we setup a supercell which includes 8 unit cells of B2 (CsCl) structure by doubling a primitive unit cell in x, y and z directions. One of the Ti or Ni is replaced with X (therefore, in the present supercell calculations, concentra-tion of X is 6.25 at%), and atomic posiconcentra-tions are optimized until the absolute value of the atomic forces become less than 0.01 eV/Å. Optimizations are performed only for the atomic positions in the unit cell and lattice constant is fixed to the optimized lattice constant of pure TiNi. Total energies of elemental crystals, which are needed to estimate Ef, are also calculated by using VASP with the same calculation conditions, except for the k-point mesh. The results for pure systems are summarized in Table 1. In addition to the calcu-lation of Ef, we perform bonding analysis by calculating the COHP proposed by Dronskowski et al.23) By showing the
energy dependence of COHP for each atomic pair, we can distinguish which part of the density of states contribute to the chemical bonding and discuss chemical trend of the for-mation energy.
3. Results and Discussion
3.1 Formation energy and site preference of X
Calculated formation energies are summarized in Table 2 and Fig. 1. It is recognized that Ef(NiTi) is smaller than Ef(TiNi) and this corresponds well to the experimental phase diagram of TiNi. As shown in the figure and the table, Sc (and trivially Ti) prefers Ti site, on the other hand Cr, Mn, Fe, Co (and trivially Ni) prefer Ni site. V and Cu are inbe-tween and the atom locations may depend on the composi-tion of Ti and Ni. According to Nakata, the preference of the atom location can be distinguished by calculating D value5,6). D value is originally defined as D = VTiX−VNiX
VTiNi , where
VTiX, VTiX and VTiNi are effective pair interaction between
Ti-X, Ni-X and Ti-Ni, respectively. Based on the similar dis-cussion, we can estimate D value from the formation
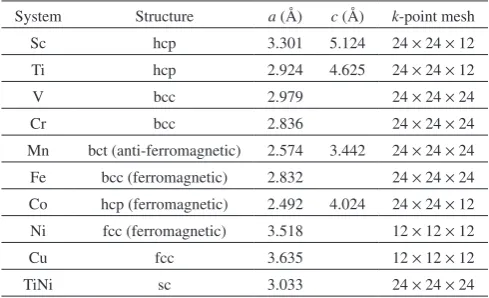
ener-Table 1 Optimized lattice constants of elemental crystals and pure TiNi. Abbreviations for the structures are simple cubic (sc), face centered cu-bic (fcc), body centered cucu-bic (bcc), Hexagonal closed pack (hcp) and body centered tetragonal (bct). Calculation conditions of k-point mesh are also tabulated.
System Structure a (Å) c (Å) k-point mesh Sc hcp 3.301 5.124 24 × 24 × 12 Ti hcp 2.924 4.625 24 × 24 × 12
V bcc 2.979 24 × 24 × 24
Cr bcc 2.836 24 × 24 × 24
Mn bct (anti-ferromagnetic) 2.574 3.442 24 × 24 × 24 Fe bcc (ferromagnetic) 2.832 24 × 24 × 24 Co hcp (ferromagnetic) 2.492 4.024 24 × 24 × 12 Ni fcc (ferromagnetic) 3.518 12 × 12 × 12
Cu fcc 3.635 12 × 12 × 12
[image:2.595.304.549.638.787.2]gies as DTi = EEff(TiNi)(XTi)−−EEff(NiNi(XNi)) for substitutional Ti and DNi =
Ef(XTi)−Ef(XNi)
Ef(NiTi)−Ef(TiTi) for substitutional Ni. The meaning of the D value is same as Nakatas definition, and the Ti site is pre-ferred for D < −1, Ni site is preferred for D > 1, and for
−1 < D < 1 X occupies both sites. In the present calcula-tions, for the denominator of the expressions of D we use the average of Ef(TiNi) and Ef(NiTi), i.e., (Ef(TiNi) + Ef(NiTi) )/2. Stable sites predicted from D are summarized in Table 2 with experimental observations. They show rea-sonable agreement with experiments, except for Cr and Mn. For those elements, in the experiments they occupy both sites, but according to our calculations they should occupy Ni site. This contradiction may arise from the strong chemi-cal interactions between neighboring Mn atoms or Cr atoms. To clarify this point, we need further calculations of site oc-cupancy including the interactions between X atoms in TiNi.
3.2 Stability of TiX and NiX
In order to consider the origin of the chemical trend of the formation energy of XTi and XNi, we try to simplify the
problem. It was pointed out that the site occupancy of X at-oms in TiNi can be related to the formation enthalpy ΔH of TiX and NiX, and Nakata et al. succeeded to understand the chemical trend of the site preferences of the third element by
using the experimental ΔH. This means that the atomic in-teractions in TiNi alloyed with X are short range and the nearest neighbor interaction dominates. Being encouraged by this observation, we calculate stability of TiX and NiX in B2 structure (so to say, 100% replacement of Ni or Ti by X, respectively) and investigate the chemical bonding in TiX and NiX, instead of treating rather big 2 × 2 × 2 supercell used for the calculations of XTi and XNi. Stability of TiX and
NiX in B2 structure is calculated as the energy difference
ΔE. ΔE(TiX) = E(TiX in B2) − E(Ti) − E(X), and
ΔE(NiX) = E(NiX in B2) − E(Ni) − E(X).
[image:3.595.46.285.430.739.2]The optimized lattice constants for TiX and NiX in B2 structure and calculated energy difference ΔE are summa-rized in Table 3 and their chemical trends are plotted in Fig. 2. For these calculations, we assume non-magnetic state for each case. As shown in the table and the figure, TiMn, TiFe, TiCo, TiNi, TiCu and NiSc are possible to be stable in B2 structure. In fact, TiFe28), TiCo28), TiNi28) and NiSc29) are
stable in B2 structure at room temperature. The behavior of
ΔE basically corresponds well to the calculation results of the formation energy of substitutional X in TiNi (Fig. 1 and Table 2), i.e., the formation energy Ef of ScTi, MnNi, FeNi,
CoNi and CuNi are relatively small, compared to the other
cases. The estimated D values from ∆E s show similar
be-Table 2 Calculated formation energy Ef of XTi and XNi in TiNi. Values
with * are from Ref. 31. Experimental results are from Refs. 5–9. 1 (eV) = 1.60218 × 10− 19 (J).
X Ef(XTi) (eV) Ef(XNi) (eV) D value Stable site Exp.
Vc 2.27, 1.74* 1.74, 1.09* - Ni
-Sc −0.31 1.85 −3.1 Ti Ti
Ti 0 0.79, 0.64* −1.1 Ti
-V 0.93 0.39 0.8 Ti, Ni Ti, Ni
Cr 1.73 0.33 2.0 Ni Ti, Ni
Mn 1.60 −0.04 2.3 Ni Ti, Ni
Fe 1.03 0.01 1.4 Ni Ni
Co 1.11 −0.02 1.6 Ni Ni
Ni 0.62, 0.65* 0 0.9 Ti, Ni
-Cu 0.61 0.61 0 Ti, Ni Ti, Ni
Fig. 1 Calculated formation energy Ef of XTi and XNi in TiNi. Vacancy
[image:3.595.310.540.435.762.2]formation energy VcTi and VcNi are also calculated. 1 (eV) = 1.60218 × 10−19 (J).
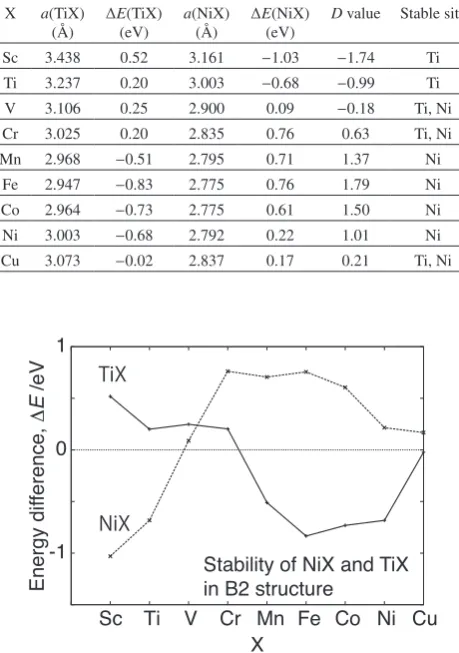
Table 3 Energy difference ∆E of TiX and NiX in B2 structure. Negative ∆E indicates that the B2 structure for Ti(Ni)X is more stable compared to the elemental crystal phases of Ti(Ni) and X. Optimized lattice con-stants and estimated D values are also tabulated. 1 (eV) = 1.60218 × 10−19 (J).
X a(TiX) (Å)
∆E(TiX) (eV)
a(NiX) (Å)
∆E(NiX) (eV)
D value Stable site Sc 3.438 0.52 3.161 −1.03 −1.74 Ti Ti 3.237 0.20 3.003 −0.68 −0.99 Ti V 3.106 0.25 2.900 0.09 −0.18 Ti, Ni Cr 3.025 0.20 2.835 0.76 0.63 Ti, Ni Mn 2.968 −0.51 2.795 0.71 1.37 Ni
Fe 2.947 −0.83 2.775 0.76 1.79 Ni Co 2.964 −0.73 2.775 0.61 1.50 Ni Ni 3.003 −0.68 2.792 0.22 1.01 Ni Cu 3.073 −0.02 2.837 0.17 0.21 Ti, Ni
[image:3.595.307.550.437.571.2] [image:3.595.63.271.586.750.2] [image:3.595.322.527.605.763.2]havior to those estimated from Ef. Thus, we can predict that Ti substitutional site is stable for Sc and Ni substitutional site is stable for Mn, Fe and Co. Similar to the results pre-sented in Table 2 and Fig. 1, Ni site is predicted to be stable for Mn contrary to the experimental observation. In the next section, we will show the calculated density of states (DOS) and discuss the origin of the stability based on the COHP analysis for typical TiX and NiX systems.
3.3 DOS and COHP analyses in TiX and NiX
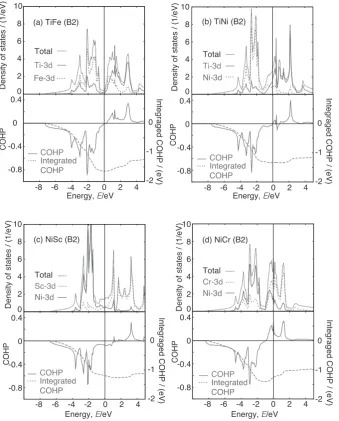
As already pointed out by Yamashita et al.30), B2 structure
is especially stable when the total number of valence elec-trons is 12 per unit cell. This is due to the characteristic band structure of bcc structure. In bcc structure, the DOS shows deep valley in the middle of the DOS and this valley is basi-cally originated from bonding-antibonding splitting of
d-bands. For the pair of two elements which realizes the to-tal valence number of 12, the lower part of DOS are com-pletely occupied and we can expect strong chemical bond-ing. Typical combinations for realizing strong chemical
bonding are TiFe (total valence 12) and NiSc (total valence 13). In fact, these ones are very stable as shown in Fig. 2 and Table 3.
In order to confirm this simple consideration, the DOS and the COHP are calculated for typical cases, such as TiFe, TiNi, NiSc and NiCr in Fig. 3. As shown in the upper panels of respective pictures in Fig. 3, calculated DOS shows char-acteristic deep valley in the middle of the DOS. The peak at lower energy side of the valley originates from d states of heavier elements. On the other hand, the peak at higher en-ergy side of the valley originates from the d states of lighter elements. The former ones are bonding states and the latter ones are anti-bonding counter parts of them. This fact is clearly shown in the plot of the COHP between nearest neighbors shown in the lower panels of respective pictures. The COHP has finite amplitude where the DOS exists, and the peaks in DOS correspond to the peaks in the COHP. Negative COHP indicates that these states have bonding contribution to the pair. As shown in Fig. 3(a), in the case of TiFe, the lower part of the DOS is completely filled up and
[image:4.595.131.471.332.754.2]all of them show bonding contribution. In the case of TiNi (Fig. 3(b)), there are two more electrons than in the case of TiFe and the tail of the upper peak is occupied. However, this tail is almost non-bonding like (very small COHP) and this is why TiNi is still stable. NiSc, which has 13 valence electrons, looks very similar to TiNi case, even though it has one less valence electron than TiNi. In the case of NiCr, there are two more valence electrons compared to NiSc. As shown in the lower panel of Fig. 3(d) the peak at the higher energy side of the DOS, those are anti-bonding states as in-dicated in the COHP plot in the lower panel of Fig. 3(d), are partially occupied and this explains the instability of this system.
So far, we have analyzed the relation between the stability and the electronic structure of TiX and NiX. As mentioned, the atomic interactions in these systems can be approxi-mated by the nearest neighbor interactions. Therefore, simi-lar discussion can be applicable to explain the stable loca-tion of X atoms in TiNi. Namely, from the view point of electron counts per atomic pair, Sc prefers Ti site because the Sc-Ni bonding is optimal. Mn, Fe and Co prefer Ni site because the Ti-Mn, Ti-Fe and Ti-Co bondings are optimal. Situations for V, Cr and Cu are inbetween and neutral, and they can occupy both sites and occupancy may depend on the experimental condition.
3.4 Comparison with the previous studies
Finally, we discuss our results above on the third ele-ments site preferences in B2 type Ti-Ni-X (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, and Cu) comparing to other prior ap-proaches based on first-principle calculations by Sheng et
al.16) and Singh et al.22) To our knowledge, the earliest work
on the current issue above approached by the electronic structure calculation was made by Sheng et al.16) They
pre-dicted the substitution behavior of third elements in TiNi compounds by the calculation using the DV-Xα cluster method, in which bond orders between X and Ti (Ni) were estimated employing Mulliken population analysis. As for the site preference trend of 3d transition metal elements in the B2-type Ti8Ni6X or Ti6Ni8X cluster consists of 15 atoms
(X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, and Cu), their results all agree with those of ALCHEMI analysis by Nakata et al.5,6)
It overall coincides with our results using VASP, except for Cr and Mn as mentioned in the previous section. This dis-crepancy may arise from the atomic coordination optimiza-tion. Namely, the atomic coordination was fixed in the Sheng cluster model, although it was relaxed in our super lattice models.
In the most recent work at this moment using VASP on the third elements site preferences in TiNi compounds, Singh and collaborators22) have reached their conclusions as
follows: (1) Sc substitutes Ti site, and (2) Cr, Mn, Fe, Co, and Cu substitute Ni site, respectively, in either super lattice model of Ti- or Ni-rich. (3) V substitutes deficient site of Ti or Ni. Among these elements, Sc, Fe, Co, and V coincide with the results of ALCEMI analysis by Nakata et al.7,8)
However, Cr, Mn, and Cu are inconsistent with those. On the one hand, our results of Cr and Mn also disagree with those of ALCEMI analysis, which may originate from temperature effect. That is, entropy term of free energy may not be
ne-glected at finite temperatures. On the other hand, the prefer-ence trend of Cu in our calculation is in good agreement with that of ALCEMI analysis, despite that Singh s result was not. The cause of this discrepancy may arise from the difference of super lattice model between ours and Singh s. In the super lattice model we used, the concentration of third element X is 6.25 at%, which is twice as that in Singh s model. However, the detail cause of the discrepancy on Cu s preference trend is unclear at this moment. As for the cause of site preference trend of third elements in TiNi, Singh et
al. pointed out the influence of the comparable size of ter-nary additions to the host atoms. According to our calcula-tion using COHP method, we are convinced that the atomic interaction between host atom (Ti or Ni) and X controls the preference trend of third elements in TiNi.
4. Summary
In this paper, we have presented ab initio prediction of the atomic location of the third elements X in TiNi. Formation energies, which are needed to replace Ti or Ni with X, were calculated for X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni and Cu, based on the density functional theory by using the VASP code. Local atomic relaxations due to the inclusion of X atom were considered by minimizing the atomic forces. It is found that Sc prefers Ti site and Cr, Mn, Fe and Co prefer Ni site. For V and Cu, both sites are possible. This prediction agrees well with the experimental observations by the ALCHEMI method, except for Cr and Mn for which both sites were observed in the experiment for atom locations. In order to clarify the reason of this discrepancy, it is necessary to consider atomic interactions between X atoms in TiNi and include temperature effects by calculating free energy, which is currently in progress. The origin of the preference of the X atom location was analyzed by calculating the DOS and the COHP. The result of our analysis suggests that the characteristic shape of the electronic DOS and occupation of the DOS control the atomic interaction between host atom (Ti or Ni) and X. Since the bonding-like states, which corre-sponds to the peak at the lower energy side of the DOS of bcc structure, are fully occupied with 12 valence electrons per unit cell, atomic pair which has this electron count real-ize strong chemical bonding. With this simple rule, we can understand that lighter elements prefer Ti site, on the other hand, heavier elements prefer Ni site.
Acknowledgement
K. S. acknowledges the financial support from Centerforspintronics research network (CSRN), Osaka University.
REFERENCES
1) K. Otsuka and X. Ren: Intermetallics 7 (1999) 511.
2) J.M. Jani, M. Leary, A. Subic and M.A. Gibson: Mater. Des. 56 (2014) 1078.
3) K.H. Eckelmeyer: Scr. Metall. 10 (1976) 667.
4) H. Hosoda, S. Hanada, K. Inoue, T. Fukui, Y. Mishima and T. Suzuki:
Intermetallics 6 (1998) 291.
580.
6) Y. Nakata, T. Tadaki and K. Shimizu: Mater. Trans., JIM 32 (1991) 1120.
7) Y. Nakata, Doctral Dissertation (1991), Graduate School of Engineering, Osaka University (in Japanese).
8) Y. Nakata: Materia Japan 32 (1993) 858 (in Japanese).
9) T. Tadaki, Y. Nakata and K. Shimizu: J. Phys. Colloq. 5 (1995) C8-81. 10) J.C.H. Spence and J. Taftø: J. Microscopy 130 (1983) 147.
11) H. Hosoda, T. Shinoda, T. Suzuki and Y. Mishima: J. Jpn. Inst. Metals
58 (1994) 483 (in Japanese).
12) H. Hosoda, A. Kamio, T. Suzuki and Y. Mishima: J. Jpn. Inst. Metals
60 (1996) 793 (in Japanese).
13) G. Bozzolo, R. D. Noebe and H. O. Mosca: J. Alloys Comp. 386 (2005) 125.
14) G. Bozzolo, J. Ferrante and J.R. Smith: Phys. Rev. B 45 (1992) 493.
15) G. Bozzolo, R. D. Noebe and H. O. Mosca: J. Alloys Comp. 389 (2005) 80.
16) X.D. Sheng, S. Yan, L. Dong and H.Z. Qi: Philos. Mag. A 75 (1997) 1185.
17) R. S. Mulliken: J. Chem. Phys. 23 (1955) 1833.
18) R. S. Mulliken: J. Chem. Phys. 23 (1955) 1841.
19) R. S. Mulliken: J. Chem. Phys. 23 (1955) 2338.
20) R. S. Mulliken: J. Chem. Phys. 23 (1955) 2343.
21) X. Sun, X. Ni, J. Shen and N. Chen: J. Alloys Comp. 509 (2011) 8323.
22) N. Singh, A. Talapatra, A. Junkaew, T. Duong, S. Gibbons, S. Li, H. Thawabi, E. Olivos and R. Arróyave: Comput. Mater. Sci. 112 (2016) 347.
23) R. Dronskowski and P.E. Bloechl: J. Phys. Chem. 97 (1993) 8617.
24) G. Kresse and D. Joubert: Phys. Rev. B 59 (1999) 1758.
25) G. Kresse and J. Furthmueller: Phys. Rev. B 54 (1996) 11169.
26) J.P. Perdew, K. Burke and M. Ernzerhof: Phys. Rev. B 77 (1996) 3865.
27) H.J. Monkhorst and J.D. Pack: Phys. Rev. B 13 (1976) 5188.
28) P. Duwez and J.L. Taylor: Trans. Am. Inst. Min. Metall. Petr. Eng. 188 (1950) 1173.
29) A.T. Aldred: Trans. Metall. AIME 224 (1962) 1082.
30) J. Yamashita and S. Asano: Prog. Theor. Phys. 48 (1972) 2119.
31) J.M. Lu, Q.M. Hu, L. Wang, Y.J. Li, D.S. Xu and R. Yang: Phys. Rev. B 75 (2007) 094108.