organic papers
o2644
Ivan Halaszet al. C16H23BrO10 doi:10.1107/S1600536805022555 Acta Cryst.(2005). E61, o2644–o2645
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
2-Bromoethyl 2,3,4,6-tetra-
O
-acetyl-b-
D-gluco-pyranoside
Ivan Halasz,a* Renata Odzˇak,a Srdanka Tomic´aand Dubravka Matkovic´-Cˇ alogovic´b
aLaboratory of Organic Chemistry, Department
of Chemistry, Faculty of Science, University of Zagreb, Strossmayerov trg 14, 10000 Zagreb, Croatia, andbLaboratory of General and Inorganic Chemistry, Department of Chemistry, Faculty of Science, University of Zagreb, Zvonimirova 8, 10002 Zagreb, Croatia
Correspondence e-mail: ihalasz@chem.pmf.hr
Key indicators
Single-crystal X-ray study
T= 295 K
Mean(C–C) = 0.006 A˚
Rfactor = 0.047
wRfactor = 0.142
Data-to-parameter ratio = 15.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
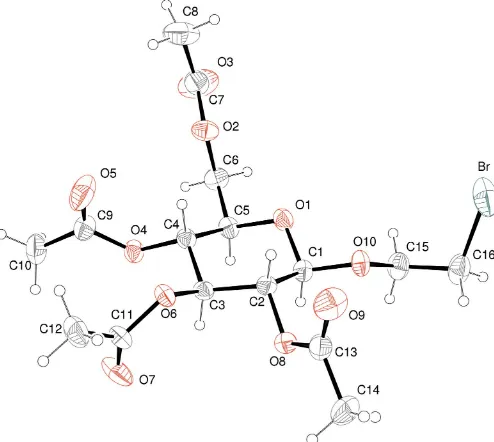
The six-membered ring of the title compound, C16H23BrO10, adopts a chair conformation. The molecules are intercon-nected by weak C—H O hydrogen bonds into a three-dimensional network.
Comment
The synthesis of oligosaccharides has played an important role in the development of carbohydrate chemistry. Today, many different procedures are known, and these are constantly being improved and further developed (Lindhorst, 2003). Neighbouring-group participation has frequently been used in organic synthesis. In carbohydrate chemistry, it is generally thought that glycosyl donors possessing anO-acyl group with a participating function at C2 give exclusively the corresponding 1,2-trans-glycoside with quite high stereoselectivity in glycosylation reactions. In our investigations of mono-saccharides, the OAc group was used as the protective group for further glycosylation reactions. The 1-OAc group in the glycosyl donor was transformed to a good leaving group using activated BF3Et2O. Thus, we synthesized the title compound, (I), and determined its crystal structure.
The six-membered ring of (I) adopts a chair conformation (Fig. 1). The absolute configuration, known from the synthetic pathway, is fully confirmed by refinement of the Flack para-meter (Flack & Bernardinelli, 2000).
Since all its hydroxyl groups are either acylated or alkyl-ated, compound (I) has many possible hydrogen-bond acceptors and, at the same time, no hydrogen-bond donors other than C—H groups. A number of possible weak hydrogen bonds can be found, but only those which satisfy the usual geometrical conditions are listed in Table 1. Six C—H O hydrogen bonds are intermolecular and form a three-dimen-sional network. Among these, four link the molecules along thebaxis, whereas the other two link the molecules along the
aandcaxes. It is interesting to note that only acetyl O atoms and the anomeric O atom are involved in hydrogen bonding in (I), even though, in general, the ester O atom is known to be involved in hydrogen bonding in small organic molecules (Molcˇanov et al., 2004). The reason for this could be steric hindrance of the ester O atoms in the crystal structure of (I).
The packing of compound (I) is different from that observed in (2-ethyl)-2,3,4,6-tetra-O-acetyl--d
-glucopyrano-side [Cambridge Structural Database (Allen, 2002; version 5.26) refcode ZETNOB (Prawat et al., 1995)]. The Br atom itself forms no contacts other than those of a van der Waals nature. It can be speculated that the differences in packing arise from the larger radius of the Br atom and its effect on the H-atom donor properties of the methylene group to which it is bonded. The C—H group of this methylene group is involved in the shortest hydrogen bond in the crystal structure of (I), whereas in ZETNOB it forms no hydrogen bonds.
Experimental
The title compound was synthesized according to the method of Dahme´net al.(1983).1H and13C NMR spectra are in agreement with data presented by Lindhorst (2003).
Crystal data
C16H23BrO10 Mr= 455.25
Monoclinic,P21 a= 9.4153 (12) A˚ b= 9.8326 (14) A˚ c= 11.2069 (19) A˚
= 96.391 (12) V= 1031.1 (3) A˚3 Z= 2
Dx= 1.466 Mg m
3
MoKradiation Cell parameters from 2233
reflections
= 2.8–18.3 = 2.04 mm1 T= 295 (2) K Prism, colourless 0.450.100.08 mm
Data collection
Oxford Xcalibur 3 CCD area-detector diffractometer
!scans
Absorption correction: analytical
3873 independent reflections 3044 reflections withI> 2(I) Rint= 0.040
max= 26.1
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.047 wR(F2) = 0.142 S= 1.03 3873 reflections 249 parameters
H-atom parameters constrained w= 1/[2
(Fo2) + (0.0929P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.39 e A˚3
min=0.58 e A˚3 Absolute structure: Flack &
Bernardinelli (2000), with 1722 Friedel pairs
Flack parameter = 0.005 (13)
Table 1
Hydrogen-bonding geometry (A˚ ,).
D—H A D—H H A D A D—H A
C1—H1 O5i
0.98 2.55 3.479 (6) 157
C6—H6B O9i
0.97 2.73 3.697 (6) 173
C10—H10B O9i 0.96 2.63 3.497 (8) 154 C8—H8A O7ii
0.96 2.60 3.459 (8) 149
C12—H12C O1iii
0.96 2.66 3.581 (6) 158
C16—H16B O3iv
0.97 2.45 3.367 (7) 157
Symmetry codes: (i) 2x;y1
2;1z; (ii) 2x; 1
2þy;1z; (iii) 1þx;y;z; (iv)
x;y;z1.
Methine and methylene H atoms were positioned geometrically, with C—H = 0.98 A˚ andUiso(H) = 1.2Ueq(C) for methine H, and C— H = 0.97 A˚ andUiso(H) = 1.2Ueq(C) for methylene H atoms. Methyl H atoms were refined using a riding model, with C—H = 0.98 A˚ and
Uiso(H) = 1.5Ueq(C).
Data collection: CrysAlisCCD (Oxford Diffraction, 2003); cell refinement:CrysAlisRED(Oxford Diffraction, 2003); data reduction:
CrysAlisRED; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:SHELXL97.
The authors thank the Ministry of Science, Education and Sports of the Republic of Croatia for financial support (grant Nos. 119632 and 119610).
References
Alcock, N. W. (1970).Crystallographic Computing, edited by F. R. Ahmed, S. R. Hall & C. P. Huber, p. 271. Copenhagen: Munksgaard.
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Dahme´n, J., Frejd, T., Gro¨nberg, G., Lave, T., Magnusson, G. & Noori, G. (1983).Carbohydr. Res.116, 303–307.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Flack, H. D. & Bernardinelli, G. (2000).J. Appl. Cryst.33, 1143–1148. Lindhorst, T. K. (2003).Essentials of Carbohydrate Chemistry and
Biochem-istry, 2nd edition, revised and updated. Weinheim: Wiley–VCH Verlag. Molcˇanov, K., Kojic´-Prodic´, B. & Raos, N. (2004).Acta Cryst.B60, 424–432. Oxford Diffraction (2003). CrysAlisCCD and CrysAlisRED. Versions
1.171.26 beta. Oxford Diffraction Ltd, Abingdon, England.
[image:2.610.47.294.72.293.2]Prawat, H., Mahidol, C., Ruchirawat, S., Prawat, U., Tuntiwachwuttikul, P., Tooptakong, U., Taylor, W. C., Pakawatchai, C., Skelton, B. W. & White, Figure 1
supporting information
sup-1
Acta Cryst. (2005). E61, o2644–o2645
supporting information
Acta Cryst. (2005). E61, o2644–o2645 [https://doi.org/10.1107/S1600536805022555]
2-Bromoethyl 2,3,4,6-tetra-
O
-acetyl-β-
D-glucopyranoside
Ivan Halasz, Renata Od
ž
ak, Srdanka Tomi
ć
and Dubravka Matkovi
ć
-
Č
alogovi
ć
2-Bromoethyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside
Crystal data
C16H23BrO10
Mr = 455.25 Monoclinic, P21
Hall symbol: P 2yb
a = 9.4153 (12) Å
b = 9.8326 (14) Å
c = 11.2069 (19) Å
β = 96.391 (12)°
V = 1031.1 (3) Å3
Z = 2
F(000) = 468
Dx = 1.466 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2233 reflections
θ = 2.8–18.3°
µ = 2.04 mm−1
T = 295 K
Prismatic, colourless 0.45 × 0.10 × 0.08 mm
Data collection
Oxford Xcalibur 3 CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: analytical (Alcock, 1970)
Tmin = 0.686, Tmax = 0.871
13961 measured reflections 3873 independent reflections 3044 reflections with I > 2σ(I)
Rint = 0.040
θmax = 26.1°, θmin = 3.6°
h = −11→11
k = −12→11
l = −13→13
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.047
wR(F2) = 0.142
S = 1.03 3873 reflections 249 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0929P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.39 e Å−3
Δρmin = −0.58 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br 0.44532 (6) 0.87439 (9) 0.10552 (6) 0.0997 (3)
O1 0.7903 (3) 0.7113 (3) 0.4259 (2) 0.0461 (6)
O4 1.1191 (3) 0.6509 (3) 0.6177 (2) 0.0455 (6)
O6 1.2101 (3) 0.8151 (3) 0.4228 (2) 0.0407 (6)
O8 1.0379 (3) 0.7442 (3) 0.2001 (2) 0.0473 (7)
O10 0.7438 (3) 0.7440 (3) 0.2230 (2) 0.0483 (7)
C1 0.8439 (4) 0.6935 (4) 0.3131 (3) 0.0428 (9)
H1 0.8612 0.5968 0.2993 0.051*
C2 0.9815 (4) 0.7729 (4) 0.3119 (3) 0.0378 (8)
H2 0.9637 0.8705 0.3195 0.045*
C3 1.0884 (4) 0.7245 (4) 0.4132 (3) 0.0382 (8)
H3 1.1198 0.6324 0.3955 0.046*
C5 0.8828 (4) 0.6483 (4) 0.5194 (3) 0.0420 (8)
H5 0.8994 0.5540 0.4963 0.050*
C4 1.0243 (4) 0.7236 (4) 0.5307 (3) 0.0390 (8)
H4 1.0113 0.8169 0.5580 0.047*
C15 0.6246 (5) 0.6522 (6) 0.1936 (4) 0.0580 (11)
H15A 0.5650 0.6497 0.2588 0.070*
H15B 0.6593 0.5610 0.1812 0.070*
C6 0.8085 (5) 0.6476 (5) 0.6302 (4) 0.0535 (10)
H6A 0.7121 0.6128 0.6119 0.064*
H6B 0.8592 0.5889 0.6902 0.064*
C7 0.7320 (5) 0.8035 (6) 0.7730 (4) 0.0601 (12)
O7 1.3641 (3) 0.6423 (4) 0.4314 (4) 0.0809 (11)
C13 1.0254 (5) 0.8379 (5) 0.1129 (4) 0.0528 (11)
C12 1.4549 (4) 0.8683 (7) 0.4627 (5) 0.0712 (13)
H12C 1.5477 0.8270 0.4768 0.107*
H12B 1.4361 0.9191 0.5324 0.107*
H12A 1.4523 0.9283 0.3949 0.107*
C9 1.2020 (5) 0.7217 (5) 0.7020 (4) 0.0550 (10)
O5 1.1933 (5) 0.8428 (4) 0.7112 (4) 0.0947 (14)
C16 0.5404 (5) 0.7028 (6) 0.0816 (4) 0.0676 (13)
H16A 0.4692 0.6353 0.0535 0.081*
supporting information
sup-3
Acta Cryst. (2005). E61, o2644–o2645
O2 0.8040 (3) 0.7848 (3) 0.6761 (2) 0.0544 (7)
C11 1.3429 (4) 0.7591 (5) 0.4376 (4) 0.0490 (10)
C10 1.3017 (5) 0.6327 (7) 0.7772 (4) 0.0733 (14)
H10A 1.3574 0.5807 0.7268 0.110*
H10B 1.2484 0.5721 0.8226 0.110*
H10C 1.3642 0.6873 0.8312 0.110*
C14 1.0672 (6) 0.7818 (7) −0.0004 (4) 0.0768 (15)
H14A 0.9949 0.7199 −0.0344 0.115*
H14B 1.1564 0.7344 0.0155 0.115*
H14C 1.0774 0.8546 −0.0558 0.115*
O3 0.6779 (6) 0.7089 (6) 0.8173 (4) 0.1114 (18)
C8 0.7405 (9) 0.9464 (7) 0.8188 (6) 0.094 (2)
H8A 0.6789 1.0035 0.7663 0.141*
H8B 0.8371 0.9784 0.8215 0.141*
H8C 0.7109 0.9491 0.8981 0.141*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Geometric parameters (Å, º)
Br—C16 1.943 (6) C6—H6A 0.9700
O1—C1 1.422 (5) C6—H6B 0.9700
O1—C5 1.427 (5) C7—O3 1.194 (7)
O4—C9 1.350 (5) C7—O2 1.356 (5)
O4—C4 1.436 (4) C7—C8 1.495 (9)
O6—C11 1.360 (5) O7—C11 1.169 (6)
O6—C3 1.446 (4) C13—O9 1.191 (6)
O8—C13 1.338 (5) C13—C14 1.477 (7)
O8—C2 1.443 (4) C12—C11 1.510 (7)
O10—C1 1.394 (5) C12—H12C 0.9600
O10—C15 1.449 (5) C12—H12B 0.9600
C1—C2 1.514 (5) C12—H12A 0.9600
C1—H1 0.9800 C9—O5 1.199 (6)
C2—C3 1.507 (5) C9—C10 1.478 (7)
C2—H2 0.9800 C16—H16A 0.9700
C3—C4 1.508 (5) C16—H16B 0.9700
C3—H3 0.9800 C10—H10A 0.9600
C5—C6 1.492 (5) C10—H10B 0.9600
C5—C4 1.517 (5) C10—H10C 0.9600
C5—H5 0.9800 C14—H14A 0.9600
C4—H4 0.9800 C14—H14B 0.9600
C15—C16 1.494 (7) C14—H14C 0.9600
C15—H15A 0.9700 C8—H8A 0.9600
C15—H15B 0.9700 C8—H8B 0.9600
C6—O2 1.446 (6) C8—H8C 0.9600
C1—O1—C5 110.6 (3) C5—C6—H6B 109.9
C9—O4—C4 119.1 (3) H6A—C6—H6B 108.3
C11—O6—C3 118.0 (3) O3—C7—O2 120.1 (5)
C13—O8—C2 119.3 (3) O3—C7—C8 126.8 (5)
C1—O10—C15 112.8 (3) O2—C7—C8 113.0 (4)
O10—C1—O1 108.8 (3) O9—C13—O8 122.9 (4)
O10—C1—C2 108.7 (3) O9—C13—C14 126.1 (5)
O1—C1—C2 109.4 (3) O8—C13—C14 111.1 (4)
O10—C1—H1 110.0 C11—C12—H12C 109.5
O1—C1—H1 110.0 C11—C12—H12B 109.5
C2—C1—H1 110.0 H12C—C12—H12B 109.5
O8—C2—C3 108.3 (3) C11—C12—H12A 109.5
O8—C2—C1 107.8 (3) H12C—C12—H12A 109.5
C3—C2—C1 109.3 (3) H12B—C12—H12A 109.5
O8—C2—H2 110.5 O5—C9—O4 122.2 (4)
C3—C2—H2 110.5 O5—C9—C10 125.8 (5)
C1—C2—H2 110.5 O4—C9—C10 112.0 (4)
O6—C3—C2 108.6 (3) C15—C16—Br 112.5 (4)
supporting information
sup-5
Acta Cryst. (2005). E61, o2644–o2645
O6—C3—H3 109.3 C15—C16—H16B 109.1
C2—C3—H3 109.3 Br—C16—H16B 109.1
C4—C3—H3 109.3 H16A—C16—H16B 107.8
O1—C5—C6 108.0 (3) C7—O2—C6 116.6 (4)
O1—C5—C4 107.9 (3) O7—C11—O6 123.5 (4)
C6—C5—C4 114.9 (3) O7—C11—C12 126.1 (4)
O1—C5—H5 108.6 O6—C11—C12 110.4 (4)
C6—C5—H5 108.6 C9—C10—H10A 109.5
C4—C5—H5 108.6 C9—C10—H10B 109.5
O4—C4—C3 108.5 (3) H10A—C10—H10B 109.5
O4—C4—C5 106.7 (3) C9—C10—H10C 109.5
C3—C4—C5 111.3 (3) H10A—C10—H10C 109.5
O4—C4—H4 110.1 H10B—C10—H10C 109.5
C3—C4—H4 110.1 C13—C14—H14A 109.5
C5—C4—H4 110.1 C13—C14—H14B 109.5
O10—C15—C16 108.0 (4) H14A—C14—H14B 109.5
O10—C15—H15A 110.1 C13—C14—H14C 109.5
C16—C15—H15A 110.1 H14A—C14—H14C 109.5
O10—C15—H15B 110.1 H14B—C14—H14C 109.5
C16—C15—H15B 110.1 C7—C8—H8A 109.5
H15A—C15—H15B 108.4 C7—C8—H8B 109.5
O2—C6—C5 109.1 (3) H8A—C8—H8B 109.5
O2—C6—H6A 109.9 C7—C8—H8C 109.5
C5—C6—H6A 109.9 H8A—C8—H8C 109.5
O2—C6—H6B 109.9 H8B—C8—H8C 109.5
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C1—H1···O5i 0.98 2.55 3.479 (6) 157
C6—H6B···O9i 0.97 2.73 3.697 (6) 173
C10—H10B···O9i 0.96 2.63 3.497 (8) 154
C8—H8A···O7ii 0.96 2.60 3.459 (8) 149
C12—H12C···O1iii 0.96 2.66 3.581 (6) 158
C16—H16B···O3iv 0.97 2.45 3.367 (7) 157