organic papers
Acta Cryst.(2006). E62, o873–o874 doi:10.1107/S1600536806003205 Shaheenet al. C
11H13NO
o873
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
(
Z
)-4-Anilinopent-3-en-2-one
Farkhanda Shaheen,a* L Marchio,bAmin Badshahaand Muhammad Kaleem Khosac
aDepartment of Chemistry, Quaid-i-Azam
University, Islamabad 45320, Pakistan,bDip. di
Chimica Generale ed Inorganica Chimica Analitica Fisica, Universita di Parma, Parco Area delle, Scienze 17/A43100, Parma, Italy, and
c
Department of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
Correspondence e-mail: fshaheenpk@yahoo.com
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.004 A˚
Rfactor = 0.048
wRfactor = 0.139
Data-to-parameter ratio = 11.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 3 January 2006 Accepted 26 January 2006
#2006 International Union of Crystallography All rights reserved
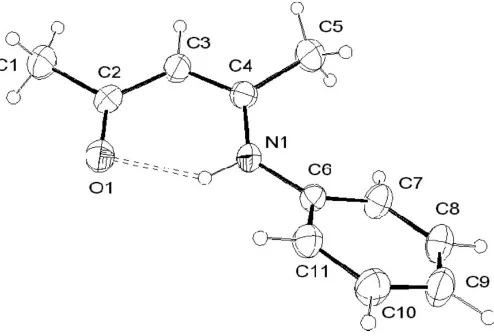
The title compound, C11H13NO, crystallizes as theZisomer of
the-enamino–ketone. An intramolecular hydrogen-bonding interaction exists between the N—H and C O groups.
Comment
The chelating tendencies of-diketones have been extensively studied because of their applications in pharmaceutical compounds, biochemistry, biomedical research and immu-nochemistry (Sandler & Karo, 1986; Chen & Rhodes, 1996) and as precursors for the preparation of a large number of heterocyclic compounds (Khosropour et al., 2004). Previous studies of some cyclizations involved the preparation of 4-methylaminopent-3-en-2-ones (Kaschereset al., 2001), (1-methylethylaminomethylene)-1,3-dioxane-4,6-diones and 5- (dimethylethylaminomethylene)-2,2-dimethyl-1,3-dioxane-4,6-diones (Zhuo, 1997). The anticonvulsant activities of various acyclic and cyclic enaminoketones have been studied in connection with their lipophilicity, steric, electronic and hydrogen-bonding effects (Edafioghoet al., 1994; Hinkoet al., 1993). The condensation products of acetylacetone with various amines exist in three forms,viz. the Schiff base, the ketamine and the enimine. The interchange between the ketamine and enimine tautomers involves a small displace-ment in the equilibrium position of the acidic proton.
In the title compound, (I), the C2 O1 and C2—C3 bond distances [1.224 (3) and 1.424 (4) A˚ , respectively] confirm the existence of the enamino–ketone. The bond distances in the C3 C4—N1 chain indicate greater electron delocalization [C3 C4 = 1.365 (3) A˚ and N1—C4 = 1.365 (3) A˚ ]. The
enamino–ketone fragment (N1—C4 C3—C2 O1) is
essentially planar, the maximum deviation being 0.011 (3) A˚ for atom C2. The dihedral angle between this fragment and the phenyl ring is 32.06 (9). This is a result of the steric
molecule is the Z isomer and an intramolecular hydrogen bond is present between the keto and imino groups [N1—H1 = 0.89 (3) A˚ , H1 O1 = 1.92 (3) A˚ , N1 O1 = 2.675 (3) A˚ and N1—H1 O1 = 140 (2)].
Experimental
The title compound was prepared by modification of a published method (Fusteroet al., 1999). Aniline (15.26 ml, 0.1 mol) was added to acetylacetone (20 ml, 0.1 mol) and two drops of H2SO4in benzene
(50 ml). The reaction mixture was refluxed for 4 h. On cooling, the product was filtered and recrystallized from a chloroform/n-hexane (9:1v/v) mixture.
Crystal data
C11H13NO
Mr= 175.22
Monoclinic,P21=c a= 9.157 (3) A˚
b= 11.040 (7) A˚
c= 10.130 (5) A˚ = 106.10 (3)
V= 983.9 (9) A˚3
Z= 4
Dx= 1.183 Mg m
3
MoKradiation Cell parameters from 24
reflections = 6.0–17.8
= 0.08 mm1
T= 293 (2) K Prism, colorless 0.600.400.40 mm
Data collection
Philips PW 1100 diffractometer !/2scans
Absorption correction: refined from
F[local program based on Walker & Stuart (1983)]
Tmin= 0.893,Tmax= 0.970
1812 measured reflections 1717 independent reflections 1120 reflections withI> 2(I)
Rint= 0.022
max= 25.0
h=10!10
k= 0!13
l= 0!12
1 standard reflections every 100 reflections intensity decay: none
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.048
wR(F2) = 0.139
S= 1.04 1717 reflections 148 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.059P)2
+ 0.3446P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.16 e A˚
3
min=0.12 e A˚
3
Methyl H atoms were located in difference Fourier syntheses and refined as rigid rotating groups, riding on their parent atoms, with C— H = 0.96 A˚ andUiso(H) =Ueq(C). All other H atoms were found in a
difference map and refined freely.
Data collection: local program; cell refinement: local program; data reduction: local program; program(s) used to solve structure:
SIR97(Altomareet al., 1999); program(s) used to refine structure:
SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997); software used to prepare material for publication:PARST(Nardelli, 1995).
For financial support of this research, we thank the Higher Education Commission Islamabad, Pakistan.
References
Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999).J. Appl. Cryst.32, 115–119.
Chen, H. & Rhodes, J. (1996).J. Mol. Med.74, 497–504.
Edafiogho, I. O., Moore, J. A., Alexander, M. S. & Scott, K. R. (1994).J. Pharm. Sci.83, 1155–1170.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Fustero, S., Torre, G. M., Pina, B. & Fuentes, S. A. (1999).J. Org. Chem.64, 5551–5556.
Hinko, C. N., Change, H., El-Assadi, A. & Nicholson, J. M. (1993).J. Med. Chem.36, 1947–1955.
Kascheres, C., Negri, G., Ferreira, M. C. M. & Sabino, L. C. (2001).J. Chem. Soc. Perkin Trans. 2, pp. 2237–2243.
Khosropour, A. R., Khodaei, M. M. & Kookhazadeh, M. (2004).Tetrahedron Lett.45, 1725–1728.
Nardelli, M. (1995).J. Appl. Cryst.28, 659.
Sandler, S. R. & Karo, W. (1986).Organic Functional Group Preparation. Vol. 11, pp. 291–319. New York: Academic Press.
Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Walker, N. & Stuart, D. (1983).Acta Cryst.A39, 158–166.
[image:2.610.317.564.71.239.2]Zhuo, J. C. (1997).Molecules,2, 31–35.
Figure 1
supporting information
sup-1 Acta Cryst. (2006). E62, o873–o874
supporting information
Acta Cryst. (2006). E62, o873–o874 [https://doi.org/10.1107/S1600536806003205]
(
Z
)-4-Anilinopent-3-en-2-one
Farkhanda Shaheen, L Marchio, Amin Badshah and Muhammad Kaleem Khosa
(Z)-4-Anilinopent-3-en-2-one
Crystal data C11H13NO
Mr = 175.22
Monoclinic, P21/c
a = 9.157 (3) Å b = 11.040 (7) Å c = 10.130 (5) Å β = 106.10 (3)° V = 983.9 (9) Å3
Z = 4 F(000) = 376
Dx = 1.183 Mg m−3
Melting point: 240 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 24 reflections θ = 6.0–17.8°
µ = 0.08 mm−1
T = 293 K Prism, colorless 0.60 × 0.40 × 0.40 mm
Data collection Philips PW 1100
diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
θ–2θ scans
Absorption correction: part of the refinement model (ΔF)
[local program based on Walker & Stuart (1983)]
Tmin = 0.893, Tmax = 0.970
1812 measured reflections 1717 independent reflections 1120 reflections with I > 2σ(I) Rint = 0.022
θmax = 25.0°, θmin = 3.2°
h = −10→10 k = 0→13 l = 0→12
1 standard reflections every 100 reflections intensity decay: none
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.048
wR(F2) = 0.139
S = 1.04 1717 reflections 148 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.059P)2 + 0.3446P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.16 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N1 0.0879 (2) 0.20512 (18) 0.3349 (2) 0.0473 (5) C5 0.1041 (3) 0.4261 (2) 0.3023 (3) 0.0668 (8) H5A 0.2126 0.4187 0.3349 0.100* H5B 0.0721 0.4974 0.3408 0.100* H5C 0.0742 0.4322 0.2039 0.100* O1 −0.1231 (2) 0.12712 (17) 0.4494 (2) 0.0677 (6) C6 0.2128 (2) 0.1669 (2) 0.2893 (2) 0.0421 (6) C3 −0.0889 (3) 0.3310 (2) 0.3985 (3) 0.0502 (6) C4 0.0312 (3) 0.3168 (2) 0.3448 (2) 0.0457 (6) C2 −0.1630 (3) 0.2349 (2) 0.4481 (2) 0.0517 (6) C11 0.2923 (3) 0.0664 (2) 0.3519 (3) 0.0541 (7) C8 0.3746 (3) 0.1763 (3) 0.1404 (3) 0.0654 (8) C9 0.4554 (3) 0.0776 (3) 0.2044 (3) 0.0666 (8) C7 0.2532 (3) 0.2207 (3) 0.1806 (3) 0.0559 (7) C10 0.4129 (3) 0.0223 (3) 0.3096 (3) 0.0648 (8) C1 −0.2942 (3) 0.2663 (3) 0.5042 (3) 0.0700 (8) H1A −0.3787 0.2141 0.4637 0.105* H1B −0.3234 0.3490 0.4826 0.105* H1C −0.2643 0.2557 0.6021 0.105* H7 0.194 (3) 0.287 (2) 0.131 (3) 0.059 (7)* H3 −0.121 (3) 0.413 (2) 0.401 (2) 0.053 (7)* H11 0.262 (3) 0.034 (2) 0.422 (3) 0.059 (8)* H10 0.464 (3) −0.049 (3) 0.353 (3) 0.073 (8)* H9 0.542 (4) 0.051 (3) 0.175 (3) 0.085 (9)* H8 0.400 (3) 0.211 (2) 0.065 (3) 0.068 (8)* H1 0.039 (3) 0.147 (2) 0.365 (2) 0.058 (8)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2006). E62, o873–o874
C2 0.0534 (15) 0.0534 (16) 0.0536 (15) 0.0034 (12) 0.0236 (12) −0.0052 (12) C11 0.0629 (17) 0.0532 (16) 0.0523 (16) −0.0013 (13) 0.0261 (14) 0.0048 (13) C8 0.0654 (18) 0.083 (2) 0.0576 (17) −0.0023 (16) 0.0341 (15) 0.0072 (16) C9 0.0532 (17) 0.082 (2) 0.074 (2) 0.0002 (16) 0.0341 (15) −0.0026 (17) C7 0.0578 (16) 0.0677 (18) 0.0471 (15) 0.0059 (13) 0.0227 (13) 0.0091 (13) C10 0.0629 (18) 0.0594 (18) 0.077 (2) 0.0109 (14) 0.0271 (16) 0.0076 (15) C1 0.0678 (18) 0.0686 (18) 0.087 (2) 0.0011 (15) 0.0439 (16) −0.0039 (16)
Geometric parameters (Å, º)
N1—C4 1.352 (3) C2—C1 1.505 (3)
N1—C6 1.412 (3) C11—C10 1.379 (4)
N1—H1 0.89 (3) C11—H11 0.90 (3)
C5—C4 1.499 (3) C8—C9 1.374 (4)
C5—H5A 0.9600 C8—C7 1.376 (4)
C5—H5B 0.9600 C8—H8 0.94 (3)
C5—H5C 0.9600 C9—C10 1.375 (4)
O1—C2 1.244 (3) C9—H9 0.96 (3)
C6—C11 1.381 (3) C7—H7 0.96 (3)
C6—C7 1.389 (3) C10—H10 0.96 (3)
C3—C4 1.365 (3) C1—H1A 0.9600
C3—C2 1.424 (4) C1—H1B 0.9600
C3—H3 0.95 (2) C1—H1C 0.9600