Neuroinflammatory changes increase the impact
of stressors on neuronal function
Alessia Piazza and Marina A. Lynch1
Trinity College Institute of Neuroscience, Trinity College, Dublin 2, Ireland
Abstract
In the last few years, several research groups have reported that neuroinflammation is one feature common to several neurodegenerative diseases and that similar, although perhaps less profound, neuroinflammatory changes also occur with age. Age is the greatest risk factor in many neurodegenerative diseases, and the possibility exists that the underlying age-related neuroinflammation may contribute to this increased risk. Several animal models have been used to examine this possibility, and it is now accepted that, under experimental conditions in which microglial activation is up-regulated, responses to stressors are exacerbated. In the present article, these findings are discussed and data are presented fromin vitroand in vivoexperiments which reveal that responses to Aβ (amyloidβ-peptide) are markedly up-regulated in the presence of LPS (lipopolysaccharide). These, and previous findings, point to a vulnerability associated with inflammation and suggest that, even though inflammation may not be the primary cause of neurodegenerative disease, its treatment may decelerate disease progression.
Neuroinflammation is a feature
of Alzheimer’s disease
In the last decade or so, there has been a consolidation of the evidence demonstrating a causal relationship between neuroinflammatory changes and age-related deterioration in neuronal function. This correlation has been observed in several neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease [1]. Since the original observation that the risk of developing Alzheimer’s disease was demonstrated to be reduced in patients on long-term non-steroidal anti-inflammatory treatment [1], a great deal of interest has focused on examining the role of inflammation in the pathogenesis of the disease, in the anticipation that appropriate anti-inflammatory therapy may alleviate or limit the symptoms of the disease. Interestingly, some of the neuroinflammatory changes characteristic of Alzheimer’s disease also occur in normal aging [2]; this is significant since age is the greatest risk factor for Alzheimer’s disease and therefore it might be speculated that this risk is related to the underlying age-related inflammation.
One indicator of neuroinflammatory change which has been demonstrated in Alzheimer’s disease is an increase in expression of pro-inflammatory cytokines, particularly IL
(interleukin)-1βwhich parallels, and probably contributes to,
development of Aβ(amyloidβ-peptide)-containing plaques
[3]. This observation, and the finding that polymorphisms in genes encoding pro-inflammatory cytokines is associated with increased risk of developing Alzheimer’s disease [3],
Key words:aging, amyloidβ-peptide (Aβ), interleukin-1β(IL-1β), long-term potentiation (LTP), microglial activation, neurodegeneration.
Abbreviations used:Aβ, amyloidβ-peptide; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; IFNγ, interferonγ; LPS, lipopolysaccharide, LTP, long-term potentiation. 1To whom correspondence should be addressed (email [email protected]).
suggest a role for IL-1βin its pathogenesis. Since microglial
cells are the primary source of IL-1β, it is not surprising
that microglial activation is a feature of Alzheimer’s disease
where IL-1β-positive microglia are found clustered around
amyloid plaques [4,5]. Increased expression of MCP-1 (monocyte chemoattractant protein-1), which is another indicator of activated microglia, has been reported in serum and peripheral blood mononuclear cells of Alzheimer’s disease patients and the evidence revealed that it correlated significantly with MMSE (Mini-Mental State Examination) score [6], whereas expression of ICAM-1 (intercellular adhesion molecule 1), which is yet another marker of
activated microglia, co-localizes with Aβ-containing lesions
[7]. It is interesting that up-regulation of inflammatory markers co-localizes with those areas of the brain which are mainly affected by the disease and are low or minimal in brain regions with very low Alzheimer’s disease susceptibility [8].
Animal models of Alzheimer’s disease
exhibit neuroinflammatory changes
Much of the data suggesting that inflammatory changes occur in Alzheimer’s disease have been supported by findings from animal models. First, in animal models, the increase in microglial activation in brain tissue is accompanied by up-regulation of pro-inflammatory cytokines such as
interleukin-1β, IL-6 and TNFα(tumour necrosis factor α)
and chemokines such as MIP-1α(macrophage inflammatory
peptide-1α), whereas nNOS (neuronal nitric oxide synthase)
has been shown to co-localize with activated glia [9,10]. Secondly, there is evidence of increased microglial activation and increased pro-inflammatory cytokine production
following injection of Aβ [11–15] and in transgenic mice
which overexpress APP (amyloid precursor protein) [16].
Thirdly, the clustering of activated microglia around Aβ
deposits which has been demonstrated in post-mortem tissue is also observed in animal models [17,18]. Finally, the evidence indicates that treatment of animals with cyclo-oxygenase inhibitors or agents which have been shown to possess
anti-inflammatory properties such as the PPARγ
(peroxisome-proliferator-activated receptor γ) agonist pioglitazone or
polyunsaturated fatty acids, such as docosapentaenoic acid or eicosapentaenoic acid, reduces markers of inflammation,
reduces plaque burden and/or ameliorates Aβ-induced
impairment in hippocampal functioning [11,13,14,16,19].
Neuroinflammatory changes are evident in
other neurodegenerative conditions
In addition to Alzheimer’s disease, neuroinflammatory changes have also been described in Parkinson’s disease [20]. It is known that the underlying cause of Parkinson’s disease is degeneration of dopaminergic neurons in the substantia nigra, and, although genetic and environmental factors are known to be major risk factors, it has been acknow-ledged that inflammatory changes are a feature of the disease. It remains to be established whether these inflammatory changes contribute to the pathogenesis of the disease or are a consequence of the cell loss. Among the indicators of neuroinflammation in Parkinson’s disease is increased mi-croglial activation in the substantia nigra [20], and polymor-phisms in genes encoding pro-inflammatory cytokines have also been reported [21]. Consistent with evidence for inflammatory changes is the finding that non-steroidal anti-inflammatory treatment has beneficial, although limited, effects in modulating the symptoms of the disease [22].
An association between inflammatory changes and prion diseases, multiple sclerosis, amyotrophic lateral sclerosis and HIV has also been established [23–26]. Consistent with a role for inflammatory changes in the pathogenesis of these diseases is the finding that anti-inflammatory treatment, at least in some cases, can be beneficial. It should be acknow-ledged that, whereas neuroinflammation is a factor common to several neurodegenerative diseases, it may not be the initiator of the disease, but rather a secondary effect. However, there is undisputed evidence that neuroinflam-matory changes trigger detrimental effects and, consequently, it is vital to consider the causes, consequences and strategies for limiting these detrimental effects.
Inflammatory changes and the aged brain
There is an increasing acceptance that normal aging is also associated with evidence of inflammatory change, and it is significant that some of the neuroinflammatory changes which have been observed in these neurodegenerative con-ditions also occur in normal aging, e.g. microglial activation,
increased IL-1 expression and increased S100βexpression [2].
The age-related increase in microglial activation [27] has been associated with evidence of morphological changes including
loss of processes, dystrophic processes and pyknotic nuclei [4], and studies in aged animals have confirmed these observations [28], where an increase in expression
of pro-inflammatory cytokines such as IL-1β or IL-6
have been consistently reported [29,30]. These age-related changes are associated with a decrease in synaptic plasticity, specifically a reduction in LTP (long-term potentiation) [14,31], and a decrease in cognitive function, specifically poorer performance in hippocampal-dependent tasks [30].
It has been consistently shown that decreasing the inflam-matory phenotype in the brain of aged animals decreases these impairments, therefore reducing the hippocampal
concen-tration of IL-1β, perhaps by increasing the concentration
of the anti-inflammatory cytokine, IL-4, ameliorates the age-related impairment in LTP [14,31] and the deficit in spatial learning [32,33]. Similarly, an impairment in the hippocampal-dependent object-recognition task was observed with age, and this impairment was attenuated by dietary supplementation with blueberries which possess anti-oxidant and anti-inflammatory properties [32,34].
Microglial activation affects synaptic
function
The probable source of the inflammatory cytokines is activated microglia, and there are several reports indicating that microglial activation accompanies increased expression of pro-inflammatory cytokines [28,33,35,36]; paired increases
have been reported in aged and Aβ-treated rats, and,
import-antly, it has been shown that when microglial activation is
decreased, IL-1βconcentration is also decreased. It has also
been shown that the inhibitor of microglial activation,
mino-cycline, restores LTP in aged rats [28] and Aβ-treated rats
[12]. These findings illustrate the importance of maintaining microglia in a resting state for the preservation of synaptic function, and they highlight the need to understand the mech-anisms involved in modulation of microglial function.
Among the most potent activators of microglia is IFNγ
(interferon γ) [37], and the evidence shows that
intra-cerebroventricular injection of IFNγincreases microglial
ac-tivation [38], and that the age-associated [31] and Aβ-induced
[12] increases in microglial activation are accompanied by
increased hippocampal concentration of IFNγ. However
Figure 1 LPS and Aβexert an additive effect on mixed glia
(a) Incubation of mixed glia in the presence of LPS (10 ng/ml), but not Aβ1−42 (2μM), significantly increased MHCII mRNA (**P<0.01;
ANOVA); co-incubation in the presence of both further increased MHCII mRNA (***P<0.001; ANOVA). (b) Co-incubation in the presence of LPS and Aβsignificantly increased IL-1βconcentration (*P<0.05; ANOVA), but neither agent alone exerted any significant effect. Results are means±S.E.M. for six observations.
Evidence that increased microglial
activation imposes vulnerability to stress
There is a growing awareness that underlying inflammation can exert a negative impact on the progression in some neurodegenerative diseases. Specifically, it has been shown that systemic infections can exacerbate symptoms and trigger rapid progression in Parkinson’s disease [40], multiple sclerosis [41] and Alzheimer’s disease [42], and that infection influences recovery in stroke [43].
It has been proposed that this increased susceptibility to a stressor is a consequence of an underlying increase in the activation state of microglia, which leads to release of inflammatory mediators, and this is borne out by findings from several laboratories using several model systems. Thus exposure of an animal to inescapable shock increases the response to LPS (lipopolysaccharide) [44], whereas, in a model of prion disease, induced by injecting mice with scrapie-infected brain homogenate, Perry and colleagues reported a more profound LPS-induced effect on temperature and locomotor activity compared with non-infected mice; these changes were paralleled by more marked changes in
IL-1βin the brain [45]. Consistently, treatment of aged mice
with LPS orEscherichia colior HIV-1 gp120 (glycoprotein
120), which stimulates the innate immune system, exacerbates depressive-like symptoms and sickness behaviour and exerts a greater effect on working memory [46–49]. In these exper-imental models, the exaggerated responses were attributed to increased microglial activation with, in some cases, increased resting concentrations of pro-inflammatory cytokines.
The evidence suggests that the effect of Aβis dependent on
inflammatory status, thus we have found that concentrations
of Aβwhich exerted no effect on IL-1βproduction or LTP
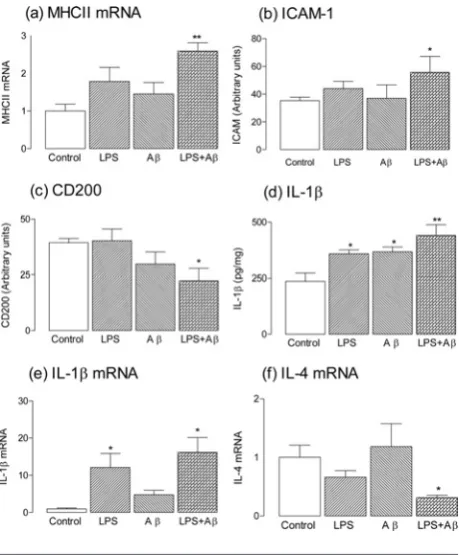
Figure 2 LPS and Aβin vivoexert an additive effect on neuroinflammatory changes in hippocampus
MHCII mRNA (a), ICAM (b), IL-1β (d) and IL-1β mRNA (e) were significantly increased in hippocampal tissue prepared from rats which received chronic intracerebroventricular administration of LPS (0.5 mg/ml), Aβ(18.9μM Aβ1−40and 26.6μM Aβ1−42) alone or in
combination for 28 days (*P<0.05; **P<0.01; ANOVA), whereas CD200 (c) and IL-4 mRNA (f) were significantly decreased (*P<0.05; ANOVA). Aβalone exerted no significant effects on any measure except on IL-1β, whereas LPS alone increased both IL-1βand IL-1β mRNA (*P<0.05; ANOVA). Results are means±S.E.M. for six to twelve observations.
in young rats enhanced the already increased IL-1β
concen-tration in hippocampus of aged rats and inhibited further the ability of these rats to sustain LTP [13]; we proposed that this was due to the underlying inflammation which is a feature of the aged brain. This additive effect of endogenous and exogenous stimuli, or even a synergism between them, has been described by several authors and has led to the proposal that, under circumstances in which microglia are in a primed state, inflammatory stimuli triggers an exaggerated
response [42,45]. In vitro analysis has also indicated that
prior exposure of glia to one inflammatory stimulus leads to a greater response to a second stimulus. For example, the data presented in Figure 1 indicate that incubation of
rat glial cells in the presence of LPS and Aβ increased
MHCII mRNA and IL-1β production in an additive or
even synergistic manner which is similar to results reported previously [50]. We have recently assessed the interaction of
Aβand LPSin vivoand show that, whereas chronic infusion
with Aβor LPS alone did not significantly affect hippocampal
[image:3.595.288.518.200.478.2]and significantly decreased CD200 (Figures 2a–2c); this inverse relationship supports the proposal that CD200 plays a role in modulating microglial activation [39]. The data also
show that IL-1β concentration in hippocampus prepared
from rats treated with Aβand LPS was significantly greater
than that treated with either alone, although both stimuli
individually increased IL-1β (Figure 2d). Consistent with
a modulatory effect of IL-4 on IL-1β production [14], we
show that the Aβ+LPS-induced increase in IL-1βmRNA
was accompanied by a decrease in IL-4 mRNA (Figures 2e and 2f). These findings are consistent with others which have also shown that coincident exposure to two stressors can result in amplification of the effect of either alone [46–49].
Conclusion
The data described here, and elsewhere [19], which
demon-strate an interaction between Aβand LPS and the finding that
systemic infections can exacerbate symptoms and/or trigger the progression of some neurodegenerative diseases [40–42], suggest that reducing underlying inflammatory changes in the brain may be beneficial in limiting responsiveness to additional stressors. However the challenges are (i) to id-entify the time at which this intervention may be most effective, and (ii) to recognize that the limited success in clinical trials to date, in which anti-inflammatory agents are assessed in late chronic neurodegenerative disease, may be due to inappropriate timing of the intervention.
Funding
This work was supported by Science Foundation Ireland.
References
1 McGeer, P.L. and McGeer, E.G. (2001) Inflammation, autotoxicity and Alzheimer disease. Neurobiol. Aging22, 799–809
2 Sheng, J.G., Mrak, R.E. and Griffin, W.S. (1998) Enlarged and phagocytic, but not primed, interleukin-1α-immunoreactive microglia increase with age in normal human brain. Acta. Neuropathol.95, 229–234 3 Griffin, W.S. and Mrak, R.E. (2002) Interleukin-1 in the genesis and
progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J. Leukocyte Biol.72, 233–238
4 Miller, K.R. and Streit, W.J. (2007) The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 3, 245–253
5 Mrak, R.E. and Griffin, W.S. (2005) Potential inflammatory biomarkers in Alzheimer’s disease. J. Alzheimers Dis.8, 369–375
6 Galimberti, D., Fenoglio, C., Lovati, C., Venturelli, E., Guidi, I., Corra, B., Scalabrini, D., Clerici, F., Mariani, C., Bresolin, N. and Scarpini, E. (2006) Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging27, 1763–1768
7 Verbeek, M.M., Otte-Holler, I., Wesseling, P., Ruiter, D.J. and de Waal, R.M. (1996) Differential expression of intercellular adhesion molecule-1 (ICAM-1) in the Aβ-containing lesions in brains of patients with dementia of the Alzheimer type. Acta Neuropathol.91, 608–615 8 Smith, M.A., Richey Harris, P.L., Sayre, L.M., Beckman, J.S. and Perry, G.
(1997) Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci.17, 2653–2657
9 Simic, G., Lucassen, P.J., Krsnik, Z., Kruslin, B., Kostovic, I., Winblad, B. and Bogdanovi (2000) nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Exp. Neurol.165, 12–26
10 Tuppo, E.E. and Arias, H.R. (2005) The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol.37, 289–305 11 Clarke, R.M., O’Connell, F., Lyons, A. and Lynch, M.A. (2007) The
HMG-CoA reductase inhibitor, atorvastatin, attenuates the effects of acute administration of amyloid-β1–42 in the rat hippocampusin vivo. Neuropharmacology52, 136–145
12 Lyons, A., Griffin, R.J., Costelloe, C.E., Clarke, R.M. and Lynch, M.A. (2007) IL-4 attenuates the neuroinflammation induced by amyloid-βin vivo
andin vitro. J. Neurochem.101, 771–781
13 Minogue, A.M., Lynch, A.M., Loane, D.J., Herron, C.E. and Lynch, M.A. (2007) Modulation of amyloid-β-induced and age-associated changes in rat hippocampus by eicosapentaenoic acid. J. Neurochem.103, 914–926 14 Lynch, A.M., Loane, D.J., Minogue, A.M., Clarke, R.M., Kilroy, D., Nally,
R.E., Roche, O.J., O’Connell, F. and Lynch, M.A. (2007) Eicosapentaenoic acid confers neuroprotection in the amyloid-βchallenged aged hippocampus. Neurobiol. Aging28, 845–855
15 Block, M.L. and Hong, J.S. (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog. Neurobiol.76, 77–98
16 Heneka, M.T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O’Banion, K., Klockgether, T., Van Leuven, F. and Landreth, G.E. (2005) Acute treatment with the PPARγagonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1−42levels in APPV717I
transgenic mice. Brain128, 1442–1453
17 Stalder, M., Phinney, A., Probst, A., Sommer, B., Staufenbiel, M. and Jucker, M. (1999) Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol.154, 1673–1684
18 Frautschy, S.A., Yang, F., Irrizarry, M., Hyman, B., Saido, T.C., Hsiao, K. and Cole, G.M. (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol.152, 307–317
19 Cakala, M., Malik, A.R. and Strosznajder, J.B. (2007) Inhibitor of cyclooxygenase-2 protects against amyloidβpeptide-evoked memory impairment in mice. Pharmacol. Rep.59, 164–172
20 McGeer, P.L., Itagaki, S., Boyes, B.E. and McGeer, E.G. (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology38, 1285–1291
21 Nishimura, M., Mizuta, I., Mizuta, E., Yamasaki, S., Ohta, M. and Kuno, S. (2000) Influence of interleukin-1βgene polymorphisms on age-at-onset of sporadic Parkinson’s disease. Neurosci. Lett.284, 73–76
22 Chen, H., Jacobs, E., Schwarzschild, M.A., McCullough, M.L., Calle, E.E., Thun, M.J. and Ascherio, A. (2005) Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol.58, 963–967 23 Cozzolino, M., Ferri, A. and Carri, M.T. (2008) Amyotrophic lateral
sclerosis: from current developments in the laboratory to clinical implications. Antioxid. Redox Signaling10, 405–443
24 Martino, G., Adorini, L., Rieckmann, P., Hillert, J., Kallmann, B., Comi, G. and Filippi, M. (2002) Inflammation in multiple sclerosis: the good, the bad, and the complex. Lancet Neurol.1, 499–509
25 Aguzzi, A. and Heikenwalder, M. (2006) Pathogenesis of prion diseases: current status and future outlook. Nat. Rev. Microbiol.4, 765–775 26 Reynolds, A., Laurie, C., Mosley, R.L. and Gendelman, H.E. (2007)
Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol.82, 297–325
27 Conde, J.R. and Streit, W.J. (2006) Microglia in the aging brain. J. Neuropathol. Exp. Neurol.65, 199–203
28 Griffin, R., Nally, R., Nolan, Y., McCartney, Y., Linden, J. and Lynch, M.A. (2006) The age-related attenuation in long-term potentiation is associated with microglial activation. J. Neurochem.99, 1263–1272 29 Godbout, J.P. and Johnson, R.W. (2004) Interleukin-6 in the aging brain.
J. Neuroimmunol.147, 141–144
30 Lynch, M.A. (2004) Long-term potentiation and memory. Physiol. Rev. 84, 87–136
31 Clarke, R.M., Lyons, A., O’Connell, F., Deighan, B.F., Barry, C.E., Anyakoha, N.G., Nicolaou, A. and Lynch, M.A. (2008) A pivotal role for IL-4 in atorvastatin-associated neuroprotection in rat brain. J. Biol. Chem.283, 1808–1817
32 Frautschy, S.A., Hu, W., Kim, P., Miller, S.A., Chu, T., Harris-White, M.E. and Cole, G.M. (2001) Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiol. Aging22, 993–1005
33 Hauss-Wegrzyniak, B., Vraniak, P. and Wenk, G.L. (1999) The effects of a novel NSAID on chronic neuroinflammation are age dependent. Neurobiol. Aging20, 305–313
35 Kullberg, S., Aldskogius, H. and Ulfhake, B. (2001) Microglial activation, emergence of ED1-expressing cells and clusterin upregulation in the aging rat CNS, with special reference to the spinal cord. Brain Res.899, 169–186
36 Perry, V.H., Matyszak, M.K. and Fearn, S. (1993) Altered antigen expression of microglia in the aged rodent CNS. Glia7, 60–67 37 Nguyen, V.T. and Benveniste, E.N. (2000) IL-4-activated STAT-6 inhibits
IFN-γ-induced CD40 gene expression in macrophages/microglia. J. Immunol.165, 6235–6243
38 Maher, F.O., Clarke, R.M., Kelly, A., Nally, R.E. and Lynch, M.A. (2006) Interaction between interferonγand insulin-like growth factor-1 in hippocampus impacts on the ability of rats to sustain long-term potentiation. J. Neurochem.96, 1560–1571
39 Lyons, A., Downer, E.J., Crotty, S., Nolan, Y.M., Mills, K.H. and Lynch, M.A. (2007) CD200 ligand receptor interaction modulates microglial activation
in vivoandin vitro: a role for IL-4. J. Neurosci.27, 8309–8313 40 Weller, C., Oxlade, N., Dobbs, S.M., Dobbs, R.J., Charlett, A. and
Bjarnason, I.T. (2005) Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunol. Med. Microbiol.44, 129–135
41 Correale, J., Fiol, M. and Gilmore, W. (2006) The risk of relapses in multiple sclerosis during systemic infections. Neurology67, 652–659 42 Holmes, C., El-Okl, M., Williams, A.L., Cunningham, C., Wilcockson, D. and
Perry, V.H. (2003) Systemic infection, interleukin 1β, and cognitive decline in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry74, 788–789
43 Palasik, W., Fiszer, U., Lechowicz, W., Czartoryska, B., Krzesiewicz, M. and Lugowska, A. (2005) Assessment of relations between clinical outcome of ischemic stroke and activity of inflammatory processes in the acute phase based on examination of selected parameters. Eur. Neurol.53, 188–193
44 Frank, M.G., Baratta, M.V., Sprunger, D.B., Watkins, L.R. and Maier, S.F. (2007) Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun.21, 47–59
45 Combrinck, M.I., Perry, V.H. and Cunningham, C. (2002) Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience112, 7–11
46 Chen, J., Buchanan, J.B., Sparkman, N.L., Godbout, J.P., Freund, G.G. and Johnson, R.W. (2008) Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun.22, 301–311
47 Godbout, J.P., Moreau, M., Lestage, J., Chen, J., Sparkman, N.L., Connor, J.O., Castanon, N., Kelley, K.W., Dantzer, R. and Johnson, R.W. (2008) Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system.
Neuropsychopharmacology33, 2341–2351
48 Barrientos, R.M., Higgins, E.A., Biedenkapp, J.C., Sprunger, D.B., Wright-Hardesty, K.J., Watkins, L.R., Rudy, J.W. and Maier, S.F. (2006) Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol. Aging27, 723–732
49 Abraham, J., Jang, S., Godbout, J.P., Chen, J., Kelley, K.W., Dantzer, R. and Johnson, R.W. (2008) Aging sensitizes mice to behavioral deficits induced by central HIV-1 gp120. Neurobiol. Aging29, 614–621 50 Gasic-Milenkovic, J., Dukic-Stefanovic, S., Deuther-Conrad, W., Gartner, U.
and Munch, G. (2003)β-Amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon-γand ‘advanced glycation endproducts’ in a murine microglia cell line. Eur. J. Neurosci. 17, 813–821