Blood and Lymphatic Cancer: Targets and Therapy
Recent developments in myelofibrosis
Raoul Tibes
James M Bogenberger Ruben A Mesa
Division of Hematology and Oncology, Mayo Clinic, Scottsdale, AZ, USA
Correspondence: Raoul Tibes Division of Hematology and Oncology, Mayo Clinic, 13400 E. Shea Blvd, Scottsdale, AZ 85259, USA Tel +1 480 301 4502 Fax +1 480 301 4675 Email [email protected]
Abstract: The myeloproliferative neoplasm of myelofibrosis (MF) is clinically constituted by individuals both with primary MF, as well as those that evolved from an antecedent polycythemia vera or essential thrombocythemia. Individuals presenting with MF have a heterogeneous phenotype which can involve significant constitutional symptoms (night sweats, fevers, weight loss, fatigue, variable but frequently problematic splenomegaly, and multifactorial cytopenias). These individuals clearly have decreased survival. Refinement of MF prognostic scores can distinguish from survival as poor as 16 months, to a median survival of 185 months. Sadly, although curative, allogeneic stem cell transplant still has sobering success rates for individuals of the standard ages for MF. Recent reports suggest less than half of patients will be alive at 3 years after allotransplant above the age of 60 years. The most important recent advancement in MF therapy has been the development of Janus kinase 2 (JAK2) inhibitors led by ruxolitinib, now Food and Drug Administration approved in the United States, and several other JAK2 inhibitors (in testing) including SAR302503, CYT387, and SB1518. In randomized, placebo-controlled studies, ruxolitinib was demonstrated to be superior for the improvement of splenomegaly and symptoms. These benefits are mirrored across other JAK2 inhibitors. Improving anemia remains an unmet need in MF and is currently being evaluated by clinical trials utilizing the JAK2 inhibitor CYT387, as well as pomalidomide. Additional areas of interest for MF therapy include the inhibition of histone/lysine deacetylases, hedgehog pathway inhibition, as well as combination strategies with JAK2 inhibitors.
Keywords: myeloproliferative neoplasm, myelofibrosis, JAK2 inhibitors
Introduction to myeloproliferative diseases
The myeloproliferative neoplasms (MPNs) are a group of clonal hematological disorders that are characterized by terminal myeloid cell expansion in the peripheral blood. According to the 2008 World Health Organization (WHO) Classification System, MPNs are divided into “classic” and “atypical” MPNs with further subdivision into Philadelphia chromosome ([9;22] chromosomal translocation resulting in the BCR-ABL1 fusion protein)-positive and -negative MPNs. The latter are comprised of polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (MF).1
The first description of MPNs as a unified concept of diseases was proposed by William Dameshek in 1951, based on clinical and bone marrow similarities of the three entities – PV, ET, and primary MF.2 For many years it was known that MPNs are hypersensitive to hematopoietic growth factors and show cytokine-independent colony growth.3–6 A breakthrough into the pathogenesis of MPNs was reported in 2005
Dove
press
R E v i E w
open access to scientific and medical research
Open Access Full Text Article
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
For personal use only.
Number of times this article has been viewed
This article was published in the following Dove Press journal: Blood and Lymphatic Cancer: Targets and Therapy
with the discovery of the JAK2V617F mutation, occurring to varying frequencies, in all three disease entities.7–10 This gain-of-function mutation in Janus kinase 2 (JAK2; a key component of cell growth and differentiation in the JAK-signal transducers and activators of transcription [STAT] pathway) results in constitutive activation of the pathway.11,12 Since its discovery, several additional genetic mutations with potential pathogenic implications have been described.13–16 However, the initial molecular genetic event leading to clonal transformation has not been determined.17,18
Clinically, it is important to distinguish PV and ET from primary MF. In PV and ET, vascular/thromboembolic events are responsible for most of the disease-associated symptoms followed by constitutional symptoms, eg, pruritus.19,20 There is a long-term risk of progressing into a post-PV/ET MF state resembling primary MF and further progression into a blast phase MPN.21,22 Primary MF mostly arises de novo and has a more rapid course with bone marrow fibrosis and extramedullary hematopoiesis with progressive splenomegaly, cytopenias (often anemia), and burdensome constitutional symptoms, as well as the occurrence of vascular/thromboembolic events.19 Patients are at higher risk of transformation into acute leukemia.21,22 Current available therapies have rarely been able to impact the natural history of primary MF beyond palliating symptoms or decreasing the risk of vascular events. This article will focus on recent insights and clinical treatment advances for primary MF and post-PV/ET MF patients.
MF: epidemiology and etiology
Epidemiology of primary MF
Primary MF is a rare disease and the least common of the MPNs, with an incidence of around 1.5 per 100,000 per year in one study for the United States.23 This incidence figure is higher than those reported from other regions, likely based on the demographic distribution (eg, the Olmsted County, MN population may have an older age demographic). Disease onset of primary MF occurs with a median age at presentation of 67 years.23 Few patients are diagnosed at a younger age, with 5% and 17% of patients before the age of 40 years and 50 years, respectively.24 Familial cases have rarely been reported.25
Etiology and pathogenesis
of primary MF/MF
The discovery of constitutively active JAK2 mutations has brought enormous opportunities for a better understanding of primary MF and MPNs, as well as for new treatment options.7–10
JAK2 is a nonreceptor tyrosine kinase and has a critical role in intracellular signaling of extracellular stimuli from hematopoietic and other cytokine receptors. The dominant
JAK2 gain-of-function mutation acquired via a guanine to tyrosine conversion in nucleotide 1849 results in the substitution of valine for phenylalanine. Disruptions within the pseudokinase domain produce a persistently activated intracellular receptor molecule.26,27 As a consequence, cells harboring the JAK2V617F mutation develop cytokine-independent growth capabilities and/or increased cytokine responsiveness.5,28 The canonical downstream effectors of JAK2 are STAT proteins, preferentially STAT3 and STAT5.29 The STATs are hierarchical regulators mediating transcriptional programs controlling proliferation and differentiation of hematopoietic progenitor cell lineages including erythrocytes, thrombocytes, leukocytes, and monocytes.
Other mutations stimulating the JAK-STAT pathway have been described including JAK2 exon 12, MPL exon 10, and
LNK (SH2B3) exon 2 mutations, which are less prevalent in
BCR-ABL-negative MPNs.15,30–32 It should be mentioned that these mutations, including JAK2V617F, are likely not the earliest transforming molecular events initiating the disease but, rather, early secondary events necessary for proliferation in the pathogenesis of primary MF and MPN cells.33
In regards to the prognostic value of JAK2 and other mutations, the overall contribution of the JAK2V617F mutation to the clinical outcome in primary MF is only starting to become delineated, despite a substantial number of scientific publications.34 Interestingly, the presence or absence of the JAK2V617F mutation by itself does not appear to be prognostic for leukemic evolution, although some studies report conflicting results. JAK2-positive PV and ET with higher allele burden may have a higher rate of disease progression to advanced stages within the PV, ET, and primary MF phenotypes.35–38 However, the data regarding progression to acute myeloid leukemia is still somewhat controversial in regards to homozygosity, heterozygosity, and level of allele burden. For example, one study found that patients harboring JAK2V617F-positive disease that were in the lowest quartile of JAK2 allele burden fared worse in regards to overall and leukemia-free survival; granted, this lowest quartile was represented by 19 patients.39 A second study described a similar finding that JAK2V617F positivity did not impact on leukemic transformation; however, survival was significantly reduced in the lower quartile
JAK2-mutated primary MF patient group compared with upper quartiles and JAK2 wild-type patients.40 The rate of Dovepress
Tibes et al
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
leukemic transformation in the latter study did not increase. The authors argue that either the treatment intervention, the purer granulocyte fraction, or the various assays used across studies are variables that may explain different findings of
JAK2 allele burden or zygosity state on outcome. The reasons are yet to be fully elucidated, and speculations at this time evolve around JAK2V617F-negative clones with a more aggressive phenotype.
MPL mutations in primary MF are associated with female gender, older age, and a more severe phenotype manifested as a lower hemoglobin level and a higher likelihood of becoming transfusion-dependent.15,41 Currently, risk stratification therapy is largely based on clinical parameters and disease evolution, which will be discussed in more detail below. The karyotype/cytogenetic risk profile is built into the Dynamic International Prognostic Scoring System (DIPSS)42 and DIPSS-Plus43 scores discussed below.
JAK-STAT downstream
and nuclear function
In addition to the cytoplasmic roles of JAK2 in mediating signaling cascades as described above, a significant proportion of JAK2 has been observed in the nuclei of hematopoietic cells. Nuclear JAK2 was shown to phosphorylate histone H3 at tyrosine residue 41 residing in a region of histone H3 known to bind heterochromatin protein 1α (HP1α). Phosphorylated histone H3 at tyrosine residue 41 excludes HP1α from chromatin, which is thought to regulate chromatin architecture around specific gene promoter regions.44 Several genes lacking canonical STAT5-binding sites were shown to be downregulated in response to JAK2 inhibitors, showing that additional activities outside of the canonical JAK-STAT pathway can be affected with JAK2 inhibition. One such gene, Lmo2, known to be involved in hematopoiesis, was demonstrated to have reciprocal HP1α binding and histone H3 phosphorylation at tyrosine residue 41 in the promoter region. Another paper expanded on these findings and demonstrated that JAK2 inhibition reduced levels of Nanog, a pluripotency stem cell regulator, by affecting the Nanog
promoter through increased HP1α binding. Thus, JAK2 and aberrant JAK-STAT signaling affects chromatin modification and genes important in embryonic stem cell self-renewal.45
Pathogenesis of fibrosis
The formation of MF ultimately results from the excess deposition of extracellular matrix components that progressively remodel and destroy normal bone marrow architecture. Production of extracellular matrix components
by myofibroblasts is mediated by different cell types that sustain the production of growth factors, proteolytic enzymes, angiogenic factors, and fibrogenic cytokines. Infiltrating lymphocytes, activated monocytes,46 and megakaryocytes47,48 have been implemented in these processes. Remissions observed after hematopoietic stem cell transplantation sug-gest that genetic aberrations in hematopoietic stem cells are central to the pathogenesis of primary MF.49 Cellular and extracellular levels of fibrogenic and angiogenic growth factors are altered in primary MF patients, while the prolif-eration of myofibroblasts is thought to be polyclonal, thus the bone marrow stromal reaction is thought to be secondary to the production of cytokines. JAK2 mediates cytoplasmic signaling from diverse cytokine receptors. To date, however, reversal of fibrosis by JAK2 inhibitors has not been dem-onstrated as will be discussed below, although early data is emerging that some of the novel inhibitors may be able to reduce fibrosis.
Primary MF is defined by an increase in bone marrow fiber content, characterized by an increased synthesis and deposition of collagen,47 and an increased deposition and synthesis of interstitial and basement proteins such as fibronectin, laminin, tenascin, and vitronectin in advanced primary MF. Neovascularization is also a predominant feature of fibrotic bone marrow.50 Several cell types have been implemented in the production of fibrogenic cytokines in primary MF, including infiltrating lymphocytes, megakaryocytes, and activated monocytes, including growth factors such as platelet-derived growth factor, basic fibroblast growth factor, erythroid differentiation factor, transforming growth factor-β, vascular endothelial growth factor, and platelet factor-4 derived from these cells.51
Symptomology and natural history
Clinical and laboratory presentation
Patients most commonly present with fatigue and other constitutional symptoms such as fever, chills, night sweats, and weight loss.19,52,53 Many patients have splenomegaly, often palpable several centimeters below the left costal margin, at times extending into the left pelvis and even crossing the midline. Hepatomegaly is found as well.54,55 Both result from extramedullary hematopoiesis, a characteristic finding in both primary and secondary MF.23,53 Extramedullary hematopoiesis in the spleen and other organs is less effective and hence patients often suffer from cytopenias. Extramedullary hematopoiesis rarely occurs in other pleural-, pericardial-, or abdominal-based organs; it can involve the gastrointestinal and genitourinary tracts,
Dovepress Recent MF developments
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
the lungs, or even the central nervous system, leading to neurological deficits.56 The incidence of arterial and venous thromboembolic complications is similar in ET and primary MF (both around two in 100 patient-years), and somewhat less in PV.56 About 10%–13% of primary MF patients have had thrombotic events in one large series.57 Often, these may be precipitated by common exogenous risk factors for thrombosis (eg, surgery, access/catheter, hormonal therapy) in #71% of cases as reported in one study.57 Although vascular and thromboembolic complications are more frequent in primary MF than PV, they constitute a main disease burden in PV and ET, whereas in primary MF concurrent constitutional symptoms rival vascular complications. Portal vein thrombosis is a feared complication in primary MF and may be a presenting symptom.58,59 Involvement of the skin or pulmonary hypertension is rare, but can occur.60–62 Apart from reticulin and collagen fibrosis of the marrow, other bone complications include osteosclerosis, described in up to 80% of patients,63 periostitis, and secondary gout with hyperleukocytosis – all of which can cause severe bone and joint pain.
Many of the symptoms of primary MF patients are due to a hypercatabolic state, complaints from hepatosplenomegaly, ie, abdominal fullness/pain, as well as progressive cytopenias, especially anemia. Over time, marrow fibrosis eventually becomes more pronounced, extramedullary hematopoiesis increases as do associated constitutional symptoms,19,53,64 and the risk of leukemic (blastic) transformation and advanced disease stages increases.22 The true incidence of leukemic transformation of primary MF is challenging to assess, some published series report ∼3%–5% of patients,65 while others state the incidence to be as high as 15%–20% or up to 31% of primary MF patients transforming to leukemic phase over their disease course.66,67 The presenting symptoms and signs for post-ET/PV MF are fairly similar to those in primary MF.67
Diagnostics
Diagnostic approach
Presentations initially can be nonspecific with constitutional symptoms; however, with additional symptoms and signs such as splenomegaly or hepatomegaly, anemia, and leukocytosis and/or thrombocytosis, the clinical suspected diagnosis of an MPN/primary MF is often made and work-up initiated. Patients can also present with cytopenias, (eg, leukopenia and thrombocytopenia), especially in the more advanced disease stages. A peripheral smear typically shows a leukoerythroblastic picture with nucleated erythrocytes and
granulocyte precursors (myelocytes, metamyelocytes, and blasts) and teardrop-shaped red blood cells from myelophthisis (replacement of normal marrow elements by fibrosis). Confirmation of the diagnosis incorporates the WHO criteria for primary MF. These require all three major criteria, which are a combination of the presence of megakaryocyte proliferation and atypia, accompanied by reticulin and/or collagen fibrosis; exclusion of other WHO-defined MPNs or myeloid neoplasm (eg, ET, PV, chronic myeloid leukemia, and myelodysplastic syndrome) and demonstration of clonality (eg, JAK2617VF or MPL515WL/K). In addition to these three criteria, two of four minor criteria – namely, leukoerythroblastosis, palpable splenomegaly, anemia, and increased serum lactate dehydrogenase level – should be present.68 Bone marrow aspiration in primary MF often yields little specimens (“dry tap”) and a biopsy is required to show reticulin (silver-based staining) and collagen fibrosis (Masson’s trichrome stain), which can often be extensive.63 Bone marrow sinusoids are expanded so that intravascular hematopoiesis and osteosclerosis can be seen. Neutrophilic and megakaryocytic hyperplasia, macromegakaryocytes, and occasionally atypical megakaryocytes as well as increased numbers of erythroid precursors may be observed. Rarely a hypercellular marrow pathology with little fibrosis is encountered. Magnetic resonance imaging or isotopic marrow imaging can be employed,69 but is rarely used by the authors’ group. Fibrosis is more commonly found with certain molecular aberrations including the JAK2V617F mutation, 13q deletion, and trisomy 9.70
The discovery of JAK2V617F and other common mutations found in MPNs has transformed the diagnostic approach in MPNs. Now, one of the first tests after considering a diagnosis of primary MF is testing for the
JAKV617F mutation, which can be found in .95% of PV
and 40%–60% of ET and primary MF patients depending on the study.7,8,10,71 If JAK2V617F is negative, testing for MPL mutations can be performed,20 which are present in up to ∼5%–10% of primary MF patients;15,31 this can confirm clonality and diagnosis of primary MF in the appropriate clinical setting. If no mutation is found, the diagnosis of primary MF is made on clinical grounds alone.
To establish the diagnosis of post-ET/PV MF, the International Working Group for MF Research and Treatment proposed a classification that includes a combination of several major and/or minor criteria. These include documen-tation of a previous diagnosis of either PV or ET as defined by WHO criteria, presence of increased bone marrow fibrosis, progressive anemia or loss of phlebotomy requirement, Dovepress
Tibes et al
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
leukoerythroblastic blood picture, increasing degree of splenomegaly, development of constitutional symptoms (ie, weight loss, night sweats, unexplained fever), and increased lactate dehydrogenase level (for post-ET MF only).72 The first two (previous ET or PV and increased marrow fibrosis) are major criteria and are both required. In addition, two of the five minor findings need to be present.
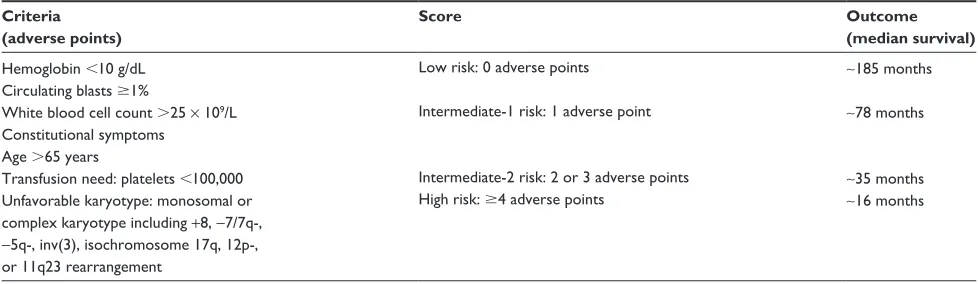
Several risk stratification scores have been developed – the most recent and widely used are DIPSS42 and DIPSS-Plus.43 For both DIPSS and DIPSS-Plus, points are assigned based on age .65 years, hemoglobin ,10 g/dL, leukocyte count .25 × 109/L, circulating blasts .1%, and presence of constitutional symptoms. An overall score predicts progression and survival and can direct choice of therapy. The DIPSS-Plus score was expanded to include the previous five criteria with the addition of platelets ,100 × 109/L, red blood cell transfusions, and unfavorable karyotype.43 DIPSS-Plus can be used at baseline and over time to assess risk of progression. Survival ranges from ∼16 months in high-risk primary MF patients to ∼185 months in low-risk primary MF patients depending on DIPSS score (Table 1).42,43
Treatments: current
and experimental
Most therapies for primary MF and post-PV/ET MF are not disease-modifying but, rather, focus on targeting symptoms including splenomegaly and hepatomegaly, cytopenias, and constitutional symptoms. Delay in progression to blast phase (leukemic phase) is a primary treatment goal and allogeneic hematopoietic stem cell transplantation is the only known cure. However, this approach is only applicable to a minority of patients.
Interpreting the literature and the differences in the “true” response rates of the different studies for the various agents
is somewhat problematic as primary MF spans a range of phenotypic presentations, and studies are often small with different study and response evaluation criteria.
Low-risk primary MF patients (DIPSS or DIPSS-Plus) are often followed by observation only. Erythropoiesis stimulating agents can be effective in earlier stage disease in nontransfusion-dependent patients.73 Clinically significant anemia (ie, hemoglobin ,10 g/dL) is present in up to 54% of patients and has been treated with a variety of agents includ-ing the immunomodulatory drugs (IMIDs) thalidomide, lenalidomide, and pomalidomide, as well as prednisone and androgens (eg, danazol).
As mentioned, anemia represents the main clinical issue in patients with primary MF and many patients become transfusion-dependent. After treatable causes (eg, iron, folic acid, or vitamin B12 deficiency and autoimmune hemolytic anemia) are ruled out, androgen therapy is often considered one of the first-line treatment interventions. The efficacy of testosterone against anemia was demonstrated in the 1960s,74,75 probably by stimulating bone marrow function. Only a few small studies with different androgens (eg, nandrolone, fluoxymesterolone, methandrostenolone, and oxymetholone) are available with overall anemia response rates generally in the range of ∼30%–60%, with an abnormal karyotype and severely compromised hematopoiesis being associated with a lower chance of response.76–78 Deeper responses are less common, probably ∼10%–20% of patients. Responses are often short-lived, although some patients respond for several years.79 Some newer studies with danazol achieved responses of 57%;80 however, no direct comparison of the various androgens has been performed.
The drug class of immunomodulatory agents – namely, thalidomide, lenalidomide, and pomalidomide – has been studied more extensively in primary MF (Table 2). Single agent lenalidomide yielded response rates of 22% for anemia,
Table 1 Dynamic international Prognostic Scoring System-Plus
Criteria (adverse points)
Score Outcome
(median survival) Hemoglobin ,10 g/dL
Circulating blasts $1%
white blood cell count .25 × 109/L Constitutional symptoms
Age .65 years
Transfusion need: platelets ,100,000 Unfavorable karyotype: monosomal or complex karyotype including +8, -7/7q-,
-5q-, inv(3), isochromosome 17q, 12p-, or 11q23 rearrangement
Low risk: 0 adverse points
intermediate-1 risk: 1 adverse point
intermediate-2 risk: 2 or 3 adverse points High risk: $4 adverse points
∼185 months
∼78 months
∼35 months
∼16 months
Dovepress Recent MF developments
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
33% for splenomegaly, and 50% for thrombocytopenia, respectively.81 In a trial of 40 MF patients treated with lenalidomide (10 mg/day for 21 days out of a 28-day cycle) in combination with prednisone starting at 30 mg/day in the first cycle, 30% of patients had anemia and 42% had a spleen response at a median follow-up of 22 months.82 The median time to response was 12 weeks and was lasting in 25% of patients at publication.82 According to the International Working Group for MF Research and Treatment consensus criteria, three (7.5%) partial responses were observed, in addi-tion to decreased levels of reticulin fibrosis and JAK2V617F.82 Grade 3–4 hematologic adverse events were neutropenia (58%), anemia (42%), and thrombocytopenia (13%). Results were not quite as good in a multicenter cooperative group study with 19% achieving anemia and only 10% with spleen response.83 Resolution of disease-related fibrosis or angiogen-esis was not observed in that study.83 Rarely, MPN/primary MF patients harbor isolated chromosome 5q deletions, and these patients are explicitly sensitive to lenalidomide84 and should be treated with lenalidomide as first-line therapy if intervention is required. An underlying myelodysplastic syndrome in these patients should be carefully excluded.
Thalidomide is effective in MF/primary MF as well, with 56% of patients having a clinical benefit.85 Overall,
anemia was improved in 42%, thrombocytopenia in 77%, and splenomegaly in 17%.85 Low-dose thalidomide (50 mg daily) combined with prednisone was better tolerated and more efficacious than the initial higher doses of 200 mg/day thalidomide. Treatment response was not affected by the baseline status of bone marrow fibrosis or other parameters (eg, angiogenesis, osteosclerosis, cytogenetics, or circulating CD34+ myeloid progenitor cells). In another study with thalidomide, 10% of patients each had a partial and complete remission.86 Improvements in anemia, thrombocytopenia, and/or splenomegaly were observed in nine patients (21%). Five of 24 patients (21%) became transfusion-independent and regression of splenomegaly was noted in nine of 29 evaluable patients (31%), with five patients experiencing a complete regression.86 Side effects were typical for thalidomide including fatigue (50%), constipation (48%), rash or pruritus (37%), sedation (35%), peripheral edema (29%), tremors (23%), peripheral neuropathy (22%), and orthostasis (16%).86 In both studies, thalidomide led to unexpected hyperproliferation (eg, reversible leukocytosis and/or thrombocytosis) in some patients, a side effect that can be seen with all IMIDs. In contrast to lenalidomide, which has some activity reducing splenomegaly, durable treatment responses with thalidomide
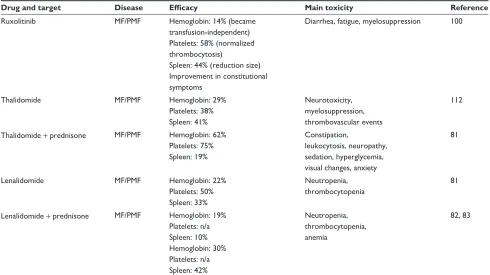
Table 2 Food and Drug Administration-approved drugs for primary myelofibrosis or any indication and use off-label for primary myelofibrosis
Drug and target Disease Efficacy Main toxicity Reference
Ruxolitinib MF/PMF Hemoglobin: 14% (became transfusion-independent) Platelets: 58% (normalized thrombocytosis)
Spleen: 44% (reduction size) improvement in constitutional symptoms
Diarrhea, fatigue, myelosuppression 100
Thalidomide MF/PMF Hemoglobin: 29%
Platelets: 38% Spleen: 41%
Neurotoxicity, myelosuppression, thrombovascular events
112
Thalidomide + prednisone MF/PMF Hemoglobin: 62% Platelets: 75% Spleen: 19%
Constipation,
leukocytosis, neuropathy, sedation, hyperglycemia, visual changes, anxiety
81
Lenalidomide MF/PMF Hemoglobin: 22%
Platelets: 50% Spleen: 33%
Neutropenia, thrombocytopenia
81
Lenalidomide + prednisone MF/PMF Hemoglobin: 19% Platelets: n/a Spleen: 10% Hemoglobin: 30% Platelets: n/a Spleen: 42%
Neutropenia, thrombocytopenia, anemia
82, 83
Note: Only full manuscripts are listed.
Abbreviations: MF, myelofibrosis; n/a, not available; PMF, primary myelofibrosis.
Dovepress
Tibes et al
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
were mostly documented for anemia and thrombocytopenia, whereas splenomegaly was not reduced in the majority of patients. Several other trials have been published showing similar response rates to thalidomide,87 depending on response evaluation criteria and patient population.
Three studies assessed the activity of the experimental IMID pomalidomide at varying dosages (0.5–3.5 mg/day) with or without prednisone. As a general tendency, pomali-domide was more active at lower dosages (0.5 mg/day) and in combination with prednisone, with up to 36% anemia responses.88 Median response duration in responding patients was 7.8 months (range 3.2–16.9 months). Hyperleukocytosis or splenomegaly .10 cm correlated with lower response rates. Improvement of thrombocytopenia was seen in 43% of patients. Bone marrow histology or JAK2V617F allele burden were unaffected by the treatment. Severe grade 3–4 adverse events were infrequent, consisting of neutropenia (8%), thrombocytopenia (11%), pneumonia/sepsis (11%), and venous thrombosis (4%). Drug-induced eosinophilia, basophilia, and even marked thrombocytosis were attributed to the treatment. A third (published) study showed similar response rates with an overall anemia response of 27%.89 These were higher (53%) in JAK2V617F-positive patients and in those where splenomegaly measured ,10 cm and circulating blasts were ,5%.89 In contrast, no responses were observed in JAK2V617F mutation-negative patients that also had splenomegaly $10 cm or $5% circulating blasts. Consistent with these “preclinical” markers, spleno-megaly was not affected in most patients. Discontinuation rate reported by these authors was high.89 The incidence of neuropathy was low. Risk adapted survival was not improved in patients not receiving pomalidomide. However, MF/ primary MF patients are complex in their disease presen-tation and evolution, and a randomized, multicenter trial is ongoing. The second published study of higher doses (#3.5 mg/day) showed poor tolerance; however, at reduced doses of 0.5 mg/day, excellent responses, mainly anemia, were seen.90 Therefore, pomalidomide is most efficacious at lower doses of 0.5 mg, possibly also in combination with prednisone, and targets mostly anemia. For all IMIDs, especially thalidomide, thromboembolic events occur at greater frequency, thus prophylaxis (eg, aspirin) needs to be considered if medically feasible.
Constitutional symptoms including fever, fatigue, bone pain, and night sweats have historically been resistant to most conventional therapeutic options. However, JAK2 inhibitors show a remarkable clinical improvement and will be discussed in more detail below.
The only curative opportunity available, hematopoietic stem cell transplant, is often restricted to a small percentage of patients due to toxicity;91,92 timing of the transplant is also important, as discussed below.
Therapy for high or intermediate-2 risk primary MF patients according to the DIPSS and DIPSS-Plus depends on clinical symptoms. For anemia, androgens (eg, danazol) or steroids (prednisone) may be given as has been discussed for lower risk primary MF in the above section. The IMIDs lenalidomide and thalidomide as well as pomalidomide can be used in the advanced setting as well, and lenalidomide in particular has a higher chance of reducing splenomegaly than thalidomide. A drug that works well against splenomegaly is hydroxyurea and ∼40%–50% of patients achieve a good response,93,94 although dose and effectiveness may be limited by side effects of hydroxyurea (eg, myelosuppression and skin ulcers). Splenectomy and splenic/hepatic radiation has found limited utility in the past given the morbidity associated with postoperative complications; nevertheless, surgical splenectomy is an option that can occasionally be effective.94 The authors’ Mayo Clinic experience has demonstrated that the surgical removal of the spleen does not compromise hematopoiesis.94 Indeed, removal of a massive spleen which acts as a sequestering reservoir for circulating erythrocytes, platelets, and leukocytes generally leads, postoperatively, to an increase in all three lineages in the circulation, sometime necessitating cytoreductive therapy. In fact, splenectomy is used on several of the authors’ patients each year with good outcome. Preferably, splenectomies should be performed in a tertiary center with surgeons experienced with these patients and, in the authors’ experience, the perioperative morbidity and mortality is negligible (Mesa and Tibes, personal observation).
Lastly, to control counts and splenomegaly, cytotoxics such as busulfan, pipobroman, chlorambucil, or radioactive phosphorous are effective in lowering counts, and these agents are suitable options especially for elderly patients.94
Interferon (IFN) has activity in PV and ET; however, for patients with advanced primary MF/MF, several studies have shown that IFN is poorly tolerated at higher doses for these advanced patients, mainly due to cytopenias.95 However, in recent reports, pegylated IFNα-2a and lower doses of standard IFN-α in 17 patients with low-risk or intermediate-risk primary MF showed two complete responses, seven partial responses, and five patients with stable disease or clinical benefit, for an overall clinical benefit rate of 80%. Improvement in marrow pathology was observed in four patients and toxicity was acceptable.95 In a retrospective analysis, somewhat lower but still encouraging results
Dovepress Recent MF developments
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
were observed,96 suggesting that low-dose standard IFN or pegylated IFNα-2a may be effective, especially in the proliferative/hypercellular phase of primary MF.
Leukemic transformation
of primary MF/MF
After many years, a fraction of patients with primary MF (∼5%–10% at 10 years),21 in some reports up to 31% (see above), progress to more aggressive stages of MPN including blast phase MPN, resembling acute myeloid leukemia. Patients present with decreased intramedullary hematopoiesis with worsening cytopenias and constitutional symptoms. The molecular events leading to leukemic transformation have not been well characterized and several mutations occur at increased frequencies in blast phase MPN (ie, IDH and EZH2).97 The JAK2V617F mutation can also be lost at leukemic transformation, suggesting that it is not obligatory for disease progression.33 Response to induction therapy and outcome in blast phase primary MF are very poor, with a median overall survival of ∼2–3 months as reported in one study.65 Allogeneic stem cell transplantation is the only curative option for leukemic transformation and should be employed early in the process of disease acceleration, ideally before transformation to frank leukemic phase. Several studies report encouraging outcomes with allogeneic transplant in these advanced stage patients, especially using reduced intensity conditioning regimens. Overall survival rates range from 43%–90%, depending on the study and length of follow-up.91 Patients should be referred to transplant centers with experience in transplanting these patients as prolonged myelosuppression and delayed resolution of marrow fibrosis, together with slow shrinkage of hepatosplenomegaly, can lead to substantial challenges posttransplant.
Data for other salvage therapies in the accelerated and leukemic phase are limited. A recent study of low-intensity treatment with 5-azacitidine in 54 patients with advanced primary MF and post-PV/ET MF showed an overall response rate of 52%, with 24% of patients achieving complete response; median response duration was 9 months.98
JAK2 inhibitors
The discovery of JAK2 mutations has enabled rapid clinical development for several JAK targeting agents, and more than ten JAK2 targeting inhibitors are being investigated clinically.29,99 Most are not specific for the mutant JAK2V617F and inhibit wild-type JAK2, as well as other kinases, possibly explaining the varying response rates and side effect profiles.29,99 As a general tenor, many of
the inhibitors show similar response rates, and 70%–90% of patients experience improvement in constitutional disease-associated symptoms. Pruritus improves or resolves in 50%–90% of patients. Spleen responses are seen in around 35%–60% of patients, leukocytosis is reduced or normalized in 50%–60%, and thrombocytosis improves in .90%, especially in early-stage MPNs.29,99
Many JAK2 inhibitors are initially tested in advanced-stage disease in primary MF and post-PE/ET MF. The JAK2 inhibitor furthest in clinical development, and recently Food and Drug Administration approved, is ruxolitinib. In a pivotal randomized phase I/II trial of 153 patients with primary MF and post-PV/ET MF treated with ruxolitinib, 71%–78% of patients experienced $50% improvement in symptoms based on the MF Symptom Assessment Form. Improvements included improved exercise capacity, decreased pruritus, weight gain, and control of counts.100
Subsequently, ruxolitinib was compared to placebo or best available therapy in two large randomized trials. Both of the COMFORT (Controlled MF Study With Oral JAK Inhibitor Treatment) trials, COMFORT-I and COMFORT-II, were conducted in patients with primary MF or secondary MF. COMFORT-I is a randomized, double-blind, placebo-controlled phase III study with 309 intermediate-2/high-risk patients assigned to twice-daily oral ruxolitinib (15 mg or 20 mg) or placebo. The primary endpoint was a reduction in spleen volume by 35% by the end of week 24 of therapy. Patients randomized to the ruxolitinib arm were statistically more likely to attain the primary endpoint (41.9%) versus placebo (0.7%). Patients were also more likely to have symptomatic improvement in abdominal discomfort, early satiety, itching, bone or muscle pain, pain under left ribs, night sweats, and inactivity. Anemia (45.2%) and thrombo-cytopenia (12.9%) were the two grade 3–4 adverse events observed more frequently in comparison to placebo.101
COMFORT-II is a randomized, open-label phase III study of ruxolitinib in comparison to best available therapy in patients with intermediate-2/high-risk primary or secondary MF.102 The primary endpoint was evaluation of the proportion of subjects achieving .35% reduction in spleen volume from baseline to week 48. The week-48 response rate was 28.5% for ruxolitinib patients versus 0% for best available therapy. Beginning at week eight, a highly significant improvement was noted in spleen volume for patients receiving ruxolitinib. Thrombocytopenia (44.5%), anemia (40.4%), diarrhea (24.0%), and peripheral edema (21.9%) remained the most common adverse effects. Thrombocytopenia and anemia were the most common toxicity-limiting side effects. Dovepress
Tibes et al
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
Pneumonia, dyspnea, thrombocytopenia, and anemia were the most frequent grade 3–4 adverse events.
Currently ongoing, the RESPONSE (Randomized Study of Efficacy and Safety in PV with JAK Inhibitor INCB018424 Versus Best Available Care) trial is a phase III, open-label trial designed to compare best available therapy to ruxolitinib in hydroxyurea-resistant PV patients requiring phlebotomy.103 A number of trials with ruxolitinib and other JAK2 inhibitors are ongoing, including combination trials.
Apart from ruxolitinib, several other JAK2 inhibitors are in preclinical or clinical development. For example, the investigational JAK2 targeting agent CYT387 led to anemia responses in ∼50% of patients,104 while 57%–69% of patients became transfusion-independent depending on drug dose.105 LY2784544,106 AZD-1480, R723,107 NS-018,108 and BMS-911543106 are other investigational agents with early clinical trial data. As all JAK2 inhibitors except ruxolitinib are still in clinical development, this review focused on ruxolitinib and other standard therapy options discussed above; other reviews and literature should be referred to for a more detailed description of experimental JAK2 inhibitors.
Interestingly, responses to most JAK2 inhibitors have been independent of underlying disease, ie, primary MF, post-PV/ET MF, or JAK2V617F mutation status. Withdrawal of JAK inhibitors predictably results in splenic growth, symp-tomatic relapse, and, at times, hemodynamic instability. With long-term safety still under investigation, patients need to be followed closely. It has been suggested that slow tapering over a 2–4-week period may mitigate a “cytokine-flare” that occurs with abrupt withdrawal.
Other experimental therapies, such as mammalian target of rapamycin or histone deacetylase inhibitors, have been investigated in small studies in PV, ET, and MF and show activity in reducing constitutional symptoms and splenom-egaly and improving cytopenias.109–111 Novel agents are being tested in MPNs and MF/primary MF, including hedgehog pathway inhibitors; however, most studies are still early and no mature results are available.
Conclusion and future perspectives
Research in and molecular understanding of PV, ET, and primary MF has rapidly increased in recent years. The discovery of the JAK2V617F mutation led to accelerated development of JAK2 inhibitors, which have shown clini-cal benefit in patients with primary MF and post-PV/ET MF. The development of JAK-targeted treatment options has provided new avenues for therapeutic intervention in mitigating the debilitating disease effects of primary and
secondary MF. The first-in-class JAK2 inhibitor ruxoli-tinib has recently been approved. Other investigational JAK2 inhibitors show encouraging clinical effectiveness and demonstrable efficacy against proinflammatory cytok-ines. Side effect profiles vary between the various JAK2 targeting agents probably because of their slightly distinct kinase inhibitory spectrums. Several other agents and treat-ment modalities compete with JAK2 inhibitors. IMIDs and especially incorporation of IFN, alone or in combination, into treatment strategies for these patients early on may show benefit. The role of epigenetic targeting agents appears promising and future trials need to confirm the response rates with 5-azacytidine as well as define the role of histone deacetylase inhibitors in primary MF; the latter class of agents likely need to be used in combination. Allogeneic stem cell transplantation is the only known cure and remains a viable option for eligible patients.
As data continues to be acquired through future inves-tigational efforts, it remains to be determined if the same benefits for morbidity are also observed in regards to reducing mortality. Despite significant advances in treatment options, many outstanding questions remain regarding optimal dos-ing strategies, rational combinations, length of therapy, and whether patients can be taken off therapy with continued response. It is the authors’ hope that continued efforts in this field will ultimately address the many questions inherent to this new therapeutic modality.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5): 937–951.
2. Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372–375.
3. Correa PN, Eskinazi D, Axelrad AA. Circulating erythroid progenitors in polycythemia vera are hypersensitive to insulin-like growth factor-1 in vitro: studies in an improved serum-free medium. Blood. 1994;83(1):99–112.
4. Dai CH, Krantz SB, Green WF, Gilbert HS. Polycythaemia vera. III. Burst-forming units-erythroid (BFU-E) response to stem cell factor and c-kit receptor expression. Br J Haematol. 1994;86(1):12–21.
5. Dai CH, Krantz SB, Means RT Jr, Horn ST, Gilbert HS. Polycythemia vera blood burst-forming units-erythroid are hypersensitive to interleukin-3. J Clin Invest. 1991;87(2):391–396.
6. Kobayashi S, Teramura M, Hoshino S, Motoji T, Oshimi K, Mizoguchi H. Circulating megakaryocyte progenitors in myeloproliferative disorders are hypersensitive to interleukin-3. Br J Haematol. 1993;83(4):539–544. 7. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the
tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061.
Dovepress Recent MF developments
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
8. James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148.
9. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17): 1779–1790.
10. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myel-oid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. 11. Klingmuller U, Wu H, Hsiao JG, et al. Identification of a novel pathway
important for proliferation and differentiation of primary erythroid progenitors. Proc Natl Acad Sci U S A. 1997;94(7):3016–3021. 12. Walz C, Crowley BJ, Hudon HE, et al. Activated Jak2 with the V617F
point mutation promotes G1/S phase transition. J Biol Chem. 2006; 281(26):18177–18183.
13. Abdel-Wahab O, Pardanani A, Patel J, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–1202.
14. Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301.
15. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270.
16. Stegelmann F, Bullinger L, Schlenk RF, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25(7):1217–1219. 17. Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced
myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129(6):1097–1110.
18. Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081–1095.
19. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011; 118(2):401–408.
20. Tibes R, Mesa RA. Blood consult: resistant and progressive essential thrombocythemia. Blood. 2011;118(2):240–242.
21. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep. 2012;7(1):78–86. 22. Noor SJ, Tan W, Wilding GE, et al. Myeloid blastic transformation of
myeloproliferative neoplasms – a review of 112 cases. Leuk Res. 2011; 35(5):608–613.
23. Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A. Population-based incidence and survival figures in essential throm-bocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976–1995. Am J Hematol. 1999;61(1):10–15.
24. Visani G, Finelli C, Castelli U, et al. Myelofibrosis with myeloid metaplasia: clinical and haematological parameters predicting survival in a series of 133 patients. Br J Haematol. 1990;75(1):4–9.
25. Bonduel M, Sciuccati G, Torres AF, Pierini A, Gallo G. Familial idiopathic myelofibrosis and multiple hemangiomas. Am J Hematol. 1998;59(2):175–177.
26. Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–47963.
27. Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10): 3387–3395.
28. Ash RC, Detrick RA, Zanjani ED. In vitro studies of human pluripotential hematopoietic progenitors in polycythemia vera. Direct evidence of stem cell involvement. J Clin Invest. 1982;69(5):1112–1118. 29. Tibes R, Mesa RA. JAK2 inhibitors in the treatment of myeloproliferative
neoplasms: rationale and clinical data. Clin Investig (Lond). 2011;1(12): 1681–1693.
30. Oh ST, Simonds EF, Jones C, et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116(6):988–992. 31. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in
myelo-proliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–3476.
32. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007; 356(5):459–468.
33. Beer PA, Delhommeau F, Le Couedic JP, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010;115(14):2891–2900.
34. Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138.
35. Silver RT, Vandris K, Wang YL, et al. JAK2(V617F) allele burden in polycythemia vera correlates with grade of myelofibrosis, but is not substantially affected by therapy. Leuk Res. 2011;35(2):177–182. 36. Barosi G, Bergamaschi G, Marchetti M, et al. JAK2 V617F mutational status
predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood. 2007;110(12):4030–4036.
37. Passamonti F, Rumi E, Pietra D, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24(9):1574–1579. 38. Larsen TS, Pallisgaard N, Moller MB, Hasselbalch HC. The JAK2
V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis – impact on disease phenotype. Eur J Haematol. 2007;79(6):508–515.
39. Tefferi A, Lasho TL, Huang J, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22(4):756–761.
40. Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009;114(8):1477–1483. 41. Guglielmelli P, Pancrazzi A, Bergamaschi G, et al. Anaemia
characterises patients with myelofibrosis harbouring Mpl mutation. Br J Haematol. 2007;137(3):244–247.
42. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708.
43. Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397.
44. Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819–822.
45. Griffiths DS, Li J, Dawson MA, et al. LIF-independent JAK signalling to chromatin in embryonic stem cells uncovered from an adult stem cell disease. Nat Cell Biol. 2011;13(1):13–21.
46. Rameshwar P, Denny TN, Stein D, Gascon P. Monocyte adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-beta. J Immunol. 1994;153(6):2819–2830.
47. Bock O, Neuse J, Hussein K, et al. Aberrant collagenase expression in chronic idiopathic myelofibrosis is related to the stage of disease but not to the JAK2 mutation status. Am J Pathol. 2006;169(2):471–481. 48. Martyre MC, Romquin N, Le Bousse-Kerdiles MC, et al.
Transform-ing growth factor-beta and megakaryocytes in the pathogenesis of idiopathic myelofibrosis. Br J Haematol. 1994;88(1):9–16.
49. Kerbauy DM, Gooley TA, Sale GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant. 2007;13(3):355–365.
Dovepress
Tibes et al
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
50. Mesa RA, Hanson CA, Rajkumar SV, Schroeder G, Tefferi A. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood. 2000;96(10):3374–3380.
51. Castro-Malaspina H, Jhanwar SC. Properties of myelofibrosis-derived fibroblasts. Prog Clin Biol Res. 1984;154:307–322.
52. Mesa RA, Kantarjian H, Tefferi A, et al. Evaluating the serial use of the Myelofibrosis Symptom Assessment Form for measuring symptomatic improvement: performance in 87 myelofibrosis patients on a JAK1 and JAK2 inhibitor (INCB018424) clinical trial. Cancer. 2011;117(21):4869–4877.
53. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1): 68–76.
54. Liu TT, Chen JB, Chen WJ, Kuo CY, Lee CT. Idiopathic myelofibrosis associated with renal extramedullary hematopoiesis and nephrotic syndrome: case report. Chang Gung Med J. 2000;23(3):169–174. 55. Merry GM, Aronowitz PB. Myelofibrosis with massive
hepatosplenom-egaly and osteolytic bone lesions. J Hosp Med. 2010;5(3):E27–E28. 56. Koch CA, Li CY, Mesa RA, Tefferi A. Nonhepatosplenic extramedullary
hematopoiesis: associated diseases, pathology, clinical course, and treatment. Mayo Clin Proc. 2003;78(10):1223–1233.
57. Elliott MA, Pardanani A, Lasho TL, Schwager SM, Tefferi A. Thrombosis in myelofibrosis: prior thrombosis is the only predictive factor and most venous events are provoked. Haematologica. 2010;95(10): 1788–1791.
58. Randi ML, Fabris F, Tison T, Barbone E, Rossi C, Girolami A. Prevalence of specif ic thrombotic accidents in patients with thrombocytosis. Haematologia (Budap). 1993;25(3):149–152. 59. Wanless IR, Peterson P, Das A, Boitnott JK, Moore GW, Bernier V.
Hepatic vascular disease and portal hypertension in polycythemia vera and agnogenic myeloid metaplasia: a clinicopathological study of 145 patients examined at autopsy. Hepatology. 1990;12(5):1166–1174. 60. Adir Y, Humbert M. Pulmonary hypertension in patients with chronic
myeloproliferative disorders. Eur Respir J. 2010;35(6):1396–1406. 61. Gupta R, Perumandla S, Patsiornik Y, Niranjan S, Ohri A. Incidence
of pulmonary hypertension in patients with chronic myeloproliferative disorders. J Natl Med Assoc. 2006;98(11):1779–1182.
62. Garypidou V, Vakalopoulou S, Dimitriadis D, Tziomalos K, Sfikas G, Perifanis V. Incidence of pulmonary hypertension in patients with chronic myeloproliferative disorders. Haematologica. 2004;89(2):245–246. 63. Thiele J, Kvasnicka HM, Fischer R. Histochemistry and
mor-phometry on bone marrow biopsies in chronic myeloproliferative disorders – aids to diagnosis and classification. Ann Hematol. 1999; 78(11):495–506.
64. Johansson P, Mesa R, Scherber R, et al. Association between quality of life and clinical parameters in patients with myeloproliferative neoplasms. Leuk Lymphoma. 2012;53(3):441–444.
65. Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3): 973–977.
66. Dupriez B, Morel P, Demory JL, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88(3):1013–1018.
67. Tefferi A. Primary myelofibrosis: 2012 update on diagnosis, risk strati-fication, and management. Am J Hematol. 2011;86(12):1017–1026. 68. Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision
of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recom-mendations from an ad hoc international expert panel. Blood. 2007; 110(4):1092–1097.
69. Courcoutsakis N, Spanoudaki A, Maris TG, et al. Perfusion parameters analysis of the vertebral bone marrow in patients with Ph1- chronic
myeloproliferative neoplasms (Ph(neg) MPN): a dynamic contrast-enhanced MRI (DCE-MRI) study. J Magn Reson Imaging. 2012;35(3): 696–702.
70. Hussein K, Van Dyke DL, Tefferi A. Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol. 2009; 82(5):329–338.
71. Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–22792. 72. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis
of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008; 22(2):437–438.
73. Cervantes F, Alvarez-Larran A, Hernandez-Boluda JC, et al. Darbepoetin-alpha for the anaemia of myelofibrosis with myeloid metaplasia. Br J Haematol. 2006;134(2):184–186.
74. Silver RT, Jenkins DE Jr, Engle RL Jr. Use of testosterone and busulfan in the treatment of myelofibrosis with myeloid metaplasia. Blood. 1964; 23:341–353.
75. Kennedy BJ. Effect of androgenic hormone in myelofibrosis. JAMA. 1962;182:116–119.
76. Hast R, Engstedt L, Jameson S, et al. Oxymetholone treatment in myelofibrosis. Blut. 1978;37(1):19–26.
77. Besa EC, Nowell PC, Geller NL, Gardner FH. Analysis of the androgen response of 23 patients with agnogenic myeloid metaplasia: the value of chromosomal studies in predicting response and survival. Cancer. 1982;49(2):308–313.
78. Brubaker LH, Briere J, Laszlo J, et al. Treatment of anemia in myelo-proliferative disorders: a randomized study of fluoxymesterone v transfusions only. Arch Intern Med. 1982;142(8):1533–1537. 79. Levy V, Bourgarit A, Delmer A, et al. Treatment of agnogenic myeloid
metaplasia with danazol: a report of four cases. Am J Hematol. 1996; 53(4):239–241.
80. Cervantes F, Hernandez-Boluda JC, Alvarez A, Nadal E, Montserrat E. Danazol treatment of idiopathic myelofibrosis with severe anemia. Haematologica. 2000;85(6):595–599.
81. Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelo-fibrosis with myeloid metaplasia. Blood. 2006;108(4):1158–1164. 82. Quintas-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide
plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009; 27(28):4760–4766.
83. Mesa RA, Yao X, Cripe LD, et al. Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase 2 trial E4903. Blood. 2010;116(22):4436–4438.
84. Tefferi A, Lasho TL, Mesa RA, Pardanani A, Ketterling RP, Hanson CA. Lenalidomide therapy in del(5)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia. 2007;21(8):1827–1828.
85. Mesa RA, Elliott MA, Schroeder G, Tefferi A. Durable responses to thalidomide-based drug therapy for myelofibrosis with myeloid metaplasia. Mayo Clin Proc. 2004;79(7):883–889.
86. Thomas DA, Giles FJ, Albitar M, et al. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106(9): 1974–1984.
87. Marchetti M, Barosi G, Balestri F, et al. Low-dose thalidomide ameliorates cytopenias and splenomegaly in myelofibrosis with myeloid metaplasia: a phase II trial. J Clin Oncol. 2004;22(3):424–431. 88. Tefferi A, Verstovsek S, Barosi G, et al. Pomalidomide is active in the
treatment of anemia associated with myelofibrosis. J Clin Oncol. 2009; 27(27):4563–4569.
89. Begna KH, Pardanani A, Mesa RA, et al. Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012;87(1): 66–68.
90. Mesa RA, Pardanani AD, Hussein K, et al. Phase1/-2 study of pomalidomide in myelofibrosis. Am J Hematol. 2010;85(2):129–130. 91. Kroger N, Mesa RA. Choosing between stem cell therapy and drugs in
myelofibrosis. Leukemia. 2008;22(3):474–486.
92. Mesa RA. Transplantation for myelofibrosis: time for a randomized trial. Leuk Res. 2011;35(8):987–988.
Dovepress Recent MF developments
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020
Blood and Lymphatic Cancer: Targets and Therapy
Publish your work in this journal
Submit your manuscript here: http://www.dovepress.com/blood-and-lymphatic-cancer-targets-and-therapy-journal
Blood and Lymphatic Cancer: Targets and Therapy is an international, peer-reviewed, open access journal focusing on blood and lymphatic cancer research, identification of therapeutic targets and the optimal use of preventative and integrated treatment interventions to achieve improved outcomes, enhanced survival and quality of life for the
cancer patient. The manuscript management system is completely online and includes a very quick and fair peer-review system. Visit http://www.dovepress.com/testimonials.php to read real quotes from published authors.
93. Martinez-Trillos A, Gaya A, Maffioli M, et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patients. Ann Hematol. 2010;89(12):1233–1237.
94. Mesa RA. How I treat symptomatic splenomegaly in patients with myelofibrosis. Blood. 2009;113(22):5394–5400.
95. Silver RT, Vandris K, Goldman JJ. Recombinant interferon-α may retard progression of early primary myelofibrosis: a preliminary report. Blood. 2011;117(24):6669–6672.
96. Ianotto JC, Kiladjian JJ, Demory JL, et al. PEG-IFN-alpha-2a therapy in patients with myelofibrosis: a study of the French Groupe d’Etudes des Myelofibroses (GEM) and France Intergroupe des syndromes Myeloproliferatifs (FIM). Br J Haematol. 2009;146(2):223–225. 97. Abdel-Wahab O, Manshouri T, Patel J, et al. Genetic analysis of
transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70(2):447–452.
98. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood. 2010;116(19):3735–3742.
99. Tibes R, Mesa RA. Myeloproliferative neoplasms 5 years after discovery of JAK2V617F: what is the impact of JAK2 inhibitor therapy? Leuk Lymphoma. 2011;52(7):1178–1187.
100. Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127.
101. Verstovsek S, Mesa RA, Gotlib JR, et al. Results of COMFORT-I, a randomized double-blind phase III trial of JAK1/2 inhibitor INCB18424 (424) versus placebo (PB) for patients with myelofibrosis (MF) [abstract]. J Clin Oncol. 2011;29 Suppl:6500.
102. Harrison CN, Kiladjian JJ, Al-Ali HK, et al. Results of a randomized study of the JAK inhibitor ruxolitinib (INC424) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera-myelofibrosis (PPV-MF) or post-essential thrombocythemia-myelofibrosis (PET-MF) [abstract]. J Clin Oncol. 2011;29 Suppl: LBA6501.
103. Verstovsek S, Kiladjian J, Waltzman RJ, et al. RESPONSE: a randomized, open label, phase III study of INC424 in polycythemia vera (PV) patients resistant to or intolerant of hydroxyurea (HU) [abstract]. J Clin Oncol. 2011;29 Suppl:TPS203.
104. Tyner JW, Bumm TG, Deininger J, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflamma-tory cytokines in murine myeloproliferative neoplasms. Blood. 2010; 115(25):5232–5240.
105. Pardanani A, George G, Lasho T, et al. A phase I/II study of CYT387, an oral JAK-1/2 inhibitor, in myelofibrosis: significant response rates in anemia, splenomegaly, and constitutional symptoms. Paper pre-sented at: 52nd American Society of Hematology Annual Meeting. December 4–6, 2010; Orlando, FL.
106. Ma L, Zhao B, Walgren RA, et al. Efficacy of LY2784544, a small molecule inhibitor selective for mutant JAK2 kinase, in JAK2 V617F-induced hematologic malignancy models. Paper presented at: 52nd American Society of Hematology Annual Meeting. December 4–6, 2010; Orlando, FL.
107. Shide K, Kameda T, Markovtsov V, et al. Efficacy of R723, a potent and selective JAK2 inhibitor, in JAK2V617F-induced murine MPD model. Paper presented at: 51st American Society of Hematology Annual Meeting. December 5–8, 2009; New Orleans, LA.
108. Shide K, Nakaya Y, Kameda T, et al. NS-018, a potent novel JAK2 inhibitor, effectively treats murine MPN induced by the Janus kinase 2 (JAK2) V617F mutant. Paper presented at: 52nd American Society of Hematology Annual Meeting. December 4–6, 2010; Orlando, FL.
109. Mascarenhas J, Wang X, Rodriguez A, et al. A phase I study of LBH589, a novel histone deacetylase inhibitor in patients with primary myelofibrosis (PMF) and post-polycythemia/essential thrombocythemia (post-PV/ET MF). Paper presented at: 51st American Society of Hematology Annual Meeting. December 5–8, 2009; New Orleans, LA.
110. Rambaldi A, Dellacasa CM, Salmoiraghi S, et al. A phase 2A study of the histone-deacetylase inhibitor ITF2357 in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Paper presented at: 50th American Society of Hematology Annual Meeting. December 6–9, 2008; San Francisco, CA.
111. Vannucchi AM, Guglielmelli P, Lupo L, et al. A phase 1/2 study of RAD001, a mTOR inhibitor, in patients with myelofibrosis: final results. Paper presented at: 52nd American Society of Hematology Annual Meeting. December 4–6, 2010; Orlando, FL.
112. Barosi G, Elliott M, Canepa L, et al. Thalidomide in myelofi-brosis with myeloid metaplasia: a pooled-analysis of individual patient data from five studies. Leuk Lymphoma. 2002;43(12): 2301–2307.
Dovepress
Dove
press
Tibes et al
Blood and Lymphatic Cancer: Targets and Therapy downloaded from https://www.dovepress.com/ by 118.70.13.36 on 20-Aug-2020