THE CHARACTERISATION OF A T CELL
RESPONSE TO HUMAN RHINOVIRUS
BY
SUNETHRA S. WIMALASUNDERA
THESIS SUBMITTED TO THE UNIVERSITY OF LONDON
IN PART FULFILMENT OF THE DEGREE OF DOCTOR OF PHILOSOPHY
DEPARTMENT OF IMMUNOLOGY
UNIVERSITY COLLEGE LONDON
GOWER STREET
LONDON WCl 6BT
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10017261
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
TITLE PAGE 1
DEDICATION 2
TABLE OF CONTENTS 3
LIST OF FIGURES 8
LIST OF TABLES 12
LIST OF ABBREVIATIONS 13
LIST OF REAGENTS 15
ACKNOWLEDGEMENTS 17
ABSTRACT 18
CHAPTER 1: INTRODUCTION 19
1.1 Ov e r v ie wo fim m u n it yt o Vir u s e s 19
1.2 Th e C D 4 T c e l l Me m o r yr e s p o n s e 23
1.21 Longevity of memory T cells 24
1.22 Naive and memory T cells differ in activation requirements and functional states 28
1.23 Migration of naive and memory T cells 33
1.24 Phenotypic markers of naive and memory T cells 34
a) The use of CD45 splice variants as markers of memory T cells 35
b) The expression of CD45RA may not always represent a naive phenotype 37
c) Other markers of activation/memory in humans 41
1.3 T CELL CYTOKINES 43
1.31 The cytokine dichotomy in CD4 positive T helper cells 44
1.32 The primary stimulus for differentiation into Thl and Th2 cells 44
1.33 Cross regulation of Thl and Th2 cells 47
1.34 Source of cytokines 50
1.4
1.41 Epidemiology 54
1.42 Infection 55
1.43 Structure 56
1.44 Physical properties 57
1.45 Cellular receptors for HRV 57
1.46 Receptor binding sites on HRV 58
1.47 Humoral Response 59
1.48 Cellular Response 59
1.5 Th er o l eo fCD4 T c ellsint h eim m u n er e s p o n s et oIFZ A a n dRSV 62
1.51IFZA 62
1.52 RSV 63
1.6 As s o c ia t io no fu p p e rr e s p ir a t o r yt r a c tv ir a lin f e c t io n sa n da s t h m a 65
1.7 Ob je c t iv e so fs t u d y 66
CHAPTER 2: MATERIALS AND METHOD 67
2.1 Ex p a n s io na n d PURIFICATION o fHRV 67
2.11 Expansion 67
2.12 Purification 67
2.13 ELISA for measuring HRV concentration 68
2.14 Origin of HRV serotype specific anti-sera 69
2.15 Origin of HRV serotypes 69
2 .2 ISOLATION OF CELLS FROM HUMAN TONSILS 7 0
2.21 Enrichment of T cells by resetting with sheep red blood cells (SRBC) 71
2.22 Enrichment of B cells by resetting with SRBC 71
2.23 Enrichment of T cells by depletion using anti-mouse IgG coated dynabeads 72
2.24 Enrichment of CD4/CD8 T cells by depletion with anti-mouse IgG coated 72
dynabeads
2.25 Enrichment of CD45RA/CD45RO T cells by depletion with anti-mouse IgG 73
coated dynabeads
2.27 Enrichment of DC by depletion with anti-mouse IgG coated dynabeads 73
2 .3 Is o l a t io no fc e l l sfr o m PB a n dUCB 74
2.4 Pr o l if e r a t io na s s a y s 75
2 .5 IMMUNOFLOURESCENT FLOW CYTOMETRY 75
2.6 An t ib o d ie su s e d inis o l a t io no f T c e l ls u b s e t s 76
2.7 Cy t o k in ea n a l y s is 77
2 .8 Ma in t e n a n c e o f Ce l ll in e s 7 8
CHAPTER 3: THE HRV RESPONSE IN TONSILAR T CELLS 80
3 .1 In t r o d u c t io n 80
3 .2 . Fr e q u e n c y o ft h e H R V spe c ificr e s p o n s eint o n s il a r T CELLS 82
3 .3 T CELLS FROM A RESPONDER CAN REACT TO MULTIPLE SEROTYPES 8 2
3 .4 Th e T c e l lr e s p o n s eisHRV spe c ific 82
3 .5 Ul t r a v io l e t (U V ) in a c t iv a t io no f H R V d o e sn o te ffe c tt h e T c e l l 83 PROLIFERATION
3.6 EFFICIENCY OF DIFFERENT METHODS OF ENRICHMENT FOR T CELLS 83
3.61 Phenotypic analysis of the various responder populations 84
3.62 Determination of the antigen concentration required to induce an optimal 85
response to HRV
3.63 The effect of altering the T cell number on the proliferative response to HRV 86
3.64 Determination of the time course of the HRV specific in vitroproliferative 87
3.65 APC requirement 87
a Titration of B cell enriched populations 87
b Comparison of B cell and DC enriched populations 88
3 .7 Dis c u s s io n 90
CHAPTER 4;T CELL SUBSETS INVOLVED IN THE HRV RESPONSE 117
4.1 INTRODUCTION 117
4 .2 HRV SPECIFIC T CELL RESPONSES ARE DEPENDENT ON THE PRESENCE 118
OF CD4 T CELLS
4 .3 De t e r m in a t io no ft h ec y t o k in ep r o fil eo ft h e H R V sp e c if ic 118
T CELL RESPONSE
4 .4 Th eHRV r e s p o n s e inCD45RA a n dCD45RO T c e l l s 119
4 .4 1 The use of magnetic beads to enrich for CD45RA, CD3 positive T cells 119
4 .4 2 Comparison of the HRV response in CD45RA and CD45RO enriched T cells 120
a. Comparison of CD45RA and CD45RO responder populations enriched 120
for T cells by depleting CD 14 and CD 19 expressing cells
b. Comparison of CD45RA and CD45RO responder populations enriched for 120
T cells by depleting CD 14, CD 19, and MHC class II expressing cells
c. Comparison of CD45RO‘*“” and CD45RO*’”^ ‘ responder populations enriched for 121
T cells by resetting with SRBC
4 .4 3 Time course of the HRV response in CD45RA enriched T cells 121
4 .4 4 Comparative titration of CD45RA and CD45RO T cells required for the HRV 122
specific response
4.5 UCBMC DO NOT RESPOND TO HRV 123
CHAPTER 5; HRV RESPONSE IN PB DERIVED T CELLS 145
5 .1 Introduction 145
5.2 Time course of the HRV response in PBMC 146
5.3 Frequency of HRV response in PBMC 146
5.4 Determination of the cytokine profile of the HRV response in PBMC 147
5.5 Comparison of cells isolated from tonsil or PB of the same individual 147
5.6 CD45RA enriched T cells from PB can respond to HRV and IFZ A 148
5.7 Di s c u s s io n 149
CHAPTER 6; GENERAL CONCLUSION 160
6 .1 Su m m a r y 160
6 .2 Fu t u r e St u d i e s 161
Figure 2.1 Isolation of cells from tonsils 79
Figure 3.1 The frequency of HRV specific responders 95
Figure 3.2 The T cell response to multiple serotypes. 96
Figure 3.3 T cell responses are HRV specific 97
Figure 3.4 The effect of UV treatment of HRV on the T cell response 98
Figure 3.5 The Phenotypic analysis of E+ HD cells 99
Figure 3.6 The Phenotypic analysis of CD 14, CD19 depleted HD cells 100
Figure 3.7 The Phenotypic analysis of CD 14, CD19, MHC class II depleted HD cells 101
Figure 3.8. Dose response of HRV specific T cells in HD responders 102
Figure 3.9 Dose response of HRV specific T cells in E+ HD cells 103
Figure 3.10 Dose response of HRV specific T cells in CD 14, CD 19, MHC class II 104
depleted HD cells
Figure 3.11 The effect of altering the number of E+ HD T cells on the proliferative 105
response to HRV
Figure 3.12 The effect of altering the number of CD14, CD 19, MHC class II depleted 106
HD cells on the proliferative response to HRV
Figure 3.13 Limiting dilution analysis of the HRV response 107
Figure 3.15 The time course of the HRV specific proliferative response in 109
CD 14, CD 19, MHC class II depleted HD cells
Figure 3.16 The APC requirement of E+ HD cells 110
Figure 3.17 The APC requirement of CD 14, CD 19 depleted HD cells 111
Figure 3.18 The APC requirement CD 14, CD19, MHC class II depleted HD cells 112
Figure 3.19 Phenotypic comparison of B cell enriched and B cell depleted LD cells 113
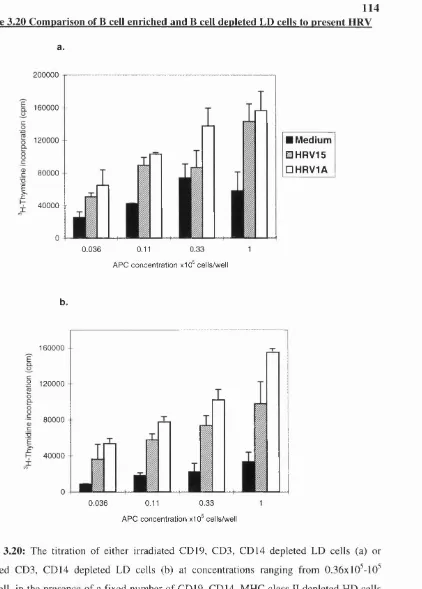
Figure 3.20 Comparison of B cell enriched and B cell depleted LD cells to present HRV 114
Figure 3.21 Phenotypic comparison of B cell enriched and DC enriched populations 115
Figure 3.22 Comparison of the ability of B cell enriched and DC enriched populations 116
to present HRV to CD45RA enriched T cells
Figure 4.1 Phenotypic comparison of CD4 and CDS depleted HD cells 130
Figure 4.2 The HRV specific T cell response is dependent on the presence of CD4 T cells 131
Figure 4.3 The cytokine profile of the HRV response 132
Figure 4.4 Phenotypic analysis of CD45RO, CD 14, CD19 depleted HD cells 133
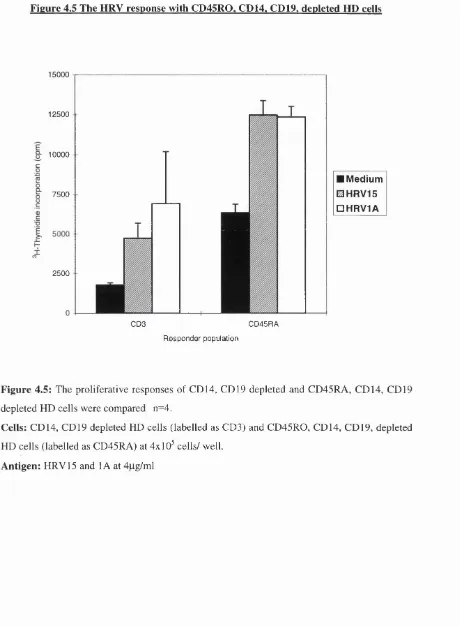
Figure 4.5 The HRV response in CD45RO, CD 14, CD 19 depleted HD cells 134
Figure 4.6 Phenotypic analysis of CD45RO, CD 14, CD 19, MHC class II depleted 135
HD cells
Figure 4.7 The APC requirement of the CD45RO, CD 14, CD 19, MHC class II depleted 136
Figure 4.8 Comparison of the HRV response in CD45RO, CD 14, CD 19 depleted and 137 CD45RA, CD14, CD19 depleted HD cells
Figure 4.9 Comparison of the phenotypic profile of CD45RO, CD14, CD19, MHC class II 138 depleted and CD45RA, CD 14, CD 19, MHC class II depleted HD cells
Figure 4.10 Comparison of the HRV response in CD45RO, CD 14, CD 19, MHC class II 139 depleted and CD45RA, CD 14, CD 19, MHC class II depleted HD cells
Figure 4.11 Comparison of the HRV response in CD45RO‘*““ and CD45RO*’'^^*’‘ T cells 140
Figure 4.12 Time course of the HRV response in CD45RO, CD 14, CD 19, MHC class II 141 depleted HD cells
Figure 4.13 Comparison of the HRV response in CD45RO, CD 14, CD 19, MHC class II 142 depleted and CD45RA, CD 14, CD 19, MHC class II depleted HD cells after 4 days in culture
Figure 4.14 Comparative titration of CD45RO, CD14, CD19, MHC class II depleted 143 and CD45RA, CD 14, CD 19, MHC class II depleted HD cells required for
the HRV specific response
Figure 4.15 UCBMC do not respond to HRV 144
Figure 5.1 Time course of the HRV response in PBMC 152
Figure 5.2 The frequency of the HRV responses in PBMC from seven healthy volunteer 153
Figure 5.3 The HRV response in CD 14, CD 19, MHC class II depleted PBMC 154
Figure 5.4 Determination of the cytokine profile of the HRV response in PBMC 155
Figure 5.6 The phenotypic profile CD45RO, CD 14, CD 19, MHC class II depleted 158
PBMC
LIST OF TABLES
TABLE 1: Origin of HRV serotypes 69
TABLE 2: Antibodies used in isolation of T cell subsets 69
LIST OF ABBREVIATIONS APC CLA Con A CTL Da DC E+HD E-LD ELISA FDC GM-CSF HD HLA HRV ICAM IFN IFZA Ig IL KD LD Leishmania Major LEA LN EPS LRT mAbs MHC mRNA NIms PB PBMC
Antigen presenting cell(s)
Cutaneous Lymphocyte associated antigen
Conconavalin A
Cytotoxic T lymphocyte
Daltons
Dendritic cells
Sheep blood rosseted HD cells
Sheep blood rosseted LD cells
Enzyme-Ihiked immunosorbant assay
Follicular dendritic cell
Granulocyte colony stimulatory factor
High density
Human lymphocyte associated antigen
Human rhinovirus
Intercellular adhesion molecule
Interferon Influenza A Immunoglobulin Interleukin Kilo daltons Low density L.Major
Lymphocyte function associated antigen
Lymph node
Lipopolysaccharide
Lower respiratory tract
Monoclonal antibodies
Major histocompatibilty complex
Messenger Ribonucleic acid
Neutralising immunogens
Peripheral blood
PCR Polymerase chain reaction
PHA Phytohaemoglutinin
RNA Ribonucleic acid
RSV Respiratory syncytial virus
SI Stimulation index
TAP Transporter associated with antigen presentation
TcR T cell receptor
Th T helper cells
UCB Umbilical cord blood
UCBMC Umbilical cord blood mononuclear cells
URT Upper respiratory tract
VCAM Vascular cell adhesion molecule
LIST OF REAGENTS
10 ml conical tubes: Nunclon, Kamstrup, Denmark
1000ml roller bottles: Falcon, Oxford, UK
1 Ox Hanks: Gibco, Paisley, UK
2-mercaptoethanol: Sigma, Dorset, UK
20ml syringe plunger: Terumo, Leven, Belgium
^H-Thymidine: ICN, Oxfordshire, UK
5-[^^^I]Iodo-2’-deoxyuridine ([^^^I]-IdUdR): Amersham, Bukinghamshire,UK
6 well tissue culture plates: Nunclon, Kamstrup, Denmark
96 well microtitre plates: Nunclon, Kamstrup, Denmark
Alkaline phosphatase conjugated rabbit anti-mouse antibody: Sigma, Dorset, UK
Ammonium chloride: Sigma, Dorset, UK
Ammonium sulphate: Sigma, Dorset, UK
Amphotericin B: Gibco, Paisley, UK
Bovine serum albumin (BSA): Sigma, Dorset, UK
Collagenase type II: Sigma, Dorset, UK
Conconavalin A (Con A): Sigma, Dorset, UK
Dulbecco’s Modified Eagle Medium (E4): Claire Hall Laboratories, Hertfordshire, UK
Ethylene diamine tetraacetate (EDTA): Sigma, Dorset, UK
Fluorescein isothiocyanate (FITC) conjugated sheep anti-mouse Ig: Dako, Glostrup,
Denmark
Foetal calf serum (FCS): Gibco, Paisley, UK
Glass Petri-dish: BDH, Lab Supplies, Upminster, UK
Glycine: Sigma, Dorset, UK
Hanks balanced salt solution (HBSS): Gibco, Paisley, UK
Hanks balanced salt solution (without phenol red) : Gibco, Paisley, UK
Histopaque 1077: Sigma, Dorset, UK
IFNy or IL-4 ELISA kits: AMS, Biotechnology, Oxon
Recombinant IL-2, IFNy or IL-4: Preprotech, London UK
Magnesium chloride: Sigma, Dorset, UK
N-2 hydroxyethyl piperazine-N’-2-ethane sulphonic acid (HEPES): Gibco, Paisley, UK
Nylon mesh (125pM pore size): Cadish, London, UK
p-Nitrophenyl phosphate: Sigma, Dorset, UK
Paraf ormlaldehy de : Sigma, Dorset, UK
Penicilin: Gibco Paisley, UK N
Percoll: Pharmacia, Hertfordshire, UK
Phosphate buffered saline (PBS): Claire Hall Laboratories, Hertfordshire, UK
Rosewell Park Memorial Institute 1640 medium (RPMI 1640): Gibco, Paisley, UK
Sarkosyl: Sigma, Dorset, UK
Sheep anti-mouse IgG coated magnetic beads: Dynal, Wirral, UK
Sheep red blood cells: Tissue Culture Services, Slough, Berkshire, UK
Sodium azide: BDH chemicals, Poole UK
Sodium carbonate buffer: Sigma, Dorset, UK
Syringe with 0.6x25mm needle size: Sabre, Berkshire, UK
Streptomycin: Gibco, Paisley, UK
Sucrose: Sigma, Dorset, UK
Tris: Sigma, Dorset, UK
Tissue culture flasks: Nunclon, Kamstrup, Denmark
Tween 20: Sigma, Dorset, UK
ACKNOWLEDGEMENTS
Firstly, I would like to express my deepest gratitude to my supervisors Professors Benny
Chain and David Katz for their invaluable guidance.
I would also like to thank Professor Peter Beverley, Dr. Luciene Lopes, Ms. Vanessa
Woodhead, Dr. Micheal Binks, Mr. Mark George, Dr. Diana Wallace, Dr. Mala Maini,
Mr. Hal Drakesmith and Mr Manminder Kambo for their assistance.
I would like to acknowledge all blood donars, the staff and patients at Middlesex and
Royal National Hospital for providing tonsils and at University College Hospital for
providing umbilical cord blood.
Finally, I would like to express a special thanks to my family and Verold West for their
ABSTRACT
Most viral infections are controlled either by neutralising antibodies and/or cytolysis of
infected cells by MHC class I restricted CDS T cells. However, CD4 T cells may also
contribute towards the defence against viral infections by the release cytokines which can either
provide help for the differentiation of effector T and B cells or have a direct anti-viral effect on
the infected cell. The T cell response induced by HRV has not yet been directly examined. The
objective of this study was to understand the nature of the T cell response and its contribution
to the pathogenesis of human rhinovirus (HRV) infection in humans. As a local lymphoid tissue
to the upper respiratory tract, the tonsil provides a site of drainage for any antigen that enters
via the nasopharyngeal surface. The high prevalence of HRV and the site of the tonsil would
predict the presence of a recall or memory response to HRV in T cells derived from tonsils. The
present study demonstrates that indeed a T cell proliferative response to HRV can be detected
in tonsil derived T cells in approximately 80% of the 132 individuals (mostly children aged
between 4-14 years of age) tested. This response was found to be dependent on the presence of
T cells and MHC class 11+ cells and mediated predominantly by the CD4 T cell subset with
underlying cytokine profile of Thl type cells. In most of the experiments only a single
representative member of each viral subgroup was used although in a few tonsils a panel of 7
different serotypes comprising members of each group were tested. Evidence to suggest a recall
or memory type response was suggested by the ability of these limited number of serotypes to
induce a response in the large majority of individuals and also by the ability to induce a
response within 3-4 days in culture. However, surprisingly HRV as well as influenza A (IFZ A)
was able to induce a recall type response in both the CD45RA and CD45RO T cell subsets but
were unable to activate umbilical cord blood derived mononuclear cells (UCBMC). In a limited
number of experiments the HRV response in peripheral blood (PB) derived T cells was
examined and shown to compare both quantitatively and qualitatively with tonsil derived T
cells. A population with antigen presenting capacity was shown to exist in an 80-90% CD3
enriched responder population which could only be removed after MHC class II depletion.
Sequential depletion of various subsets from the stimulator population suggested that these may
be dendritic cells (DC). Thus, the present study suggests that in contrast to most viral
infections, HRV induces predominantly a MHC class II restricted CD4 T cell response in vitro.
The induction of CD4 Thl cells could provide defence against primary infection by controlling
viral replication via cytokines such as IFNy and TNFa and provide protection against re
infection by supporting the production of neutralising antibodies by B cells. The ability to
induce comparable HRV specific responses in both CD45RA and CD45RO T cells suggest that
CHAPTER 1 INTRODUCTION
1.1 OVERVIEW OF IMMUNITY TO VIRUSES
Primary exposure of vertebrates to a viral pathogen results in the induction of a number
of com plet mechanisms by the immune system that are geared towards eradicating the
virus initially and then secondly developing specific protection against reinfection by the
virus. Although immune responses vary from virus to virus, certain fundamental
common features can be observed in many infections. The primary defence is mediated
by the non-specific mechanisms of the innate immune system, such as mucociliary
responses at mucosal surfaces, activation of complement, natural killer cells, and
phagocytic cells (macrophages and granulocytes) and their products (cytokines).
However, the innate system is not always very effective against viral pathogens and the
virus may survive this initial onslaught to continue replication. More specific
mechanisms are therefore required to combat the ever increasing virus load. The
expansion of effector B and T cells bearing antigen specific receptors for the virus can
effectively destroy the virus and/or virus infected cells and in addition, generate memory
cells to prevent reinfection.
Although both the T cell and B cell numbers are increased after primary exposure to the
virus or vaccine, the initial response is mediated by effector T cells. In contrast, the
antibody response requires longer periods to reach maximum levels. On eradication of
the virus, the numbers of effector T cells and B cells rapidly decreases. However, a
subpopulation of these primed T cells and B cells may survive so that on re-exposure to
the virus (or reactivation of latent virus), a more rapid expansion of these cells occurs
than observed on the primary exposure. The factors governing the survival of these T
cells are discussed in more detail in section 1.2.
T cells and B cells recognise different forms of virus. B cells recognise mostly structural
epitopes on a free virus, whereas T cells can recognise proteolytic fragments of
regulatory and structural proteins of the virus, that are presented on the surfaOe of
\
y
molecules MHC class I and MHC class n. Different T cell populations have evolved to
recognise presentation by either MHC class I or class II molecules. Whilst peptides
presented by MHC class II molecules are recognised by CD4 T cells, MHC class I
associated peptides are recognised by CDS T cells.
Different effector mechanisms are induced by these T cell subsets. On activation, the
main effector mechanism of CDS T cells is cytolysis of the targeted cell, although they
are also capable of producing a number of cytokines. However, cytokine production is
the main effector mechanism of CD4 T cells which allows these cells to provide help for
activation and proliferation of other cells such as B cells, macrophages and CDS T cells,
although CD4 T cells are also capable of cytolysis under certain conditions. The
preferential induction of the different subsets of CD4 T cell and the different cytokines
produced has a profound effect on the course of the viral infection and is discussed in
more detail in section 1.3.
The nature of the virus determines the mode of entry into the host cell. Following
binding of the virus to specific receptors on the host cell membrane, enveloped viruses
gain entry into the host cell by either fusion of the viral lipid membrane and the host cell
membrane (paramyxoviruses) or by receptor mediated endocytosis (orthomyxoviruses),
whilst non enveloped viruses can only enter via receptor mediated endocytosis
(picomaviruses) (reviewed in Knipe et al. 1996). The fusion pathway is promoted by
virion surface proteins and releases the viral nucleocapsid directly into the cytoplasm of
the cell. Receptor mediated endocytosis is the normal mechanism used by cells to take
up antigens from the extracellular environment. It occurs by the invagination of the
section of the plasma membrane bound to the virus (via a receptor) to form a clathrin
coated vesicle called an endosome. Once in the endosome the nucleocapsid of the
enveloped or the viral capsid of the non enveloped virus fuses with the endosomal
membrane to release the viral genome into the cytoplasm. In the cytoplasm, the newly
synthesised viral proteins will be subjected to proteolytic degradation by proteosomes
and subsequently transported to the endoplasmic reticulum by a peptide transporter
consisting of multi-membrane spanning proteins TAPI and TAP2 (Neefjes et al. 1993).
MHC class n loading occurs in the endosome whilst MHC class I loading occurs within
Although viruses can load both MHC class I and class II molecules, the viral proteins
encountered in the different compartments will differ for MHC class I and class II
molecules. Thus viral peptides presented by MHC class II molecules will be mainly
proteins of the virion capsid/nucleocapsid encountered in the endosome but fusogenic
viruses can also load MHC class II if viral membrane proteins can enter the endocytic
pathway. Proteins available for MHC class I presentation will include structural and
non-structural proteins, such as enzymes which are synthesised within the infected cell.
Virtually all cells express MHC class I molecules but MHC class II molecules are only
constitutively expressed on professional APC such as B cells and DC, although they can
also be induced on other cell types by cytokines released by T cells. Activated B cells
expressing both specific antibodies to the virus and MHC class II molecules can provide
a very efficient means of acquiring and presenting viral peptides to CD4 T cells.
Enveloped and non enveloped viruses also differ in the mode of exit from the infected
cell (reviewed in Knipe 1996). Enveloped viruses exit either by budding from the
plasma membrane or via secretory vesicles containing the virus which fuses with the
host plasma membrane. In general, non-enveloped viruses are released by lysing the host
cell, but there may be exceptions such as simian virus 40 and poliovirus.
The pathology that may result from a viral infection is due to a combination of the direct
effect of the pathogen, and the immunopathology associated with the host’s response
against the virus. The strength and kinetics of the response are critical to the outcome. A
weak or slow response may not be sufficient to control virus load, whilst an excessive
immune response can itself kill the host. Thus the immune system has a very delicate
balancing act to perform in order to control the virus and prevent excessive
immunopathology.
The nasopharynx region of the upper respiratory tract (URT) provides a port of entry to
a number of viruses. Although these viruses differ in their mode of infection and the
protective immunity induced, they induce similar symptoms generally recognised as a
‘common cold’, but may be combined with fever in the case of IFZ A infection. Similar
lower respiratory tract (LRT) complications can ensue from all these viruses, such as
illnesses are caused by rhinoviruses (HRV), about 15-20% by coronoviruses, 10% by
IFZ A and influenza B, whilst respiratory syncytial virus (RSV), adenovirus and
parainfluenza viruses together comprise 10% of URT illnesses in adults (Larson 1996).
All these viruses except for HRV and adenovirus are enveloped viruses and use receptor
mediated endocytosis as a route of entry to the host cell, although RSV can also enter by
fusion with the cell membrane. However, whilst HRV and adenovims are released by
cytolysis of the infected cell, the others are released by budding from the cell membrane.
Although considerable knowledge on the epidemiology, structure and biology of these
viruses is known, the immune response induced has only been well characterised for a
few, such as IFZ A and RSV. In order to gain an insight into the local cellular immune
response induced by HRV in vivo, the cellular immune responses induced by IFZ A and
RSV are discussed in more detail in section 1.5.
Human rhinovirus induces a well characterised protective humoral immune response.
However a few studies have also documented the presence of an HRV specific CD4 T
cell response. These studies suggest that T cells are involved in the immune response to
HRV, although their exact role is not known. In order to understand any pathological
condition or to develop effective therapeutic interventions, it is important to determine
all the key players and their exact role in the process.
The main objective of this study was to characterise the cellular response induced to
HRV in humans and to elucidate it’s contribution to HRV associated immunopathology.
More specifically, to determine the T cell subsets used in order to gain an insight to the
effector mechanisms induced by HRV in vivo. The cells were derived from tonsils
removed from mainly children aged between 4 and 14 undergoing routine tonsillectomy.
The tonsils form a major part of the lymphoid tissue draining the URT and it’s removal
is usually carried out after either an acute or chronic infection due to a number of
bacterial and viral pathogens which include HRV. Thus T cells derived from tonsils
would be expected to have previously been exposed to HRV and therefore reflect a
memory response to HRV.
The following in vitro study provides evidence to suggest that HRV is a highly
the preferential activation of CD4 T cells and release of Thl type cytokines, IL-2 and
IFNy.
1.2 T h e CD4 T c e l l M e m o r y r e s p o n s e
Immunological memory is the most important function of the adaptive immune
system in terms of survival, as it allows the host to remember a previous encounter
with an invading pathogen such that on secondary exposure, as a consequence of
developing a more efficient defence mechanism, little or no symptoms are suffered.
The efficiency comes from the increased rate and magnitude of the secondary
response due to the clonal expansion of the lymphocytes, and the increased sensitivity
due to the upregulation of a vast array of adhesion and costimulatory molecules
following the primary stimulus. Historically, there have been several recorded
examples of long-lived immunological memory (reviewed in Mackay 1993a). In 1781,
in the Foroe islands an outbreak of measles resulted in a severe reduction of the
population. Sixty five years later in 1846, a second epidemic of measles hit the
islands. A survey showed that only people older than 64 resisted the infection
suggesting that these individuals had acquired immunity from the first epidemic. In
1949, it was reported that immunity to yellow fever can be detected 50 years after the
primary exposure. Many other more recent studies have provided evidence to support
the idea of long-lived immunological memory. In mice, influenza specific antibodies
could be found 18 months after infection (Jones et al. 1987) and, for some viral
infections, a high antibody titre can be found for the entire life of the animal
(Zinkemagel 1990 ).
Immunological memory against viruses has been shown to correlate with an increase
in the frequency of specific serum antibody levels against the virus and an increase in
the frequency of circulating cytotoxic T cells (CTL) against the virus. However, CD4
T helper cells (Th) also play an important role in the induction of both humoral and
CTL memory by providing help in the form of cytokines required to initiate the
functions, both CTL and Th cells are subjected to similar differentiation and
activation requirements thus sharing many characteristics such as phenotypic markers.
The main emphasis in this discussion will be on the memory response induced in the
CD4 T cell subset because of it’s involvement in the response to HRV, although the
CDS T cells will be discussed where relevant.
There are still many questions to unravel with regard to the memory T cell. The
mechanisms involved in maintaining immunological memory remains contentious,
and debates continue as to whether the presence of the primary stimulus is required
for longevity of memory. Although three phases or functional states are recognised (as
defined in the murine system), specifically the unprimed naive, activated or effector
and the resting memory state, it is unclear as to whether the effector cell and the
memory cell are different stages of the same pathway, or the end products resulting
from the divergence of one pathway.
As a cell progresses from a naive unprimed state through an effector stage and or
memory state, it acquires specific phenotypic and functional characteristics, by which
these states can be defined, for example, memory cells have enhanced expression of
many adhesion and costimulatory molecules. One of the most commonly used
distinctions of naive and memory cells is the differential expression of CD45 (the
common leukocyte antigen) splice variants, with the low molecular weight isoforms
being associated with a memory phenotype whilst the high molecular weight isoforms
are associated with a naive phenotype in humans, mice, rats and sheep. In humans
these are detected by the anti-CD45RO and CD45RA monoclonal antibodies (mAbs)
respectively.
1.21 Longevity of memory T cells
Although there is evidence to suggest that the maintenance of memory for B cells may
be due to long lived cells (Schittek et al. 1990), memory T cells have been found to
have a relatively short life span compared to naive T cells. The use of mAbs that can
distinguish between naive and memory T cells has enabled the fate of these cells to be
was followed in cells with chromosomal dicentric lesions induced by radiation therapy
(Michie et al. 1992). This study showed that the CD45RA cells could survive for up to
10 years, whilst cells with the CD45RO memory phenotype disappeared after
approximately 1 year. The disappearance of the CD45RO cells may have been due to
either the death of these cells on entering cell division or due to a reversion to the
CD45RA phenotype. In either case, this study does provide evidence for the longevity
of the naive phenotype compared to the memory phenotype. In a more recent study,
the re-analysis of this data with stable dicentric lesions (as opposed to the unstable
lesions studied previously), estimated the rate of cell division in unprimed T cells as
once every 3.5 years, whereas the primed phenotype divided once every 22 weeks
(Mclean et al. 1995). Similarly in a murine study, the naive T cells were found to have
slower rate of turnover than those with memory phenotype as measured by DNA
labelling with bromodeoxyuridine in thymectomised mice (Tough et al. 1994).
The higher incidence of apoptosis in effector and memory T cells compared to naive T
cells also provides further evidence to support the relatively shorter life span of the
memory T cell compared to a naive T cell. Thus after an acute viral infection, the
CD45RO T cell subset has been shown to be more susceptible to apoptosis than the
CD45RA T cell subset (Uehara et al. 1992). The bcl-2 gene product has been shown
to block apoptosis (reviewed in Korsmeyer et al. 1992; King et al. 1993) and its
increased expression in B cells and thymocytes rescues these cells from cell death
(Liu et al. 1991a; Sentman etal. 1991). PB derived T cells isolated from an individual
during the effector phase of a viral infection (Epstein-Barr virus, varicella zoster
virus and human immuno-deficiency virus) were found to have reduced expression of
the bcl-2 gene product in both the CD4 and CDS, CD45RO T cells, and were highly
susceptible to apoptosis (Akbar et al. 1993). Furthermore, a number of studies have
shown that the differentiation of T cells from CD45RA to CD45RO results in the
down regulation of bcl-2 and the concomitant upregulation of the CD95 (Fas/APO-1
antigen), which is associated with induction of apoptosis of activated T cells (Salmon
et al. 1994; Uehara et al. 1992; Brunner et al. 1996; Dhein et al. 1996; Ju et al. 1996).
This suggests that the reciprocal expression of bcl-2 and Fas/APO-1 antigen regulates
the life span of an individual cell. Recently it has been found that the cytokines IL-2
26
In addition, other cytokines such as IL-4, IL-5, IL-6, IL-7 and IL-15 (Uehara et al.
1992; Akbar et al. 1996) have also been shown to inhibit apoptosis and this was found
to be due to their ability to signal via the XL-2y chain receptor which can promote the
expression of bcl-2 as well as bcl-x (another bcl-2 related gene involved in preventing
apoptosis) (Akbar et al. 1996).
In order to reconcile the longevity of immunological memory with the relatively short
life span of a single memory cell, it is now believed that it is the clonal specificity that
survives the length of time, and not the individual cell. However the mechanisms
involved in maintaining this clonal specificity remains a controversial issue. Several
theories have been proposed to explain how immunological memory can persist for
such long periods of time.
The theory that antigen is a prerequisite for the maintenance of memory was tested by
adoptively transferring highly purified memory T cells into irradiated or unirradiated
hosts (Gray et al. 1991a). Under these conditions it was found that immunological
memory was dependent on the presence of the antigen and in its absence decayed with
time and returned to a “primary” type of response. This was true for Th, CTLs and B
cells. There is good evidence to suggest that antigen can be stored in the form of
antibody-anti gen complexes on the surface of follicular dendritic cell (FDC) (Tew et
al. 1978, 1979; Mandel et al. 1980) and this is thought to be a means of restimulating
B cells directly (Tew et al. 1980; Gray et al. 1988; Bachman et al. 1994) and also Th
cells indirectly by presentation of the antigenic peptides processed and presented by
the same B cell (Kosco et al. 1988; Gray et al. 1991b, 1993, 1994). However, CTL
memory cannot be maintained via FDC associated antigen and consequently several
other mechanisms for maintaining memory have been suggested.
A number of very convincing studies have shown that antigen is not required to
maintain CTL memory. These studies examined the CTL memory to Sendai and
influenza viruses, where great measures were taken to ensure the absence of priming
antigen when transferring memory T cells to naive recipients. The recipients were
continuously monitored for viral RNA by polymerase chain reaction and
1994). Thus it was shown that Sendai virus specific CTLs could be detected even in
the absence of MHC-peptide complexes when transferred into MHC class I deficient
recipients (Hou et al. 1994). However, a recent study has shown that CDS T cell
memory can only be transferred and maintained in naive recipient in the absence of
antigen if the recipient is irradiated (Kundig et al. 1996). Therefore, these authors
suggest that the immunosuppressive techniques used in the above experiments may
have caused non-specific activation of the T cells and thereby have maintained these
cells over the examined period. Clearly the above studies would have to be re
examined in light of these findings.
A theory that is applicable to maintenance of both Th and CTL memory in the absence
of original antigen, is that of cross reactivity to similar environmental antigens
(Beverley et al. 1990). It was suggested that the memory T cell may have a lower
threshold of activation due to the increased expression of adhesion molecules which
would allow lower affinity interactions such as those that occur for recognition of
cross reactive epitopes (Beverley et al. 1990). There is a considerable amount of
experimental evidence to substantiate this theory. For example T cells specific for
malaria in humans previously unexposed to the pathogen have been observed (Fern et
al. 1992; Good et al. 1991). Furthermore, these malaria specific T cells responded to
cross reactive epitopes on various environmental antigens such as tetenus toxoid
(Currier et al. 1992). Cross reaction between serotypes of the same virus is commonly
observed (Hastings et al. 1991) and T cell clones specific for a given antigen often
shows promiscuity for allo-antigens (Ashwell et al. 1986).
Non-specific activation of bystander cells via cytokines has been suggested as another
mechanism for maintaining memory T cells (reviewed in: Tough et al. 1996). Thus for
example, the T cell activation induced by lymphocytic choriomeningitis virus or IFZ
A, far exceeds the percentage of virus specific T cells in primary and secondary
lymphoid tissue, as well as peripheral tissue (Lau et al. 1994; Carding et al. 1993). In
addition, a number of viruses can generate CTLs capable of killing uninfected
allogeneic class I expressing cells (Yang et al. 1989). However, not all of these cells
were capable of killing viral infected cells (Nahill et al. 1993). Evidence to suggest
the finding that a combination of the cytokines IL-2, TN Fa and IL-6 can activate PB
derived CD45RO T cells (but not CD45RA T cells) to induce the expression of
mRNA for IFNy and IL-4 and mediate effector functions such as B cell help (Unutmaz
et al. 1994).
Another possibility is that the memory cells retain their viability by reverting to a
quiescent state but still require periodic stimulation to maintain clonal specificity.
Thus in the absence of IL-2, the majority of CD45RO T cells were able to survive for
at least 6 days by culturing on fibroblasts (Akbar et al. 1993). In another study T cell
clones were also maintained for over a month on feeder cells, in the absence of
exogenous IL-2 (Cheever et al. 1986). There is evidence to suggest that in vivo, T
cells may revert from an activated CD45RO to a less active CD45RA state (see
section 1.24b).
Whatever the mechanism for maintaining memory, it is evident that many external
factors can affect the longevity of memory. In natural infections, the frequency of
infection, the time between each episode and the severity of the infection will affect
any measure of T cell memory. In experimental systems, in addition to time and
frequency of boosts, other factors such as adjuvants and route of entry will also come
into play.
1.22 Naive and memory T ceils differ in activation requirements and functional states.
The assessment of the activation requirement and the functional state of a T cell
provides an indication of the stage of development of that cell. This is because the
immune system has imposed stringent activation requirements for naive T cells
compared to those required at later stages of development. The murine system has
provided the major source of information regarding the activation requirements of
naive and memory T cells. The advantage in using animal models is that it allows
more control over the differentiation state of the T cell so that naive T cells (either
from transgenic mice or unprimed mice kept in pathogen free conditions) can be
the basis of the expression of mainly CD45RB). In contrast to the murine studies,
most human studies rely only on the expression of phenotypic markers (mainly
CD45RA and CD45RO; see section 1.24). In this regard only the murine system can
provide an accurate analysis of the activation requirements of naive and memory T
cells, and therefore will be the focus of this section. The human studies will only be
discussed where comparisons can be made with the murine system.
According to the two signal model proposed by Jenkins and colleagues (Mueller et al.
1989), it has been found that TcR ligation alone is insufficient to activate naive T
cells in both humans and mice, and they have an absolute requirement for
costimulatory signals. Many costimulatory receptor ligand pairs have been described,
but the most important costimulatory requirement for naive T cells has been shown to
be mediated via the interaction between B7 on ARC and its counter receptor CD28 on
the T cells in mice (Croft et al. 1995; Ho et al. 1994; McKnight et al. 1994a;
Sagerstrom et al. 1993; Janeway et al. 1994) and in humans (Horgan et al. 1990;
Azuma et al. 1993). However, costimulation can also be provided by ICAM-1 alone
for presentation of superantigens to human naive T cells (Fischer et al. 1992). In most
studies, if sufficient costimulation is provided, naive T cells can respond to various
non specific stimuli or alloantigen, at equivalent or higher levels than activated
memory T cells in both mice (Croft et al. 1994a) and in humans (Horgan et al. 1990;
Merkenschlager et al. 1991; Fischer et al. 1992; Unutmaz et al. 1994). Furthermore, a
recent study has shown that the provision of CD28 costimulation can also induce
CD45RA T cells to respond to recall antigens (although at suboptimal levels
compared to CD45RO T cells) (Pilling et al. 1996). However in one study, CD28
costimulation was not sufficient to activate naive human T cells in response to anti-
CD3 induced stimulation (Kuiper et al. 1994).
Consequently, the activation of naive T cells in vivo is restricted to AFC that have
constitutive expression of the receptors for these costimulatory molecules. The
superiority of DC as AFC has been well documented (Steinman et al. 1978, 1980;
Inaba et al. 1984) and indeed they are the most potent AFC for stimulating naive T
cells in mice (Croft et al. 1994, 1992b; Macatonia et al. 1993; Seder et al. 1992;
Thus DC and activated B cells are capable of providing sufficient costimulation for
naive T cell to allow levels of stimulation comparable to memory cells (Croft et al.
1994), although with activated B cells a lower frequency of activated T cells and IL-2
production was observed compared to DC (Cassel et al. 1994). However resting B
cells, unstimulated macrophages (Croft et al. 1994, 1992b) and resting monocytes
(Byrne et al. 1988; Horgan et al. 1990) cannot stimulate naive T cells and may in fact
induce a tolerogenic signal if used as APC (Finkelman et al. 1992; Eynon et al. 1992;
Miyazaki et al. 1993; Fuchs et al. 1992).
Examination of the costimulatory molecules expressed on the APC explains their
efficiency at activating naive T cells. The DC has the exclusive advantage of having a
high constitutive expression of CD80 (B7-1), CD86 (B7-2), CD54 (ICAM-1) (Prickett
et al. 1992; Xu et al. 1992; Young et al. 1992; Larsen et al. 1992; Lenschow et al.
1993). Their potency as APC can be further increased upon activation by upregulation
of these and many other molecules involved in the interaction with the T cell. Resting
B cells, macrophages and monocytes have to be activated in order to induce
expression of these molecules to a level required for optimal T cell activation.
Activation of B cells with lipopolysaccharide (LPS) or polyinosinic-polycytidylic acid
can induce the expression of B7-2, B7-1, B7-3, ICAM-1 and many other molecules
(Liu et al. 1991b; Lenschow et al. 1993; Freeman et al. 1993). In order to induce
expression of B7-1 and ICAM-1, macrophages have to be activated by either LPS,
Zymosan or IFNy which then allows them to costimulate naive T cell proliferation
(Ding et al. 1993). The stimulatory capacity of monocyte containing populations has
been found to be enhanced by subjecting them to an adherence step (30 minutes-1
hour) on day 1 (Horgan et al. 1990). However, it is likely that this enhancement may
be due to an enrichment of DC.
It is possible that naive T cells can be partially activated by T cell receptor (TcR)
ligation alone since studies have shown that under these conditions, murine naive T
cells can exit from a Go state (Swain et al. 1996) and also induce expression of CD40
ligand (Jaiswal et al. 1996). There also appears to be a specific requirement for TcR
ligation since in contrast to anti-CD3 stimulation, anti-TcRVP mAb was unable to
In the murine system, if the cells are analysed during the effector phase, large blastoid
cells expressing high levels of many activation markers are found (Swain et al. 1996).
These cells are less dependent on costimulatory signals than naive T cells and TcR
ligation alone can induce optimal activation (Inaba et al. 1984; McKnight et al.
1994a).'Indeed the promiscuity of these cells is demonstrated by the finding that
resting B cells, macrophages, and even T cells can act as APC to effector T cells
(Croft et al. 1994; Inaba et al. 1984). This is also supported by the finding that
bystander APC expressing B7 can provide costimulation for CD45RO T cells but not
CD45RA T cells (Van de Velde et al. 1993) and the ability of CD45RO T cells but not
CD45RA T cells, to be activated to mediate effector functions by cytokines alone
(Unutmaz et al. 1994). Furthermore, the concentration of anti-CD3, anti-CD2 or
nominal antigen required to activate effector/memory cells is also considerably lower
than that required for naive cells in mice (Swain et al. et al. 1996) and in humans
(Byrne et al. 1988; Horgan et al. 1990). This ensures that in vivo, low concentrations
of an antigen, as for example an endogenous antigen, cannot induce primary
activation.
If the cells are analysed after the effector phase has subsided, small resting memory
cells are found (Swain et al. 1996), which are far less promiscuous than effector cells
but have a less stringent requirement for costimulation than naive cells. Thus although
these cells can be activated by just TcR ligation, costimulation with anti-CD28 can
enhance the response and maximal stimulation can be achieved in the presence of
activated DC or B cells (Croft et al. 1994a).
The activation state of the CD4 T cell has profound effect on its function, particularly
in their ability to help B cells, which is related to differences in the ability to produce
and secrete different cytokines as well as expression of surface molecules. Upon
primary stimulation, resting naive cells only secrete IL-2 and do not produce any other
cytokines in mice (Croft et al. 1995) and in human neonatal and adult T cells (Salmon
et al 1989; Ehlers et al. 1991). Consequently these cells cannot immediately support
antibody production by both murine (Lee et al. 1991; Croft et al. 1995) and human
(Sleasman et al. 1990) B cells. However, if allowed time to differentiate, they can
secrete high concentrations of IL-4, IL-5, IFNy and IL-2 in mice (Croft et al. 1995)
and in human neonatal and adult T cells (Elhers et al. 1991; Kristensson et al. 1992).
Resting memory T cells secrete high levels of IL-2 and very small amounts of the
other cytokines on stimulation (Bradley et al. 1992). However, if these cells are
allowed time to differentiate into effector cells, other cytokines can be secreted
(Weinberg et al. 1990) and at a higher concentrations than their naive counterparts
(Swain et al. 1996). Thus activated naive and resting memory T cells can only support
IgM responses, whilst primary and memory effector cells can support production of
many antibody isotypes in mice (Croft et al, 1995, 1992a, 1991; Bradley et al. 1993)
and human T cells (Martensson et al. 1994).
It has been suggested that the difference in activation requirements of naive and
memory T cells may be due to a more efficient signalling in memory cells. Indeed
there are several reported examples of differences in signalling between naive and
memory T cells. For example, the TcR/CD3 complex on the membrane of naive cells
moves independently from other signal transduction molecules such as CD4 and CDS,
whereas in memory cells, many of the other signal transduction molecules are
associated with the TcR/CD3 complex (Dianzani et al. 1992); protein kinase C
activation is also increased in murine (Robinson et al. 1993a) and human memory
cells (Hollsberg et al. 1993); naive T cells show a higher intracellular Ca^"^ flux than
memory T cells in response to ionophores (Miller et al 1991); and an IL-2 silencer has
been found in CD45RA T cells from UCB and adult PB but not CD45RO T cells
obtained after in vitro activation of PB or in T cell clones (Mouzaki et al. 1993).
However, there is also evidence to suggest that these differences in intracellular
signalling are merely a reflection of the different requirements of naive and memory T
cells for costimulation. Thus functional and intracellular differences could only be
observed when the T cells were stimulated by TcR ligation and costimulatory
molecules. If however, the requirement for costimulation is bypassed using phorbol
ester to activate protein kinase C together with TcR ligation, no differences could be
observed in terms of IL-2 production, proliferation or tyrosine phosphorylation
(Schwinzer et al. 1994). Furthermore, both CD45RA+ and CD45RA- T cells express
p59^^" (associated with the CD3/TcR) and shared similar tyrosine kinase substrates
(Rothstein et al. 1993).
1.23 Migration of naive and memory T cells.
In order to ensure a system that can rapidly respond to previously encountered
infectious organisms and at the same time ensure that primary responses to novel
antigens can occur in optimal environments, the immune system has developed
specific migratory pathways for naive and memory cells. This is achieved by way of
the differential expression of adhesion molecules on both the lymphocytes and
endothelial cells across which the lymphocytes have to traverse in order to enter
various lymphoid and non lymphoid tissues.
Naive cells on leaving the thymus express high levels of CD62L (L-selectin), the
“peripheral lymph node (PLN) homing receptor”, which allows preferential entry to
PLN (Bradley et al. 1994; Picker et al. 1990). Specialised endothelial cells called high
endothelial cells in lymphoid tissues regulate the entry of these naive lymphocytes by
the differential expression of ligands for receptors expressed on the lymphocytes. The
lymph nodes (LN) are situated in strategically located positions in the body so that an
antigen can be delivered to these LN by way of the lymphatic system within a short
period of time. Unless the naive cell encounters l^ ^ p e c ific antigen it will leave the
LN within 10-20 hours via the efferent lymphatic duct, through the thoracic duct to
renter the blood where they continue to recirculate through the various secondary
lymphoid tissues unless activated with specific antigen (Springer 1994; Bradley et al.
1996; Mackay et al. 1991)
If the naive T cell is activated by encounter with its specific antigen on professional
APC in the LN, the output of lymphocytes from the LN dramatically decreases for a
few days (Mackay et al. 1992). This is called the shut down phase and during this time
the T cells undergoes vast number of changes that alters the migratory pattern of these
cells. This includes the down regulation of the LN homing receptor L-selectin in all
activated T cells in mice (Bradley et al. 1992; Mackay et al. 1992) and in a
molecules such as CD49d/CD29 (VLA-4), CD lla/C D 18 (LFA-1), ICAM-1 and CD44
are upregulated (Horgan et al. 1992; Springer 1994; Picker et al. 1993). Some of the
activated T cells remain in the LN, where they relocate to follicles to participate in B
cell differentiation (somatic hypermutation and isotype switching) within germinal
centres (MacLennan et al. 1994). Others exit via the efferent lymphatic, back to the
blood; from blood these memory cells can now traverse normal and inflamed
endothelium and thereby survey peripheral tissues for invading pathogens. These
antigen primed T cells are subjected to tissue tropic mechanisms, which directs these
cells back to the same type of tissue where they first encountered antigen so as to
ensure that they are at sites where they are most likely to encounter the specific
antigen which primed them. It is thought that the T cells are conditioned in the tissue
environment due to cytokines such as TGFp, IL-2 and IL-6 (Picker et al. 1993) or
other factors unique to that tissue. Tissue tropism has been demonstrated for the gut,
the skin, epithelial surface of the lung and inflamed synovium.
The preferential homing to different anatomical sites, is due to the expression of a
combination of molecules which includes homing receptors specific for the given
tissues and adhesion molecules on the lymphocytes which interact with their ligands
on endothelial cells of the tissues. Thus for example preferential homing to the skin is
confered by the interaction between cutaneous lymphocyte associated antigen (CLA)
and CD62E (E-selectin), and the absence of L-selectin; to inflamed skin and
peripheral inflammatory sites by interaction between CD49d/CD29 (a 4 p l) and
CD 106 (VCAM-1), LFA-1 and ICAM-1; and to mucosal tissues by the interaction
between a 4 p ? and MAdCAM-1, CD44 and hyaluronate, LFA-1 and ICAM-1
(reviewed in: Bradley et al. 1996; Mackay et al. 1991, 1993; Picker et al 1994;
Springer et al. 1994).
1.24 Phenotypic markers of naive and memory T cells.
As discussed above, on activation of resting T cells a vast number of molecules are up
regulated on the T cells which consequently allows a distinction to be made between
1.24a The use of CD45 splice variants as markers of memory
The most commonly used distinctions of naive and memory T cells is based on the
differential expression of the leukocyte common antigen CD45 expressed on all
haemopoietic cells. Several lines of evidence suggested its importance in T cell
activation. Thus crosslinking CD45 could either costimulate (Marvel et al. 1989) or
inhibit T cell activation (Prickett et al. 1990; Ledbetter et al. 1988a). Furthermore,
CD45 has tyrosine phosphotase activity in its cytoplasmic tail (Charbonneau et al.
1988; Fischer et al. 1991) and CD45 deficient cell lines were found to be defective in
signal transduction, and this defect was restored by transfection of the cell line with
wild type CD45 (Koretzky et al. 1991, 1990).
Alternative splicing of the exons 4, 5, and 6 (generally referred to as A, B and C)
located at the distal region of the extracytoplasmic domain of CD45, can encode for at
least 8 potential different isotypes (Streuli et al. 1987; Trowbridge et al. 1994;
Janeway et al. 1992). Monoclonal antibodies that recognise epitopes dependent on the
expression of the exons A, B, and C were generated. Thus for example, the mAbs
SN130 (Munro et al. 1988) and 2H4 (Morimoto et al. 1985a; Streuli et al. 1987)
recognise an epitope expressed on exon A of human CD45 and are consequently
termed CD45RA antibodies. These mAbs recognise all isoforms expressing exon A
which includes ABC, AB, AC and A isoforms. Similarly, the CD45RB antibodies will
recognise the isoforms ABC, AB, BC and B, whilst the CD45RC antibodies will
recognise ABC, BC, AC, and C isoforms. However the mAh UCHLl only recognises
an epitope expressed on the human CD45 isoform with all the exons spliced out
(Smith et al. 1986a; Terry et al. 1988). In humans the mAbs to CD45RA and
CD45RO isoforms, revealed the existence of heterogeneous populations of cells of
approximately equivalent proportions within both the CD4 and CD8 T cell subsets in
PB (Smith et al. 1986a; Morimoto et al. 1985a). In other species there are many mAbs
that recognise the CD45RA, RB and RC isoforms and are collectively referred to as
CD45R mAbs, however there are no mAbs that recognise the CD45RO isoform. In
mice, the most commonly used mAh 16A, identifies the CD45RB isoforms which can
separate the CD4 T cells into high (65%) and low (35%) expressing cells (Bottomly et
al. 1989; Dianzani et al. 1990). The murine CD45RA antibodies, such as RA3-2C2
al. 1988, 1989). In the rat, the mAh (0X22) recognises high molecular weight
isoforms (Woollett et al 1985), (which is likely to be through the recognition of the
RC exon) and identifies 50-75% of the CD4 T cells (Powrie et al 1990). Thus in
animal studies the CD45R mAbs isolates different cell populations to those isolated in
the human studies with the CD45RO and/or CD45RA antibodies.
The association of CD45 splice variants with the functional state of the T cell was
suggested by the finding that the CD45 subsets within CD4 T cell population differed
in their ability to respond to soluble antigen and help B cells and were consequently
referred to as helper/inducer or suppresser/inducer (Smith et al. 1986a; Morimoto et
al. 1985). It was also shown that on activation of T cells, the high molecular weight
isoforms were down regulated whilst the low molecular isoforms were upregulated
(Akbar et al. 1988; Serra et al. 1998; Byrne et al. 1988; Sanders et al. 1988; Ledbetter
et al. 1985). On re-exposure of these primed T cells to the primary stimulus, an
increase in the rate of the proliferative response could be observed but only in the
CD45RO population (Akbar et al. 1988). Furthermore the acquisition of the CD45RO
phenotype correlated with an acquisition of the ability to help B cells produce
antibodies (Clement et al. 1988).
However the association of the CD45RO phenotype with T cell memory was
suggested by the finding that proliferative responses to previously encountered antigen
such as IFZ A were predominantly in the CD45RO population, whereas responses to
alloantigen were found to have similar precursor frequencies in both the CD45RO+
and CD45RO- T cells (Merkenschlager et al. 1988). In these earlier studies, because
the expression of CD45RO was found to be more stable than the other known markers
of activation such as MHC class n or the lL-2 receptor, it was concluded that
CD45RO expression was also irreversible.
This theory was further substantiated by the finding that immunologically immature T
cells isolated from human UCB were predominantly CD45RO- and only acquired
CD45RO after activation (Sanders et al. 1988); UCBMC or CD45RA T cells from PB
did not respond as well as the CD45RO phenotype from PB to superantigens (Horgan
suppressive for the B cell helper function of the minority CD45RO T cells present in
cord blood, although activation of the T cells could inhibit this suppressive function
(Clement et al. 1990); and the levels of T cells expressing CD45RA were found to
decrease whilst CD45RO expressing cells increased with age and only reached adult
levels after 20 years of age (Hayward et al. 1989).
From these very convincing experiments it was concluded that this unidirectional
expression of CD45RO on T cells represented the loss of naive and acquisition of a
memory phenotype. Following these studies, many other in vitro and in vivo examples
in man, mice and other animals provided evidence to show that priming of T cells
resulted in the loss of the high molecular weight isoforms and the gain of the low
molecular weight isoforms. Thus despite the different populations isolated by the
CD45R antibodies in the different species, T cells expressing the high molecular
weight isoforms, in all species gave very poor responses to recall antigens and were
ineffective at providing help for B cells. Conversely, cells expressing the low
molecular weight isoforms, gave very good responses to recall antigens and provided
help for B cells.
1.24b The expression of CD45RA mav not alwavs represent a naive phenotvpe.
There are however, a number of studies in both man and mouse in which the results
are not consistent with the above theory and suggests that the expression of high
molecular weight isoforms and low molecular weight isoforms may not always
represent naive and memory phenotypes.
Although the activation of CD45RA T cells with mitogens does down regulate the
expression of CD45RA, expression is not completely lost (Morimoto et al. 1986) even
after 21 days in culture (Rothstein et al. 1990) and the retention of CD45RA
correlated with an inability to help B cells (Rothstein et al. 1990). There has also been
a report of transgenic unprimed mice bearing a percentage of cells with low molecular
weight isoforms (Lightstone et al. 1993). Furthermore, priming of these mice resulted
in an increase in percentage of the high molecular weight isoforms, which eventually