The NLR family of proteins is recognized for its roles in inflamma-some-mediated responses to both pathogen-associated molecular pat-terns and damage-associated molecular patpat-terns1. However, several NLR proteins, including NLRX1, NLRC3, NLRC5 and NLRP12, act as negative regulators of innate immunity with the ability to check type I interferon responses or NF-KB-induced pro-inflammatory cytokines2–5. NLRX1 is distinguished from other members of the NLR family by its localization to mitochondria, where it interacts with MAVS through its unique amino-terminal X domain and nucleotide-binding–oligomerization domain, sequesters MAVS and suppresses virus-induced interferon responses mediated by the pathogen sensor RIG-I6. NLRX1 also negatively regulates lipopolysaccharide-induced activation of NF-KB, interacting with the adaptor TRAF6 in unstimu-lated cells and being recruited to the NEMO–IKK signaling complex following lipopolysaccharide stimulation via its leucine-rich-repeat domain3. Deletion or functional knockdown of NLRX1 results in heightened interferon responses to the synthetic RNA duplex and Toll-like receptor 3 (TLR3) agonist poly(I:C) or RNA viruses, as well as increased inflammatory responses3,6,7. Acting like a Swiss Army knife, NLRX1 also interacts with STING through its nucleotide-binding–oligomerization domain and thereby inhibits interferon
responses to DNA viruses mediated through the cGAS–cGAMP sig-naling pathway8. Abundant evidence thus supports the concept that NLRX1 functions as a checkpoint inhibitor of early innate immune responses to both DNA viruses and RNA viruses.
However, not all studies have shown that NLRX1 exerts negative regulatory effects on innate immune responses to viruses. Sendai virus (SeV)-induced RIG-I- and MAVS-dependent phosphorylation of IRF3 and production of interferon-B (IFN-B) and the chemok-ine IP10 (CXCL10) are reported to be unchanged in Nlrx1−/− mouse embryonic fibroblasts (MEFs), relative to that of wild-type MEFs, as are cytokine IL-6 and IFN-B responses to poly(I:C)9. Although inflam-matory responses to infection with influenza virus are enhanced in the lungs of Nlrx1−/− mice relative to those in the lungs of of wild-type mice7, their macrophage-mediated interferon responses are impaired, secondary to enhanced apoptosis10. Also, rather than inhibiting NF-KB signaling, overexpression of NLRX1 enhances such signaling by amplifying the production of reactive oxygen species in response to several stimuli11. Such findings are puzzling given that the preponder-ance of evidence favors a negative regulatory role for NLRX1. While they might reflect different experimental conditions, they have fueled controversy about the regulatory role of NLRX1.
1Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA. 2Department of Medicine, The University
of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA. 3Department of Microbiology & Immunology, The University of North Carolina at Chapel Hill,
Chapel Hill, North Carolina, USA. 4Department of Genetics, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA. 5Department of Cell
Biology and Physiology, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA. 6Department of Pharmacology, The University of North
Carolina, Chapel Hill, North Carolina, USA. 7Present address: Department of Microbiology and Cell Biology, Tokyo Metropolitan Institute of Medical Science,
Setagaya-ku, Tokyo, Japan. 8These authors contributed equally to this work. Correspondence should be addressed to S.M.L. ([email protected]).
Received 20 January; accepted 11 September; published online 2 October 2017; doi:10.1038/ni.3853
NLRX1 promotes immediate IRF1-directed antiviral
responses by limiting dsRNA-activated translational
inhibition mediated by PKR
Hui Feng
1,2, Erik M Lenarcic
1,3,8, Daisuke Yamane
1,2,7,8, Eliane Wauthier
1,4, Jinyao Mo
1,2, Haitao Guo
1,5,
David R McGivern
1,2, Olga González-López
1,2, Ichiro Misumi
1,4, Lola M Reid
1,5, Jason K Whitmire
1,3,4,
Jenny P-Y Ting
1,4, Joseph A Duncan
1,2,6, Nathaniel J Moorman
1,3& Stanley M Lemon
1–3NLRX1 is unique among the nucleotide-binding-domain and leucine-rich-repeat (NLR) proteins in its mitochondrial localization and ability to negatively regulate antiviral innate immunity dependent on the adaptors MAVS and STING. However, some studies have suggested a positive regulatory role for NLRX1 in inducing antiviral responses. We found that NLRX1 exerted opposing regulatory effects on viral activation of the transcription factors IRF1 and IRF3, which might potentially explain such contradictory results. Whereas NLRX1 suppressed MAVS-mediated activation of IRF3, it conversely facilitated virus-induced increases in IRF1 expression and thereby enhanced control of viral infection. NLRX1 had a minimal effect on the transcription
of IRF1 mediated by the transcription factor NF-KB and regulated the abundance of IRF1 post-transcriptionally by preventing
Here we sought to understand how NLRX1 influences innate immune responses to viral infection of human hepatocytes. Hepatocytes are targeted for infection by several medically impor-tant viruses, including hepatitis A virus (HAV) and hepatitis C virus (HCV), which are RNA viruses that cause inflammatory diseases of the liver12. We found that NLRX1 competed with dsRNA-activated PKR for binding to viral RNA and that it promoted early innate immune antiviral responses by protecting NF-KB-driven increases in the expression of IRF1 from translational suppression mediated by PKR. Hepatocytes deficient in NLRX1 expression had diminished accumulation of IRF1 but more formation of IRF3 dimers in response to viral infection, relative to that of hepatocytes sufficient in NLRX1; this revealed opposing actions of NLRX1 on key signaling pathways. Our data identify a previously unknown and sophisticated regulation of early innate immune responses by NLRX1 that, overall, promotes immediate antiviral defense in hepatocytes.
RESULTS
NLRX1 is a positive immunoregulator in hepatocytes
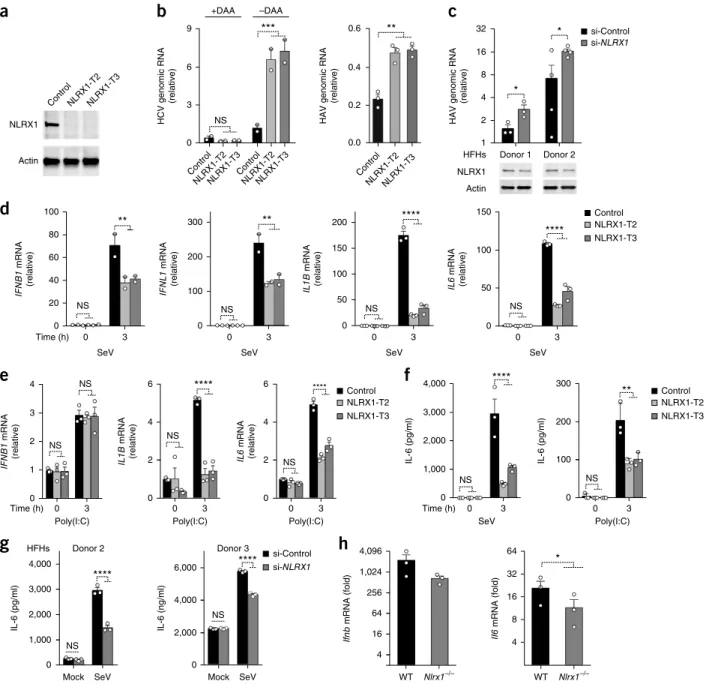
To determine how NLRX1 influences antiviral responses in hepato-cytes, we used CRISPR-Cas9 gene editing to eliminate its expression in PH5CH8 cells, which are T antigen–transformed primary human hepatocytes with functional signaling via RIG-I and TLR3 (refs. 13,14). NLRX1 expression was detected in PH5CH8 cells transduced with a nontargeting single guide RNA (sgRNA) with a scrambled sequence (called ‘control PH5CH8 cells’ throughout) but not in either of two independent PH5CH8 cell lines (NLRX1-T2 and NLRX1-T3) transduced with different NLRX1-specific sgRNAs (Fig. 1a and
Supplementary Table 1). We initiated HCV replication in these cells
by electroporating synthetic viral RNA and the duplex microRNA miR-122, an essential HCV host factor that PH5CH8 cells lack15, together into the cells. We demonstrated that subsequent increases in viral RNA were due to true viral replication because they were blocked by a direct-acting antiviral inhibitor (Fig. 1b). We also infected the cells directly with HAV16. Replication of each virus was enhanced in NLRX1-deficient (NLRX1-T2 and NLRX1-T3) cells relative to that of control PH5CH8 cells (Fig. 1b). We similarly assessed HAV replica-tion in primary human fetal hepatoblasts (HFHs). Partial deplereplica-tion of NLRX1 via RNA-mediated interference boosted viral replication in cells from two donors (Fig. 1c), which confirmed an antiviral role for NLRX1 in human liver cells.
Although innate immune responses restrict infection with HAV or HCV in PH5CH8 cells, it is difficult to document the induction of antiviral cytokines in these cells following viral challenge. We thus employed a classic agonist of RIG-I signaling, SeV, to define the effect of NLRX1 deficiency on cytokine responses. We observed sig-nificantly lower IFNB1, IFNL1, IL1B and IL6 mRNA responses early (at 3 h) in SeV-infected deficient (T2 and NLRX1-T3) cells than in SeV-infected control PH5CH8 cells (Fig. 1d). This effect was no longer evident at 8 h, by which time these responses had substantially subsided (Supplementary Fig. 1a). NLRX1 defi-ciency also impaired the accumulation of IL1B and IL6 mRNA, but not that of IFNB1 mRNA, in response to poly(I:C) added to the medium (Fig. 1e). NLRX1 deficiency consistently reduced the amount of IL-6 protein induced in response to stimulation with SeV or poly(I:C) (Fig. 1f). Likewise, small interfering RNA (siRNA)-medi-ated depletion of NLRX1 significantly impaired the increase in IL-6 protein induced by infection of primary HFHs with SeV (Fig. 1g). Neither HAV nor HCV replicates in mouse cells, but we observed a reduction of approximate fourfold in the early (3-hour) intrahepatic
Ifnb and Il6 mRNA responses to synthetic HAV RNA administered
intravenously to Nlrx1−/− mice, relative to the responses in their wild-type counterparts (Fig. 1h).
Supplementing PH5CH8 cells with the antioxidant NAC (N-acetyl-l-cysteine) did not abolish the positive effect of NLRX1 deficiency on the replication of an HCV reporter virus, nor did treating cells with an inhibitor of endoplasmic reticulum stress (TUDCA) or autophagy (3-MA) (Supplementary Fig. 1b). Thus, the increased HCV rep-lication in NLRX1-deficient cells was probably not due to dimin-ished production of reactive oxygen species11,17 or to the influence of NLRX1 depletion on autophagy or the response to endoplasmic reticulum stress18,19.
To ascertain whether the proviral effect of NLRX1 deficiency resulted from a reduction in early innate immune response, we designed a Transwell assay for soluble antiviral factors (Supplementary Fig. 1c). We assessed replication of an HCV reporter virus in indicator cells separated by a permeable membrane from NLRX1-deficient cells or control PH5CH8 cells, each stimulated by replication-competent HCV RNA, and found greater replication of the reporter virus in indi-cator cells exposed to NLRX1-deficient cells than in those exposed to control PH5CH8 cells (Supplementary Fig. 1c). Inhibition of JAK-STAT signaling with ruxolitinib (an inhibitor of JAK1 and JAK2) or tofacitinib (an inhibitor of JAK3) also substantially reduced the difference in HCV replication in NLRX1-T3 cells relative to that in control PH5CH8 cells (Supplementary Fig. 1d), consistent with the suppression of viral replication by cytokine responses involving JAK signaling. Depleting cells of NLRX1 had no effect on the replica-tion of HAV or HCV in RIG-I-deficient Huh-7.5 human hepatoma cells20 (Supplementary Fig. 1e) or in MAVS-deficient PH5CH8 cells (Supplementary Fig. 1f). Collectively, these results defined NLRX1 as a positive regulator of soluble RIG-I- and MAVS-mediated antiviral responses in human hepatocytes.
NLRX1 regulates IRF3 and IRF1 responses differentially
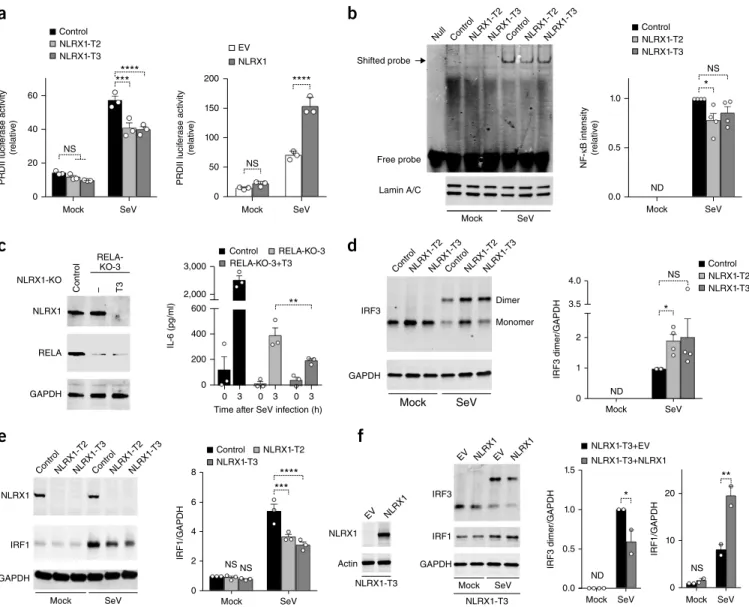
We carried out a series of dual-luciferase-promoter assays to deter-mine if NLRX1 directly regulated the transcription of cytokine-encod-ing genes. NLRX1 deficiency had little effect on the basal activity of the NF-KB-responsive promoter PRDII but modestly reduced its activation by SeV (Fig. 2a). Consistent with that, overexpression of NLRX1 enhanced the activation of PRDII triggered by SeV (Fig. 2a). In contrast, NLRX1 deficiency minimally affected activation of the
IFNB1 promoter and had no effect on the IRF3-responsive promoter 4*PRD(I/III) (Supplementary Fig. 2a,b). We also assessed the effect of NLRX1 deficiency on the stability of IL6 mRNA, as IL-6 expression is regulated in part through 3` untranslated region (UTR) sequences programmed for rapid mRNA turnover21. NLRX1 deficiency had no effect on IL6 mRNA decay in cells treated with actinomycin D
(Supplementary Fig. 2c), nor did overexpression of NLRX1 alter
luciferase expression from mRNA transcripts containing the IL6 3` UTR (Supplementary Fig. 2d). In aggregate, these data suggested that NLRX1 had a positive but limited effect on the activation of NF-KB-responsive promoters by SeV and had no influence on the stability of IL6 mRNA.
To more directly investigate the influence of NLRX1 deficiency on the activation of NF-KB, we assessed SeV-induced signaling via the NF-KB subunit RELA (p65). NLRX1 deficiency minimally affected phosphorylation of the inhibitory cytoplasmic NF-KB chaperone NFKBIA (IKBA) or RELA in SeV-infected PH5CH8 cells
(Supplementary Fig. 2e). An electrophoretic mobility-shift assay
9
6
3
0 NS
***
HCV genomic RNA
(relative)
+DAA –DAA
Control NLRX1-T2 NLRX1-T3
Control NLRX1-T2 NLRX1-T3
Control NLRX1-T2 NLRX1-T3 0.6
0.4
0.2
0.0
32
16
8
4
2
1
HFHs Donor 1 Donor 2
si-Control si-NLRX1 *
* **
HAV genomic RNA
(relative)
HAV genomic RNA
(relative)
100
80
60
40
20
0
Time (h) 0 3
SeV NS
** 300
200
100
0
0 3
SeV SeV SeV
0 3
NS 200
150
100
50
0
150
100
50
0
0 3
NS
****
Control NLRX1-T2
NLRX1-T3
IL6
mRNA
(relative)
**** **
NS
IL1B
mRNA
(relative)
IFNL1
mRNA
(relative)
IFNB1
mRNA
(relative)
4
3
2
1
0 NS
Time (h) 0 3
Poly(I:C)
0 3
Poly(I:C)
0 3
Poly(I:C)
IFNB1
mRNA
(relative)
NS
IL1B
mRNA
(relative)
6
4
2
0 NS
**** ****
NS 6
4
2
0
4,000
3,000
2,000
1,000
0
Time (h) 0 3
SeV Poly(I:C)
0 3
NS 300
200
100
0
IL-6 (pg/ml)
****
NS
**
IL6
mRNA
(relative)
IL-6 (pg/ml)
a
b
c
d
e
f
g
HFHs Donor 2h
4,000
3,000
2,000
1,000
0 NS
Mock SeV
**** 6,000
4,000
2,000
0
Mock SeV
NS
**** si-Control
si-NLRX1 Donor 3
IL-6 (ng/ml)
4,096
1,024
256
64
16
4
WT Nlrx1–/– WT Nlrx1–/– 64
32
16
8
4
Ifnb
mRNA (fold)
II6
mRNA (fold)
*
IL-6 (pg/ml)
Control NLRX1-T2
NLRX1-T3 Control
NLRX1-T2
NLRX1-T3 ControlNLRX1-T2NLRX1-T3
NLRX1
Actin
NLRX1
Actin
Figure 1 NLRX1 is a positive regulator of innate immunity and an antiviral factor in hepatocytes. (a) Immunoblot analysis of NLRX1
and actin (loading control throughout) in control PH5CH8 cells (Control), NLRX1-T2 cells and NLRX1-T3 cells (above lanes). (b) qRT-PCR
analysis of genomic RNA from HCV (left) and HAV (right) in control PH5CH8 cells, NLRX1-T2 cells and NLRX1-T3 cells (horizontal axis) transfected for 24 h with HCV RNA, in the presence (+DAA) or absence (−DAA) of a direct-acting antiviral inhibitor (left) or
infected for 72 h with HAV (right); result are presented relative to the abundance of viral RNA at 6 h (left) or in control PH5CH8 cells at 24 h (right).
(c) qRT-PCR analysis (top) of HAV genomic RNA in primary HFHs (from two different donors; horizontal axis) at 72 h after treatment with control
siRNA (si-Control) or partial depletion of NLXR1 with NLRX1-specific siRNA (si-NLRX1) (key), presented relative to the abundance of viral RNA at
5 h; below, immunoblot analysis of NLRX1 in the cells above. (d,e) qRT-PCR analysis of IFNB1, IFNL1, IL1B and IL6 mRNA in control PH5CH8
cells, NLRX1-T2 cells and NLRX1-T3 cells (key) left uninfected (0) or infected for 3 h with SeV (d) or left untreated (0) or stimulated with poly(I:
C) added to medium for 3 h (e); results were normalized to those of ACTB. (f,g) ELISA of IL-6 in control PH5CH8 cells, T2 cells and
NLRX1-T3 cells (key) infected with SeV as in d or stimulated with poly(I:C) as in e (horizontal axis) (f), and in HFHs treated with siRNA as in c (key) and
mock infected or infected with SeV (horizontal axis) (g). (h) qRT-PCR analysis of Ifnb and Il6 mRNA in the liver of wild-type (WT) and Nlrx1−/− mice
(horizontal axis) 3 h after hydrodynamic injection of HAV RNA; results were normalized to those of Actb. Each symbol (b–h) represents an individual
technical replicate (b–d (IL1B and IL6), e–g), experiment (d, IFNB1 and IFNL1) or mouse (h) (mean + s.e.m in each). NS, not significant
(P > 0.05); *P < 0.05, ** P < 0.01, ***P < 0.001 and ****P < 0.0001, control cells versus cells depleted of NLRX1 (except where indicated
otherwise) (two-way analysis of variance (ANOVA) (b (left), d–f) or t test (b (right), c,g,h)). Data are representative of three experiments (a), three
independent experiments with n = 2 (left) or n = 3 (right) technical replicates (b), two experiments with n = 3 (donor 1) or n = 4 (donor 2) technical
replicates (c), three experiments with n = 3 technical replicates in each (d (IL1B and IL6), e), three experiments (f) or two experiments with two
donors (g), with n = 3 technical replicates in each (f,g) or one experiment with n = 3 mice per genotype (h) or are from two independent experiments
in control PH5CH8 cells (Fig. 2b). We concluded from these data that NLRX1 deficiency minimally suppressed SeV-induced NF-KB signaling in PH5CH8 cells and that this subtle impairment of NF-KB signaling probably did not explain the marked reductions we observed in cytokine expression.
To further test our hypothesis, we assessed how depleting RELA influenced IL-6 production after SeV infection and the modula-tion of IL-6 expression by NLRX1 in PH5CH8 cells. As anticipated, RELA deficiency caused a large decrease in the SeV-induced expres-sion of IL-6 protein (Fig. 2c). Notably, however, eliminating NLRX1
Shifted probe
Free probe
Lamin A/C
Mock SeV
ControlNLRX1-T2NLRX1-T3ControlNLRX1-T2NLRX1-T3
Monomer Dimer IRF3
GAPDH
Control – T3
RELA-KO-3
Mock SeV
ControlNLRX1-T2NLRX1-T3ControlNLRX1-T2NLRX1-T 3
NLRX1
IRF1
GAPDH
NLRX1-T3 EV NLRX1EV NLRX1
IRF3
IRF1
GAPDH EV NLRX1
NLRX1
Actin
Mock SeV
60
40
20
Mock SeV
200
150
100
50
0
Mock SeV
NS Control
a
c
e
f
d
b
NLRX1-T2 NLRX1-T3
Control RELA-KO-3
RELA-KO-3+T3 3,000
2,000
600
400
200
0
0 3 0 3 0 3
Time after SeV infection (h)
IL
-6 (pg/ml)
PRDII lucif
erase activit
y
(r
elativ
e)
PRDII lucif
erase activit
y
(r
elativ
e)
NF
B intensity
(relative)
1.0
0.5
0.0
Mock SeV
ND
NS EV
NLRX1
NS 4.0
3.5
2
1
0
Mock SeV
ND
Control NLRX1-T2
NLRX1-T3
IRF3 dimer/G
APDH
8
6
4
2
0
Mock SeV
NS NS
IRF1/G
APDH
1.5
1.0
0.5
0.0
Mock SeV Mock SeV
NS 20
10
0 ND
IRF1/G
APDH
NLRX1-T3+EV
NLRX1-T3+NLRX1
IRF3 dimer/G
APDH
****
*** **** *
**
*
**** ***
*
**
NS
Control NLRX1-T2 NLRX1-T3
Control NLRX1-T2 NLRX1-T3 0
Null ControlNLRX1-T2NLRX1-T3ControlNLRX1-T2NLRX1-T3
NLRX1-KO
NLRX1
RELA
GAPDH
NLRX1-T3
Mock SeV
Figure 2 Effect of NLRX1 deficiency on NF-KB signaling and the activation of IRF1 and IRF3 in SeV-infected PH5CH8 cells. (a) Activity of a (firefly) luciferase reporter for the promoter PRDII in control PH5CH8 cells, NLRX1-T2 cells and NLRX1-T3 cells (key) (left) or PH5CH8 cells transfected with empty vector (EV) or transfected to overexpress NLRX1 (key) (right), all mock infected or infected with SeV (horizontal axis); results are presented
relative to renilla luciferase activity. (b) Electrophoretic mobility-shift assay of NF-KB without nuclear extracts (Null) or with nuclear extracts from
control PH5CH8 cells, NLRX1-T2 cells and NLRX1-T3 cells (above lanes) mock infected or infected with SeV (below blot) (top left), together with immunoblot analysis of lamin A/C as a loading control (bottom left), and mean infrared fluorescence intensity of the shifted probe (assessed as at
left), presented relative to that in SeV-infected control PH5CH8 cells (right). ND, not detected. (c) Immunoblot analysis of NLRX1, RELA and GAPDH
(loading control throughout) in NLRX1- and RELA-sufficient PH5CH8 cells (Control), RELA-deficient PH5CH8 cells (RELA-KO-3, NLRX1-KO −) and PH5CH8 cells doubly deficient in NLRX1 and RELA (RELA-KO-3, NLRX1-KO T3) (above lanes) (left), and ELISA of IL-6 in cells as at left (key), left
uninfected (0) or infected for 3 h with SeV (horizontal axis) (right). (d,e) Immunoblot analysis (left) of IRF3 monomers and dimers (right margin)
(d) and of NLRX1 and IRF1 (left margin) (e) in control PH5CH8 cells, NLRX1-T2 cells and NLRX1-T3 cells (above lanes) mock infected or infected with
SeV (below blots); right, quantification of the intensity of IRF3 dimers (d) or IRF1 (e), relative to that of GAPDH. (f) Immunoblot analysis of NLRX1 (far
left) or IRF3 and IRF1 (middle left) in NLRX1-T3 cells transfected with empty lentivirus vector (EV) or with lentivirus vector encoding NLRX1 (above lanes) and left uninfected (far left) or mock infected or infected with SeV (below blots) (middle left); right, quantification of the intensity of IRF3
dimers (middle right) or IRF1 (far right), relative to that of GAPDH. Each symbol represents an individual technical replicate (a,c) or experiment (b,d–f)
(mean + s.e.m. throughout). *P < 0.05, **P < 0.01,***P < 0.001 and ****P < 0.0001, control cells versus cells depleted of NLRX1 (two-way ANOVA
(a,e) and t test (b–d,f)). Data are representative of three independent experiments with n = 3 technical replicates (a), four (b,d), three (e) or two (f)
expression resulted in a further reduction in IL-6 production in RELA-deficient cells (Fig. 2c). Thus, the negative effect of NLRX1 deficiency on the IL-6 response that we observed in RELA-replete cells was preserved in RELA-deficient cells. Collectively, these results
indicated that NLRX1 regulated IL-6 production downstream and independently of NF-KB.
Although our data showed that IL-6 was strongly regulated by NF-KB in PH5CH8 cells (Fig. 2c), CRISPR-Cas9-mediated depletion of
500 400 300 200 100 0 IRF3-KO
a
b
d
e
c
IRF1-KO SeV (h)NLRX1-KO – T3 – T3
NLRX1 NLRX1 IRF1 IRF1 GAPDH IRF3 Actin Actin 0 3 SeV (h)
NLRX1-KO – T3 – T3
0 3 2,000 0.4 0.2 0.0 72 IRF3-KO IRF3-KO +T3 IRF1-KO
IRF1-KO +T3 si-Control
si-NLRX1
siRNA
Control NLRX1
ControlNLRX1ControlNLRX1 NLRX1
GAPDH
SeV (h) 0 3
siRNA 1,500 1,000 IL -6 (pg/ml) IL -6 (pg/ml) HA
V genomic RNA
(r
elativ
e)
HA
V genomic RNA
(r elativ e) 500 4,000 9 15 10 5 IRF1/G APDH 0 Mock 1 2
Time after SeV infection (h)
Time after SeV infection (h)
3 4 0 NS SeV 6 3 0 48
Time after infection (h) 72 3,000
2,000
1,000
WT Nlrx1–/–
WT Nlrx1–/– WT Nlrx1–/– BMDMs 0 10 8 6 4 2 0
1 2 3 4
120 SeV (h) SeV (h) BMDMs MEFs MEFs
0 3 4 5 6
BMDMs
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/–
WT Nlrx1
–/– 90 60 30 0 0 3 NS 60 40 20 0 6 25 20 15 10 5 0 0 3 4 2 0 0 3 NLRX1 IRF1 Actin NLRX1 15 12 9 6 3 0
0 3 4 5 6
IRF1 Actin Ifnb mRNA (r elativ e) II6 mRNA (r elativ e) II6 mRNA (r elativ e) Tnf mRNA (r elativ e) Tnf mRNA (r elativ e) Ifnb mRNA (r elativ e) IRF1/A ctin IRF1/A ctin 40 30 20 10 NS NS NS 0 0 3
0 3 0 3
Time after SeV infection (h)
MEFs
Time after SeV infection (h)
NS NS
NS 0
Mock SeV
Mock SeV
Time after infection (h) NS NS *** *** *** * **** **** * *** **** *** * 0
Figure 3 NLRX1–IRF1 signaling dominates the antiviral cytokine response in hepatocytes. (a,b) ELISA of IL-6 (middle) and qRT-PCR analysis of HAV
genomic RNA (right) in IRF3-deficient PH5CH8 cells (IRF3-KO) and PH5CH8 cells doubly deficient in IRF3 and NLRX1 (IRF3-KO + T3) (a), or in
IRF1-deficient PH5CH8 cells (IRF1-KO) and PH5CH8 cells doubly deficient in IRF1 and NLRX1 (IRF1-KO + T3) (b), mock infected or infected for
3 h with SeV (a,b, middle) or for 72 h with HAV (a, right) or infected for 48 or 72 h with HAV (b, right) (horizontal axes), and immunoblot analysis (left)
of NLRX1 and IRF1 (a) or of NLRX1 and IRF3 dimers (b) in cells as at right, left uninfected (0) or infected for 3 h with SeV (above lanes) (immunoblot
analysis of IRF1-deficient and IRF3-deficient cells, Supplementary Fig. 2f); qRT-PCR results are presented relative to those of cells at 4 h.
(c) Immunoblot analysis of NLRX1 and IRF1 in HFHs treated with control siRNA or partially depleted of NLRX1 via NLRX1-specific siRNA (above
lanes) (top) or treated with siRNA as above and left uninfected (0) or infected for 3 h with SeV (below blot) (middle), and quantification of IRF1
(infrared fluorescence intensity) in immunoblot analysis as above, relative to that of GAPDH. (d,e) qRT-PCR analysis of Ifnb, Il6 and Tnf mRNA (d),
immunoblot analysis of NLRX1 and IRF1 (e, left) and quantification of IRF1 as in c (e, right) in wild-type and Nlrx1−/− BMDMs (top) or MEFs (bottom)
left uninfected (0) or infected for 3 h (d) or 1–6 h (e) with SeV. Each symbol (a–d) represents an individual technical replicate (a,b,d) or donor (c) (mean
+ s.e.m in each). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 (t-test). Data are representative of two to three independent experiments
with n = 3 technical replicates in each (a,b), two experiments with two donors (c) or four (BMDM) or two (MEF) independent experiments with n = 3
either of two members of the IRF family of transcription factors, IRF1 and IRF3, suppressed early (3-hour) IL1B and IL6 mRNA and IL-6 protein responses to SeV (Supplementary Fig. 2f–h). Because IRF1 and IRF3 regulate the induction of IFNB1 and IFNL1 tran-scripts in human hepatoma cells22, we assessed the effect of NLRX1 deficiency on IRF3 and IRF1 responses in PH5CH8 cells. IRF3 is constitutively expressed and is activated by phosphorylation, which leads to its dimerization and cytoplasmic–nuclear translocation23. NLRX1 deficiency significantly enhanced this response and led to an increase in the SeV-induced formation of IRF3 dimers (Fig. 2d). Basal expression of IRF1 protein was low in mock-infected cells but was substantially induced by infection with SeV (Fig. 2e). In contrast to the enhancement observed in the activation of IRF3, NLRX1 deficiency significantly reduced the SeV-triggered increases in the abundance of IRF1 (Fig. 2e). Reconstituting NLRX1 expression in NLRX1-T3 cells with recombinant lentivirus reversed both of those changes (Fig. 2f). Collectively, these data revealed that NLRX1 regulated SeV-induced IRF3 signaling and IRF1 signaling in hepato-cytes differentially, suppressing the activation of IRF3 but enhancing increases in IRF1 expression.
NLRX1’s promotion of IRF1 dominates in hepatocytes
Because NLRX1 deficiency suppresses innate immunological con-trol of viral replication in hepatocytes, we reasoned that the positive regulation of IRF1 by NLRX1, rather than its negative regulation of IRF3, probably dominates cytokine responses in hepatocytes. To test our hypothesis, we depleted IRF1- and IRF3-deficient PH5CH8 cells of NLRX1 expression and assessed responses to SeV challenge in the resulting NLRX1–IRF3 or NLRX1–IRF1 doubly deficient cells. In IRF3-deficient cells, the IRF1 and IL-6 protein responses to infec-tion with SeV were suppressed and HAV replicainfec-tion was enhanced by depletion of NLRX1 (Fig. 3a). Thus, the effect of NLRX1 depletion in IRF3-deficient PH5CH8 cells was similar to that in unmodified PH5CH8 cells. In contrast, depleting IRF1-deficient cells of NLRX1 enhanced the formation of IRF3 dimers (as in IRF1-replete cells) but enhanced the SeV-induced production of IL-6 and also suppressed HAV replication (Fig. 3b). Thus, the effects of NLRX1 depletion on cytokine expression and viral replication were reversed in IRF1-deficient cells, which suggested that the NLRX1–IRF1 signaling axis is dominant in hepatocytes. Consistent with that conclusion, the reduced production of IL-6 in SeV-infected HFHs depleted of NLRX1
(Fig. 1g) was accompanied by a reduction in the accumulation of
IRF1 protein (Fig. 3c).
Most reports suggest that rather than enhancing such responses, NLRX1 suppresses, early innate immune responses in bone-mar-row-derived macrophages (BMDMs) and primary MEFs6–8. Thus, we sought to determine whether IRF1 signaling was negatively or positively regulated by NLRX1 in these cell types. Consistent with a suppressive effect on innate immunity, we found that loss of NLRX1 enhanced the SeV-mediated Ifnb, Tnf and Il6 mRNA responses in mouse BMDMs and MEFs (Fig. 3d). Despite that, NLRX1 deficiency reduced the SeV-triggered increase in the abundance of IRF1 protein in both cell types (Fig. 3e). Thus, the positive regulation of IRF1 signaling by NLRX1 was not specific to hepatocytes or human cells. The finding that cytokine responses were enhanced, while IRF1 induction was decreased, in BMDMs and MEFs depleted of NLRX1 indicated that IRF1 did not have a dominant role in determining cytokine responses in these cells. Thus, the ultimate effect of NLRX1 on early antiviral responses was determined by whether IRF1 or IRF3 dominated in driving the response.
NLRX1 enhances IRF1 protein synthesis in infected cells
IRF1 is the ‘founding member’ of the IRF family, and its expression is induced rapidly by viral infection24,25. However, the mechanisms that mediate this response are poorly understood. Given its role in initiating IRF3-directed responses to SeV in hepatocytes20, we rea-soned that RIG-I and its adaptor MAVS might mediate the IRF1 response. Consistent with that, depleting cells of MAVS significantly suppressed both IRF3 responses and IRF1 responses to SeV infection in PH5CH8 cells (Supplementary Fig. 3a). The loss of SeV-induced dimerization of IRF3 and expression of IRF1 protein was not reversed by additional depletion of NLRX1 in MAVS-deficient PH5CH8 cells (Supplementary Fig. 3a).
Consistent with published studies showing that increased IRF1
transcription drives the IRF1 protein response to viral infection25, globally inhibiting transcription with actinomycin D completely abol-ished the IRF1 protein response to SeV (Supplementary Fig. 3b). To better understand how NLRX1 might regulate this IRF1 response, we assessed the roles of IRF3 and NF-KB. Notably, depleting cells of IRF3 had no effect on either the transcription of IRF1 or the expression of IRF1 protein (Supplementary Fig. 3c). In contrast, depleting cells of RELA significantly suppressed both the transcription of IRF1 and the increase in expression of IRF1 protein in response to SeV in PH5CH8 cells (Supplementary Fig. 3d). siRNA-mediated silencing of NFKB1, which encodes the NF-KB subunit p50, modestly diminished the IRF1 protein response to SeV infection, and in combination with RELA deficiency, it substantially reduced the SeV-induced abundance of
IRF1 transcripts (Supplementary Fig. 3e,f). Collectively, these data demonstrated that the immediate (3-hour) increase in the expres-sion of IRF1 protein in response to infection with SeV was driven by signaling via MAVS and NF-KB in PH5CH8 cells.
Notably, although NLRX1 deficiency modestly enhanced the SeV-induced abundance of IRF3 transcripts (Supplementary Fig. 4a), it had no effect on either the abundance of IRF1 transcripts (Fig. 4a) or the stability of IRF1 mRNA (Fig. 4b). These data were in agreement with our earlier observations indicating an only modest impairment of NF-KB signaling in NLRX1-deficient cells (Fig. 2b) and collectively indicated that NLRX1 regulated IRF1 expression post-transcrip-tionally. To gain insight into whether this negative regulation of the IRF1 response was due to enhanced degradation of IRF1 or reduced synthesis of IRF1 protein, we globally inhibited protein synthesis in SeV-infected cells with cycloheximide and monitored the subse-quent decay of IRF1 protein. Despite the greater initial abundance of IRF1 in control PH5CH8 cells than in NLRX1-T3 cells, the half-life (stability) of IRF1 protein in NLRX1-T3 cells was indistinguishable from that in control PH5CH8 cells (Fig. 4c and Supplementary Fig. 4b). Collectively, these data suggested that the reduced abundance of IRF1 protein in NLRX1-deficient cells resulted from impaired translation of IRF1 mRNA. Consistent with that, globally blocking protein synthesis with puromycin completely abolished the effect of NLRX1 deficiency on the IRF1 response to infection with SeV (Supplementary Fig. 4c).
12
a
e
f
g
h
b
c
d
150
120
90
60
30
0
120
150
120
90
50
0
0 3 0 3
90
60
60
40
20
0
0 3 0 3
30
0 0
Time after Act D (min) Time after CHX (min) Time after SeV infection (h)
Time after SeV infection (h)
40 80 120 0 40 80 120
9
6
3
0
0.6
40S 60S
80S
40S
40S
60S 60S
80S 80S
40S 60S
80S 0.4
0.2
0.0
0.6
0.4
0.2
0.0
0.6
0.4
0.2
0.0 0.6
0.6
IRF1 IRF3 ACTB IRF1 IRF3 ACTB 90
Control NLRX1-T3
80
70
60
30
0
0.4
0.2
0.0 0.4
0.2
0.0
1 3 5 7 9 11 13 1 3 5 7 9 11 13
1 3 5 7 9 11 13
1 3 5 7 9
Fraction Fraction
11 13 30
20
10
0
30
20
10
0
30
20
10
0
30
20
10
0 Mock
Cells above threshold (%)
SeV
80S/40S
Polysome-associated
mRNA (%)
mRNA (%)
mRNA (%)
0 NS
NS
***
** *
** ** **
**
NS
NS NS
3
Time after SeV infection (h)
IRF1
mRNA
(relative)
Remaining
IRF1
mRNA (%)
Remaining IRF1 protein (%) 35[
S]-Met-Cys incorporated
(%)
Control
Control
Puromycin
Control NLRX1-T3
IRF1 IRF3 ACTB
DAPI
Mock SeV Mock SeV
Mock SeV Control
Control Control
NLRX1-T2 NLRX1-T3
NLRX1-T3 NLRX1-T3
NLRX1-T3 NLRX1-T3
Control NLRX1-T3
Control NLRX1-T3
Mock
A254
(
)
SeV
A254
(
)
Control NLRX1-T3
Figure 4 NLRX1 facilitates immediate IRF1 antiviral responses by promoting global protein synthesis in SeV-infected cells. (a) qRT-PCR analysis of
IRF1 mRNA in control PH5CH8 cells, NLRX1-T2 cells and NLRX1-T3 cells (key) left uninfected (0) or infected for 3 h with SeV (horizontal axis);
results were normalized to those of ACTB. (b) Stability of IRF1 mRNA in SeV-infected control PH5CH8 cells and NLRX1-T3 cells (key), assessed as
IRF1 mRNA remaining at various times (horizontal axis) after treatment with actinomycin D (Act D). (c) Stability of IRF1 protein in SeV-infected control
PH5CH8 cells and NLRX1-T3 cells (key), assessed by quantification of band intensity (immunoblot analysis, Supplementary Fig. 4b) at various times
(horizontal axis) after treatment with cycloheximide (CHX). (d) Nascent protein synthesis in control PH5CH8 cells and NLRX1-T3 cells (key) mock
infected (0) or infected for 3 h with SeV, assessed as the incorporation of [35S]-Met-Cys into cellular proteins (precipitated from cells by trichloroacetic
acid), relative to that in uninfected control PH5CH8 cells, set as 100% (immunoblot analysis, Supplementary Fig. 4d). (e) Confocal microscopy (left)
of control PH5CH8 cells and NLRX1-T3 cells mock infected (0) or infected for 3 h with SeV (left margin) and pulse-labeled with puromycin, assessing global protein synthesis by staining of puromycin with specific antibody, with nuclei counterstained with the DNA-binding dye DAPI (below images),
and frequency of cells with puromycin labeling exceeding an arbitrary threshold (far right). Scale bar (left), 40 Mm. (f) Polysome profile of IRF1, IRF3
or ACTB mRNA (key) in mock- or SeV-infected (left margin) control PH5CH8 cells and NLRX1-T3 cells (above plots), plotted as absorbance at 254 nm
(A254) (left vertical axis) and qRT-PCR analysis of mRNA (presented as percent of total mRNA in each fraction; right vertical axis); red vertical lines
distinguish polysome-associated mRNA (fractions 7–14) from non–polysome-associated mRNA (fractions 1–6). (g) Proportion of IRF1, IRF3 or ACTB
mRNA (horizontal axis) associated with translationally active polysomes (fractions 7–14 in f) in control PH5CH8 cells and NLRX1-T3 cells (above
plot) mock infected or infected with SeV (key). (h) Ratio of 80S to 40S in mock- or SeV infected (horizontal axis) control PH5CH8 cells and NLRX1-T3
cells (key), calculated from the area under the curve of 40S and 80S peaks of A254 traces. Each symbol (a,d,e,g,h) represents an individual technical
replicate (a) or experiment (d,e,g,h); small horizontal lines (d,e) indicate the mean (o s.e.m.). *P < 0.05, **P < 0.01 and ***P < 0.001 (two-way
ANOVA (a,d,e,g) or t-test (h)). Data are representative of three experiments with n = 3 technical replicates in each (a; mean + s.e.m.), two experiments
with n = 3 technical replicates (b; mean o s.e.m.), six experiments (c; mean o s.e.m.), three experiments with n = 1-3 technical replicates (d), three
present in the lysates of uninfected cells (Supplementary Fig. 4d) indicated that this increase was due to enhanced cellular protein synthesis, not viral protein synthesis. As an independent meas-ure of protein synthesis, we pulse-labeled NLRX1-T3 cells with a low concentration of puromycin and used confocal microscopy to monitor its incorporation into nascent protein at a single-cell level
(Fig. 4e). Infection with SeV induced an increase in nascent protein synthesis in a large proportion of control PH5CH8 cells but not in NLRX1-T3 cells (Fig. 4e). We obtained similar results with NLRX1-T2 cells, at both a whole-cell-culture level and a single-cell level of analysis (Supplementary Fig. 4e), which excluded the possibility that this reflected an off-target effect of the sgRNA used to create the
Mock
a
d
g
h
e
f
b
c
Contr ol Contr ol PKR-K O-1 Contr ol EV PKR-KO-1 PKR-K O-1 PKR-K O-3 PKR-K O-3 PKR-KO-1 PKR-KO-3 PKR-KO-1 PKR-KO-3 PKR-KO-1 PKR-KO-3 PKR-KO-3 PKR-KO-1 PKR-KO-1 +T3 Contr ol NLRX1 -T3 NLRX1 NLRX1 Low HAV RNA– NLRX1 Biotin HAV RNA Biotin Poly(I:C) High NLRX1-KO NLRX1-KO NLRX1 NLRX1-KO IRF1 p-PKR PKRSeV (h) 0
Ctr
l
T3
Ctr l
T3 T3 T3 Ctrl T3 T3 T3
0 50 250 1,250
Ctr l T3 – – Ctr l
T3 – T3 – T3
– – –
– – –
T3 T3 T3 T3
3 0 3 0 3
IRF1 IRF3 p-PKR (r elativ e) p-eIF2 (r elativ e) Actin Actin Actin Actin Actin
Time after SeV infection (h) Actin p-eIF2 NLRX1 -T3 Control Control Input IP NLRX1-T3 NLRX1-T3 NLRX1-T3 NLRX1 (ng) NLRX1 (ng) HA V RNA -bound PKR NLRX1 PKR
0 20 100500
0 50 250 1,250 NLRX1 (ng) NLRX1 PKR NLRX1 -T3 NLRX1 SeV Mock Mock NS NS NS NS NS NS NS NS *** **** **** * ** ** * * SeV SeV Mock SeV 2.0 1,500 IL -6 (pg/ml) [ 35 S]-Met-C y s incor porat ed (%) HA
V genomic RNA
(r
elativ
e)
HA
V RNA (r
elativ e) 1,000 500 0 2 3 8 6 4 2 0 140 1.2 0.9 0.6 0.3 0.0 NLRX1 (ng) P ol y(I:C)-bound PKR 0 20 100 500 NS *** ** 1.2 0.9 0.6 0.3 0.0 110 80 0
0 3 0 3
2 1 0 1 0 1.5 1.0 0.5 NLRX1-T3 NLRX1-T3
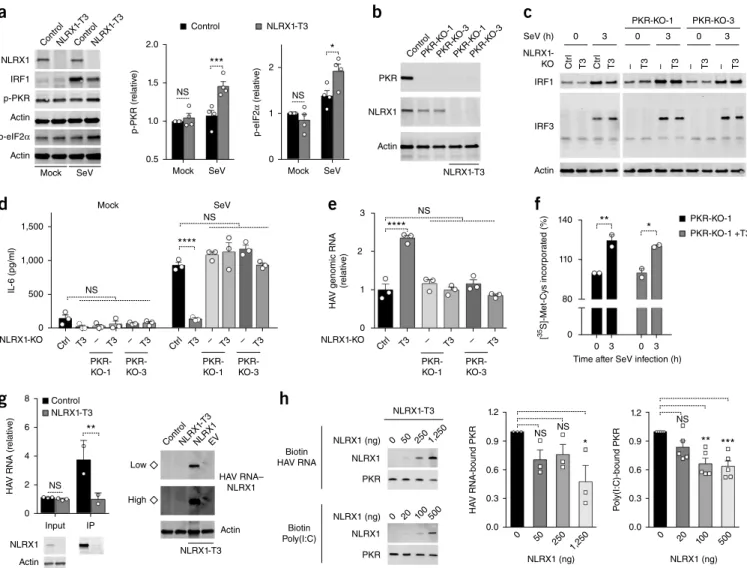
Figure 5 NLRX1 reduces the activation of PKR and subsequent inhibition of protein synthesis by competing for binding to viral RNA. (a) Immunoblot
analysis of total NLRX1 and IRF1 and phosphorylated (p-) PKR and eIF2A (left margin) in control PH5CH8 cells and NLRX1-T3 cells (above lanes)
mock infected or at 4.5 h after infection with SeV (below blots), assessing PKR–eIF2A signaling (left), and quantification (infrared fluorescence
intensity) of phosphorylated PKR or eIF2A, normalized to that of actin (right). (b) Immunoblot analysis of PKR and NLRX1 in control PH5CH8 cells (far
left) or PKR-deficient PH5CH8 cells (PKR-KO-1 or PKR-KO-3 (above lanes); middle) or PH5CH8 cells deficient in both NLRX1 and PKR (PKR-KO-1 or
PKR-KO-3 (above lanes) plus NLRX1-T3 (below blot); right). (c) Immunoblot analysis of IRF1 (top) and IRF3 dimers (middle) in control PH5CH8 cells
(Ctrl) and NLRX1-T3 cells (T3) (left blots) or PKR-deficient PH5CH8 cells (PKR-KO-1 or PKR-KO-3 (above blots) and – (above lanes); right) or PH5CH8
cells deficient in both NLRX1 and PKR (PKR-KO-1 or PKR-KO-3 (above blots) and T3 (above lanes); right). (d,e) ELISA of IL-6 (d) and qRT-PCR
analysis of HAV RNA (e) in cells as in c (below plots); qRT-PCR results are presented relative to those of control PH5CH8 cells, set as 1.
(f) Global protein synthesis in PKR-deficient PH5CH8 cells (PKR-KO-1) and PH5CH8 cells deficient in both NLRX1 and PKR (PKR-KO-1 + T3) (key)
mock infected (0) or infected for 3 h with SeV (horizontal axis), assessed as incorporation of [35S]-Met-Cys (as in Fig. 4d). (g) qRT-PCR analysis of HAV
RNA (top left) and immunoblot analysis of NLRX1 (bottom left) in control PH5CH8 cells and NLRX1-T3 cells (key) transfected for 3 h with HAV RNA, assessed as input samples or after immunoprecipitation (IP) with antibody to NLRX1 (horizontal axis or above lanes). Right, immunoblot analysis of NLRX1 precipitated together with biotin-tagged HAV RNA (HAV RNA–NLRX1; right margin) in lysates of control PH5CH8 cells or NLRX1-T3 cells (left half) or NLRX1-T3 cells (below blot) transfected with empty vector or vector expressing NLRX1 (above lanes; right half); left margin (◊), endogenous
NLRX1 associated with HAV RNA, with high or low detection intensity. (h) Immunoblot analysis (left) of NLRX1 and PKR in lysates of NLRX1-T3 cells
supplemented with various doses (above lanes) of purified recombinant NLRX1 and with biotin-tagged HAV RNA (top) or poly(I:C) (bottom); right,
quantification of the binding of HAV RNA (near right) or poly(I:C) (far right) to PKR. Each symbol (a,d,e,f,g,h) represents an individual experiment
(a,f,g,h) or technical replicate (d,e) (mean + s.e.m in each). *P < 0.05, **P < 0.01 and ***P < 0.001 (two-way ANOVA (a,d,f,g) or one-way ANOVA
(e,h)). Data are representative of four independent experiments (a), two independent experiments (b,c), two experiments with n = 3 technical replicates
in each (d,e), two experiments with n = 2 technical replicates (f), two experiments with n = 2–3 technical replicates (g) or three (HAV RNA) or five
NLRX1-T3 cells. siRNA-mediated depletion of NLRX1 expression in primary HFHs also substantially suppressed the SeV-induced increases in nascent protein synthesis (Supplementary Fig. 4f).
Although NLRX1 exerted distinctly different regulatory effects on SeV-induced cytokine expression in MEFs relative to that human hepatocytes, depleting cells of NLRX1 expression reduced the SeV-induced increase in the abundance of IRF1 in both cell types (Figs. 2e and 3e). Consistent with that finding, the incorporation of puromycin into nascent protein was also significantly lower in SeV-infected MEFs from Nlrx1−/− mice than in those from wild-type mice
(Supplementary Fig. 4g). Thus, the negative effect of NLRX1
defi-ciency on protein synthesis in virus-infected cells was not limited to human hepatocytes.
To directly characterize the effect of NLRX1 deficiency on the translation of mRNA by ribosomes, we used sucrose-density-gradient fractionation to profile polysome formation following the infection of NLRX1-T3 cells or control PH5CH8 cells with SeV (Fig. 4f). The deletion of NLRX1 resulted in a significant shift in the gradient distri-bution of IRF1, IRF3 and ACTB mRNA from SeV-infected cells, with a significantly lower proportion of each mRNA associated with trans-lationally active polysomes (fractions 7–13) (Fig. 4g). Moreover, the formation of translationally competent 80S ribosomes was lower in SeV-infected NLRX1-T3 cells than in SeV-infected control PH5CH8 cells, as shown by the much lower ratio of 80S to the 40S subunit in SeV-infected NLRX1-T3 cells than in SeV-infected control PH5CH8 cells (Fig. 4h and Supplementary Fig. 4h); this suggested a defect in initiation of translation. Collectively, these three separate measures of protein synthesis—incorporation of [35S]-Met-Cys, puromycin labeling and polysome profiling—indicated that the translation of mRNA was globally suppressed by the absence of NLRX1 expression in SeV-infected cells.
NLRX1 limits PKR-mediated global translational shutdown
The dsRNA-activated kinase PKR is triggered by viral infection to phosphorylate the translation-initiation factor eIF2A and thereby globally shuts down host-cell translation26. To ascertain whether NLRX1 influences that PKR response, we characterized the activation of PKR in NLRX1-deficient cells. NLRX1 deficiency enhanced the SeV-induced autophosphorylation of PKR, as well as phosphoryla-tion of the PKR substrate eIF2A (Fig. 5a and Supplementary Fig. 5a). To determine whether NLRX1 facilitated virus-induced increases in IRF1 protein synthesis by suppressing PKR responses, we gener-ated PH5CH8 cells doubly deficient in NLRX1 and PKR (Fig. 5b). The negative effect of the deletion of NLRX1 on SeV-triggered IRF1 responses was completely abolished by PKR deficiency (Fig. 5c). PKR deficiency also fully restored the SeV-triggered immediate IL-6 pro-tein response (Fig. 5d) and abolished the beneficial effect conferred on HAV replication by NLRX1 deficiency (Fig. 5e). We also assessed the effect of SeV infection on nascent protein synthesis in the cells doubly deficient in NLRX1 and PKR. In contrast to the lack of increase in the incorporation of [35S]-Met-Cys or puromycin into NLRX1-deficient cells, relative to that in control PH5CH8 cells (Fig. 4d,e), protein synthesis increased over time in response to SeV infection in both cells doubly deficient in NLRX1 and PKR and those deficient in PKR alone (Fig. 5f and Supplementary Fig. 5b).
To explore the mechanism by which NLRX1 suppressed PKR responses, we investigated whether NLRX1 physically interacted with PKR in hepatocytes. However, no co-immunoprecipitation of PKR protein with either endogenous NLRX1 or overexpressed NLRX1 was detectable in PH5CH8 cells (Supplementary Fig. 5c,d). Since NLRX1 binds both viral RNA and poly(I:C), a common surrogate
for viral dsRNA27,28, we reasoned that NLRX1 might suppress the activation of PKR by competing for viral RNA in infected cells. To test our hypothesis, we first confirmed that NLRX1 bound HAV genomic RNA, which is known to have extensive secondary structure29, in bidirectional precipitation assays (Fig. 5g). Next, we designed a cell-free competition assay in which lysates of NLRX1-deficient cells were used as a source of PKR, supplemented with purified recombinant NLRX1 protein and biotin-tagged HAV RNA or poly(I:C). The addi-tion of increasing amounts of recombinant NLRX1 protein led to a progressive reduction in the binding of PKR to viral RNA or poly(I:C)
(Fig. 5h), which indicated that NLRX1 competed with PKR for
bind-ing to viral RNA. Together these results demonstrated that NLRX1 suppressed the activation of PKR, in part by competitively binding viral RNA, and thereby prevented the PKR-mediated translational shutdown that would otherwise attenuate IRF1-mediated antiviral responses (Supplementary Fig. 5e).
DISCUSSION
Although some studies have reported NLRX1 to be a negative regu-lator of innate immunity3,6,8,30, others have not confirmed that9,31 or have even suggested that NLRX1 enhances antiviral responses10. Consistent with the latter notion, we found NLRX1 to be an antiviral factor and a positive regulator of early innate immune responses in human hepatocytes. HAV and HCV, both common causes of viral hepatitis in humans, are unrelated, positive-strand hepatotropic RNA viruses that share many similarities in their replication cycles and in their interactions with innate immunity12. Both produce dsRNA rep-lication intermediates that induce signaling via RIG-I and TLR3 and thereby activate NF-KB and members of the IRF family, which leads to antiviral defense12,32. To counter those host defenses, both HAV and HCV express proteases that target for degradation MAVS and TRIF, key adaptors in the RIG-I and TLR3 signaling pathways33–36. Adding to those similarities, we found here that NLRX1 acted to restrict the replication of both HAV and HCV.
Surprisingly, we observed that NLRX1 had distinct, opposing effects on the SeV-induced activation of two members of the IRF family. NLXR1 was required for maximal increases in the abundance of IRF1 but suppressed the dimerization of IRF3. As key factors in inducing cytokine transcription, members of the IRF family are trig-gered by various stimuli in all cell types. Whereas IRF3 and IRF7 are activated by viral infection in most cell types37,38, other family mem-bers, including IRF1, as well as IRF5, IRF8 and IRF9, are selectively activated by different stimuli in a cell-type-dependent manner39–44. This complexity in IRF activation provides the diversity of responses required for effective host defense. However, it also allows a single regulatory molecule, such as NLRX1, to have different functional consequences when expressed in different cell types or pathogen con-texts or, potentially, at different points of time in the response to an invading virus. The opposing regulatory activities of NLRX1 on early IRF3 and IRF1 responses might account for much of the controversy surrounding this protein7,8,10. We found that NLRX1 positively regu-lated IRF1 signaling in both BMDMs and primary MEFs. However, NLRX1 is known to attenuate innate immunological control of viral replication in these cell types6–8. We infer from this that IRF1 does not have a dominant role in determining the outcome of infection in these cells, at least not under the conditions tested, whereas our data showed that it did so in hepatocytes. Thus, whether NLRX1 functions as a pro-viral factor or an antiviral factor, or neither9,31, probably reflects which IRFs dominate in inducing antiviral responses.
in macrophages and HEK293T human embryonic kidney cells3,7, we found that NLRX1 had a slight, positive effect on RIG-I-mediated NF-KB signaling in hepatocytes. However, this effect was not critical for the promotion of early cytokine responses by NLRX1, as our data showed that NLRX1 regulated the IL-6 response downstream of the influence of NF-KB.
Negative regulation of MAVS-mediated IRF3 responses by NLRX1 has been described previously6, but how NLRX1 affects IRF1 responses has not been studied. We found that depleting cells of NLRX1 did not alter SeV-induced transcription of IRF1 mRNA in PH5CH8 cells, but it significantly reduced the synthesis of IRF1 protein. Reductions in protein synthesis were not specific to IRF1 but instead reflected a global shutdown of protein synthesis due to enhanced activation of PKR, a phenomenon that would probably have its greatest effect on proteins that turn over rapidly, such as IRF1, which has a half-life of about 60 min.
Emerging evidence is increasingly linking translational control to the regulation of immediate responses of the innate immune system. Both IRF7 and IRF8 seem to be controlled via global, regulation of translation dependent on the translation-initiation factor eIF4E45–47. PKR restricts protein synthesis globally by phosphorylating eIF2A and thereby blocking the production of virus48. However, activation of PKR also reduces the synthesis of effector proteins encoded by inter-feron-stimulated genes49 and, as we have shown here, the production of IRF1 driven by an early, NF-KB-mediated response to SeV. PKR is thus a double-edged sword, potentially weakening the cell’s antiviral defenses as well as presenting a hurdle to be overcome by the virus. The negative effect of the activation of PKR on host defense might be particularly important when cells are infected with viruses such as HCV that can continue to translate their RNAs despite such activa-tion49. Our data suggest that NLRX1 moderates the potentially nega-tive consequences of PKR activation by competing with it for binding to viral RNA. This is distinct from how other members of the NLR family, including NLRP3, NLRP1 and NLRC4, physically interact with PKR to maximize activation of the inflammasome50.
We speculate that the opposing actions of NLRX1 on early IRF1 and IRF3 antiviral responses might underlie much of the controversy surrounding the biological function of this member of the NLR family. Loss of NLRX1 expression might result in contradictory effects on out-come of infections by different viruses in different tissues, depending on which IRF protein dominates in driving the antiviral response.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Note: Any Supplementary Information and Source Data files are available in the
online version of the paper.
ACKNOWLEDGMENTS
We thank B. Glaunsinger (University of California, Berkeley) for the human IL6 3` UTR expression vector, and L. Hensley, W. Lovell, M. Deng, and M. Chua for technical support. Supported by the US National Institutes of Health (U19-AI109965 to S.M.L., J.A.D. and J.P-Y.T.; R01-AI103083 to S.M.L.; R01-AI131685 to S.M.L. and J.K.W.; R01-AI103311 to N.J.M.; and R21-AI117575 to J.K.W.) and the University of North Carolina Cancer Research Fund (S.M.L. and J.P-Y.T.).
AUTHOR CONTRIBUTIONS
H.F. and S.M.L. designed the experiments, analyzed the data and wrote the manuscript; H.F., E.M.L., D.Y., D.R.M., O.G -L. and I.M. performed experiments; E.W., J.M., H.G., L.M.R., J.K.W., J.P -Y.T., J.A.D. and N.J.M. provided critical reagents and intellectual input; S.M.L. supervised the study; and all authors reviewed the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/ reprints/index.html. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
1. Guo, H., Callaway, J.B. & Ting, J.P. Inflammasomes: mechanism of action, role in
disease, and therapeutics. Nat. Med.21, 677–687 (2015).
2. Schneider, M. et al. The innate immune sensor NLRC3 attenuates Toll-like receptor
signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-KB. Nat. Immunol.13, 823–831 (2012).
3. Xia, X. et al. NLRX1 negatively regulates TLR-induced NF-KB signaling by targeting
TRAF6 and IKK. Immunity34, 843–853 (2011).
4. Benko, S., Magalhaes, J.G., Philpott, D.J. & Girardin, S.E. NLRC5 limits the
activation of inflammatory pathways. J. Immunol.185, 1681–1691 (2010).
5. Allen, I.C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through
the negative regulation of noncanonical NF-KB signaling. Immunity36, 742–754
(2012).
6. Moore, C.B. et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature
451, 573–577 (2008).
7. Allen, I.C. et al. NLRX1 protein attenuates inflammatory responses to infection by
interfering with the RIG-I-MAVS and TRAF6-NF-KB signaling pathways. Immunity
34, 854–865 (2011).
8. Guo, H. et al. NLRX1 Sequesters STING to negatively regulate the interferon
response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host
Microbe19, 515–528 (2016).
9. Rebsamen, M. et al. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell
Death Differ.18, 1387 (2011).
10. Jaworska, J. et al. NLRX1 prevents mitochondrial induced apoptosis and enhances
macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc. Natl. Acad. Sci. USA111, E2110–E2119 (2014).
11. Tattoli, I. et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-KB
and JNK pathways by inducing reactive oxygen species production. EMBO Rep.9,
293–300 (2008).
12. Qu, L. & Lemon, S.M. Hepatitis A and hepatitis C viruses: divergent infection outcomes marked by similarities in induction and evasion of interferon responses. Semin. Liver Dis.30, 319–332 (2010).
13. Ikeda, M. et al. Human hepatocyte clonal cell lines that support persistent replication
of hepatitis C virus. Virus Res.56, 157–167 (1998).
14. Li, K., Chen, Z., Kato, N., Gale, M. Jr. & Lemon, S.M. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem.280, 16739–16747 (2005).
15. Yamane, D. et al. Differential hepatitis C virus RNA target site selection and host
factor activities of naturally occurring miR-122 3´ variants. Nucleic Acids Res.45,
4743–4755 (2017).
16. Feng, Z. et al. A pathogenic picornavirus acquires an envelope by hijacking cellular
membranes. Nature496, 367–371 (2013).
17. Abdul-Sater, A.A. et al. Enhancement of reactive oxygen species production and
chlamydial infection by the mitochondrial Nod-like family member NLRX1. J. Biol.
Chem.285, 41637–41645 (2010).
18. Lei, Y. et al. The mitochondrial proteins NLRX1 and TUFM form a complex that
regulates type I interferon and autophagy. Immunity36, 933–946 (2012).
19. Soares, F. et al. The mitochondrial protein NLRX1 controls the balance between
extrinsic and intrinsic apoptosis. J. Biol. Chem.289, 19317–19330 (2014).
20. Sumpter, R. Jr. et al. Regulating intracellular antiviral defense and permissiveness
to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol.
79, 2689–2699 (2005).
21. Masuda, K. et al. Arid5a controls IL-6 mRNA stability, which contributes to elevation
of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA110, 9409–9414 (2013).
22. Odendall, C. et al. Diverse intracellular pathogens activate type III interferon
expression from peroxisomes. Nat. Immunol.15, 717–726 (2014).
23. Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol.
Chem.282, 15325–15329 (2007).
24. Mboko, W.P. et al. Interferon regulatory factor 1 restricts gammaherpesvirus
replication in primary immune cells. J. Virol.88, 6993–7004 (2014).
25. Fujita, T., Kimura, Y., Miyamoto, M., Barsoumian, E.L. & Taniguchi, T. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor,
IRF-1. Nature337, 270–272 (1989).
26. Dauber, B. & Wolff, T. Activation of the antiviral kinase PKR and viral
countermeasures. Viruses1, 523–544 (2009).
27. Hong, M., Yoon, S.I. & Wilson, I.A. Structure and functional characterization of the
RNA-binding element of the NLRX1 innate immune modulator. Immunity36,
337–347 (2012).
28. Unger, B.L. et al. Nod-like receptor X-1 is required for rhinovirus-induced barrier
dysfunction in airway epithelial cells. J. Virol.88, 3705–3718 (2014).
29. Brown, E.A., Zajac, A.J. & Lemon, S.M. In vitro characterization of an internal
ribosomal entry site (IRES) present within the 5` nontranslated region of hepatitis
A virus RNA: comparison with the IRES of encephalomyocarditis virus. J. Virol.68,
1066–1074 (1994).
30. Barouch, D.H. et al. Rapid inflammasome activation following mucosal SIV infection
31. Soares, F. et al. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun.19, 438–448 (2013).
32. Hirai-Yuki, A. et al. MAVS-dependent host species range and pathogenicity of human
hepatitis A virus. Science353, 1541–1545 (2016).
33. Yang, Y. et al. Disruption of innate immunity due to mitochondrial targeting of a
picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 104, 7253–7258
(2007).
34. Qu, L. et al. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis
A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 7,
e1002169 (2011).
35. Li, K. et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage
of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA102,
2992–2997 (2005).
36. Li, X.D., Sun, L., Seth, R.B., Pineda, G. & Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to
evade innate immunity. Proc. Natl. Acad. Sci. USA102, 17717–17722 (2005).
37. Honda, K. & Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors
and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658
(2006).
38. Ivashkiv, L.B. & Donlin, L.T. Regulation of type I interferon responses. Nat. Rev.
Immunol.14, 36–49 (2014).
39. Tailor, P. et al. The feedback phase of type I interferon induction in dendritic cells
requires interferon regulatory factor 8. Immunity27, 228–239 (2007).
40. Li, P. et al. IRF8 and IRF3 cooperatively regulate rapid interferon-B induction in
human blood monocytes. Blood117, 2847–2854 (2011).
41. Lazear, H.M. et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN
response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog.
9, e1003118 (2013).
42. Weiss, G. et al. MyD88 drives the IFN-B response to Lactobacillus acidophilus in
dendritic cells through a mechanism involving IRF1, IRF3, and IRF7. J. Immunol.
189, 2860–2868 (2012).
43. Ousman, S.S., Wang, J. & Campbell, I.L. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system
during viral infection. J. Virol.79, 7514–7527 (2005).
44. Scherbik, S.V., Stockman, B.M. & Brinton, M.A. Differential expression of interferon (IFN) regulatory factors and IFN-stimulated genes at early times after West Nile
virus infection of mouse embryo fibroblasts. J. Virol.81, 12005–12018 (2007).
45. Xu, H. et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote
inflammatory macrophage polarization. Nat. Immunol.13, 642–650 (2012).
46. Colina, R. et al. Translational control of the innate immune response through IRF-7.
Nature452, 323–328 (2008).
47. Lee, M.S., Kim, B., Oh, G.T. & Kim, Y.J. OASL1 inhibits translation of the type I
interferon-regulating transcription factor IRF7. Nat. Immunol. 14, 346–355
(2013).
48. Elde, N.C., Child, S.J., Geballe, A.P. & Malik, H.S. Protein kinase R reveals an
evolutionary model for defeating viral mimicry. Nature457, 485–489 (2009).
49. Garaigorta, U. & Chisari, F.V. Hepatitis C virus blocks interferon effector function by
inducing protein kinase R phosphorylation. Cell Host Microbe6, 513–522 (2009).
50. Lu, B. et al. Novel role of PKR in inflammasome activation and HMGB1 release.