0095-1137/10/$12.00 doi:10.1128/JCM.00614-09
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Emergence of a New Multidrug-Resistant Serotype X Variant in an
Epidemic Clone of
Shigella flexneri
䌤
†
Changyun Ye,
1‡ Ruiting Lan,
2‡ Shengli Xia,
3‡ Jin Zhang,
3‡ Qiangzheng Sun,
1‡ Shaomin Zhang,
1‡
Huaiqi Jing,
1‡ Lei Wang,
4‡ Zhenjun Li,
1Zhemin Zhou,
4Ailan Zhao,
1Zhigang Cui,
1Jingjing Cao,
1Dong Jin,
1Lili Huang,
3‡ Yiting Wang,
1Xia Luo,
1Xuemei Bai,
1Yan Wang,
1Ping Wang,
1Qiang Xu,
5and Jianguo Xu
1*
State Key Laboratory for Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, China CDC, P.O. Box 5, Changping, Beijing, China1; School of Biotechnology and Biomolecular Sciences,
University of New South Wales, Sydney, NSW 2052, Australia2; Henan Center for Disease Control and Prevention,
Zhengzhou, Henan Province, China3; College of Life Sciences, Nankai University, Tianjin, China4; and
Suixian Center for Disease Control and Prevention, Suixian County, Henan Province, China5
Received 26 March 2009/Returned for modification 27 March 2009/Accepted 19 November 2009
Shigellaspp. are the causative agent of shigellosis withShigella flexneriserotype 2a being the most prevalent
in developing countries. Epidemiological surveillance in China found that a new serotype ofS. flexneriappeared
in 2001 and replaced serotype 2a in 2003 as the most prevalent serotype in Henan Province. The new serotype also became the dominant serotype in 7 of the 10 other provinces under surveillance in China by 2007. The serotype was identified as a variant of serotype X. It differs from serotype X by agglutination to the monovalent anti-IV type antiserum and the group antigen-specific monoclonal antibody MASF IV-I. Genome sequencing of
a serotype X variant isolate, 2002017, showed that it acquired aShigellaserotype conversion island, also as an
SfX bacteriophage, containinggtr genes for type X-specific glucosylation. Multilocus sequence typing of 15
genes from 37 serotype X variant isolates and 69 isolates of eight other serotypes, 1a, 2a, 2b, 3a, 4a, 5b, X, and Y, found that all belong to a new sequence type (ST), ST91. Pulsed-field gel electrophoresis revealed 154 pulse
types with 655S. flexneriisolates analyzed and identified 57 serotype switching events. The data suggest that
S. flexneri epidemics in China have been caused by a single epidemic clone, ST91, with frequent serotype switching to evade infection-induced immunity to serotypes to which the population was exposed previously. The clone has also acquired resistance to multiple antibiotics. These findings underscore the challenges to the current vaccine development and control strategies for shigellosis.
Shigellosis or bacillary dysentery is one of the major infec-tious diseases in developing countries, where it mainly affects economically poor populations. A multicenter shigellosis sur-veillance study involving six Asian countries found that the overall annual incidence of culture-confirmed shigellosis was 13.2 per 1,000 in children under 5 years of age and 2.1 per 1,000 in other age groups (35). The incidence in developing countries is approximately 100 times higher than that in indus-trialized countries (35). There is no sign that the incidence of diarrheal infectious diseases, the diseases of the poorest, is decreasing (26). Since the Chinese National Infectious Disease Internet Reporting System began operation in 2005, the re-ported annual incidence of shigellosis in China was one of the top four notifiable infectious diseases for four consecutive years from 2005 to 2008, with close to half a million cases each year (http://www.moh.gov.cn), which is now widely believed to be an underestimate. (36, 37).
The causative organisms for shigellosis are Shigella spp., which are in fact clones ofEscherichia coli(24).Shigella flexneri
is the predominant species in developing countries. Identifica-tion ofS. flexneriis based on biochemical and serological prop-erties. AsShigellastrains are all negative for H antigen,Shigella
serotyping is based on O antigen only.S. flexneriis divided into 15 serotypes. Some S. flexneri serotypes are more prevalent than others, with the three most prevalent being serotypes 2a, 3a, and 1a in Asian countries including China (6, 35, 37), but there are exceptions. Serotype 1c has been reported recently as the most prevalent serotype in Vietnam (28).
AllS. flexneriserotypes except serotype 6 share the backbone of the basic O-antigen repeat unit, which is a tetrasaccharide consisting of a singleN-acetylglucosamine and three rhamnose residues (3). Glucosylation of any of the four sugars and/or O acetylation of the last rhamnose gives rise to more than 13 known serotypes (3). Both processes are performed by genes carried by bacteriophages (3). Glucosylation involves 3 gtr
genes with one being type specific while O acetylation involves only one gene,oac(3). This O-antigenic variation is a major strategy used by the organism to evade host immunity (3, 39). These bacteriophage-encoded modifications allowS. flexnerito change its O antigenicity rather simply.
In this study we report a new serotype, a serotype X variant, which was more prevalent than serotype 2a in Henan Province for a 5-year period from 2002 to 2007 and spread to several * Corresponding author. Mailing address: State Key Laboratory for
Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, China CDC, P.O. Box 5, Changping, Beijing, China. Phone: 61739579. Fax: 8610-61730233. E-mail: xujianguo@icdc.cn.
† Supplemental material for this article may be found at http://jcm .asm.org/.
‡ These authors contributed equally.
䌤Published ahead of print on 2 December 2009.
419
on May 16, 2020 by guest
http://jcm.asm.org/
other provinces of China, and our findings of O-antigen switch-ing as the underlyswitch-ing mechanism of persistence of a multidrug-resistant epidemic clone ofS. flexneri.
MATERIALS AND METHODS
Bacterial isolates and conventional methods.Stool specimens from patients
with either diarrhea or dysentery were collected and screened forShigellaspp. by
conventional biochemical methods in local hospitals in Henan and other prov-inces. Isolates were identified to species and serotype levels by provincial centers for disease control. The serotypes were reconfirmed in the State Key Laboratory for Infectious Disease Prevention and Control in Beijing. Serological identifica-tion was performed by slide agglutinaidentifica-tion with polyvalent somatic (O) antigen grouping sera, followed by testing with monovalent antisera (Denka Seiken, Japan) for specific serotype identification. A selected set of serotype X variant isolates which were initially identified as serotype 4c were confirmed using monoclonal antibodies (MASF IV-1 and MASF IV-2) (Reagensia AB, Sweden). Antimicrobial susceptibility testing against ampicillin, co-trimoxazole, nalidixic acid, and ciprofloxacin was performed by the disk diffusion method following standardized Clinical and Laboratory Standards Institute (CLSI) methods (2).
Genome sequencing and analysis.Isolate 2002017, anS. flexneriX variant, obtained from a 2-year-old patient in 2002 in Henan Province, was selected for genome sequencing. Chromosomal DNA from 2002017 was prepared using standard protocols and was sequenced by a combination of pyrosequencing using a 454/Roche FLX machine, according to the manufacturer’s protocols, and Sanger sequencing using an ABI 3730 automated DNA analyzer (Applied Bio-systems). The 454 sequencing run produced 226,995 reads with an average length of 223 bp for approximately 50.7 Mb of data, representing a theoretical 10.6-fold coverage of the genome. Ninety-one percent (206,567) of the 454 sequence reads werede novoassembled or partially assembled into 701 nonredundant contigs with an average of 10.5-fold coverage, using the 454/Roche Newbler assembly
program. These contigs were reordered based on BLAST alignments withS.
flexnerigenomes of Sf301 (GenBank accession number AE005674.1) and 2457T (GenBank accession number AE014073.1). A total of 25,014 paired end
se-quences (giving 3⫻coverage) derived from pUC18 (insert size, 1.5 kb) using an
ABI BigDye Terminator v3.1 cycle sequencing kit and an ABI 3730 automated DNA analyzer (Applied Biosystems) were used to verify the orders of the contigs, as well as the sequence quality. The 454 contigs and the ABI results are hybrid assembled in Phred/Phrap (9). The gaps between these contigs were closed by PCR, and the PCR products were sequenced using BigDye terminator chemistry. All bases different from 2457T were verified by the coverage of ABI 3730 reads, the recalling of the 454 reads from the Newbler assembly, or further resequencing of the region by PCR sequencing.
The colinear blocks of the 2002017 and 2457T genomes were determined using BLASTN. Then the alignment within each of the blocks was obtained using Mauve (7). The final plot and identification of recombination segments were
generated similarly to the method used in the study ofVibrio choleraeby Feng
et al. (8).
MLST analysis. The 15 housekeeping genes used for multilocus sequence
typing (MLST) wereaspC,clpX,fadD,icdA,lysP,mdh,uidA,arcA,aroE,cyaA,
dnaG,grpE,mtlD,mutS, andrpoS. The primers used were synthesized
commer-cially (Shanghai Sangon Biological Engineering Technology & Services, China), and PCR was done based on the MLST protocol obtained from the EcMLST website (http://www.shigatox.net/ecmlst). PCR products were verified on a 1% agarose gel and purified for sequencing, commercially done at Shanghai Sangon Biological Engineering Technology & Services (China). Both directions were sequenced, and sequences were edited using SeqMan 7.0. Sequence types were allocated by the EcMLST curator.
PFGE analysis.Genomic DNA for pulsed-field gel electrophoresis (PFGE) was prepared in agarose plugs using the method described by Ribot et al. (25) with the following modifications. Slices of agarose plugs were digested with 20 U NotI for 2 h, and electrophoresis was carried out in a 1% agarose SeaKem Gold gel with the CHEF DR III system (Bio-Rad, Hercules, CA) with the following run parameters: switch time of 3 to 30 s and run time of 18 h. The PFGE patterns were imported into Bionumerics (Applied Maths, Belgium) for further analysis and manually edited for accuracy. The bands which were less than the smallest band (20.5 kb) of the molecular size standard were not included in the analysis. All patterns were visually inspected after computer analysis. Patterns identified as indistinguishable by computer and visual inspection were assigned the same pattern designation. The plasmid band was removed from comparison as there is no NotI cut site based on the plasmid sequence data published so far (4, 33, 38, 40), and in some cases the band is barely visible, likely as a result of the
supercoiled form migrating out of the detection range. The PFGE patterns belonging to the genome-sequenced strains (2002017 and Sf301) were the same as those predicted using genome sequence data, which also allowed us to discern the plasmid band on the PFGE gel.
Biochemical tests and antibiotic sensitivity testing of the serotype X variant isolates.The isolates were biochemically tested using the Autoscan system (Neg Combo Panel Type 31, Dade Behring Microscan Walkaway 40 SI; Siemens) following the manufacturer’s instructions. Antimicrobial susceptibility testing was also performed using the Autoscan system (Neg Combo Panel Type 31, Dade Behring Microscan Walkaway 40 SI; Siemens) following the
manufactur-er’s instructions. The antibiotics on the panel are ampicillin (8 to 16g/ml),
amoxicillin-clavulanic acid (8 and 4 to 16 and 8g/ml, respectively),
ampicillin-sulbactam (8 and 4 to 16 and 8g/ml, respectively), ticarcillin-clavulanic acid (16
and 2 to 64 and 2g/ml, respectively), piperacillin-tazobactam (8 and 4 to 64 and
4g/ml, respectively), piperacillin (16 to 64g/ml), aztreonam (8 to 16g/ml),
cefepime (2 to 16g/ml), cefotaxime (4 to 32g/ml), ceftazidime (2 to 16g/ml),
ceftriaxone (4 to 32g/ml), ciprofloxacin (1 to 2g/ml), gatifloxacin (2 to 4
g/ml), levofloxacin (2 to 4g/ml), imipenem (4 to 8g/ml), and
trimethoprim-sulfamethoxazole (2 to 38g/ml). The quality control organism used wasE. coli
ATCC 25922.
Detection of SDS-PAGE-separated lipopolysaccharide (LPS) by silver
stain-ing.Isolates were grown overnight with aeration at 37°C in Luria broth. Bacteria
were pelleted by centrifugation and were lysed in sodium dodecyl sulfate-poly-acrylamide gel electrophoresis (SDS-PAGE) sample buffer containing 4% 2-mercaptoethanol. The sample was boiled for 5 min, treated with proteinase K for 1 h, and analyzed on a 15% SDS-polyacrylamide gel. Gels were silver stained as described by Hitchcock and Brown (12).
Nucleotide sequence accession numbers. GenBank accession numbers re-ported in this study are CP001383 to CP001388 for the genome sequence of 2002017 and GQ480774 to GQ480794 for MLST sequences.
RESULTS
Emergence of serotype X variant, a new serotype ofS.
flex-neri. Surveillance of Shigella infections conducted by the Henan Provincial Center for Disease Control (CDC) of China since 2000 found a novel serotype ofS. flexneriwhich could not be typed according to current diagnostic serotyping criteria for theShigellaspecies. The novel serotype was initially identified as serotype 4c, described previously by Pryamukhina and Khomenko (23) as it agglutinated with monovalent anti-IV type antiserum and monovalent anti-7,8 group antisera. Using monoclonal antibodies, the serotype also agglutinated with group antigen-specific monoclonal antibody MASF IV-1 but not with serotype IV type-specific antibody MASF IV-2 and thus can be identified as serotype 4x, described previously by Carlin and Lindberg (5). To resolve the identity of this novel serotype, we sequenced the genome of one clinical isolate, 2002017 (see below for detailed genome analysis). The type IV-specific glucosyltransferase gene, gtr type IV (1), for the serotype IV antigenicity, was not found. Instead, the genegtrX, encoding the type X-specific glucosyltransferase which involves in the addition of a glucosyl group to the first rhamnose of the O-antigen tetrasaccharide backbone (34), was found in the 2002017 genome. No other type-specific glucosyltransferase genes were found. We then found that the serotype conversion bacteriophage SfX of strain 2002017 was detected by genome analysis, which could convert a serotype Y strain (27) which carries the unmodified O antigen to serotype X. In addition, we usedgtrtype-specific (I, II, IV, V, and X) andoac gene-specific PCR for serotypes 1a, 2a, 2b, 3a, 4a, 5b, and X (Q. Sun et al., unpublished data) to determine the presence of any of these serotype-specific genes. All 30 serotype X variant isolates tested were found to have only thegtrtype X gene amplified. Serologically, the new serotype differs from serotype X by the
on May 16, 2020 by guest
http://jcm.asm.org/
agglutination with monovalent anti-IV type antisera and the group antigen-specific monoclonal antibody MASF IV-1. Structurally, it differs from serotype X in mobility in the LPS ladder in the silver-stained polyacrylamide gels (data not shown), indicating that the serotype X variant O-antigen struc-ture was modified. However, we could not identify the gene(s) potentially involved in this modification from the 2002017 ge-nome sequence. Therefore, we named this serotype X variant as a new serotype ofS. flexneri.
Isolation frequency and multiantibiotic resistance ofS.
flex-neriserotype X variant. In the 9-year surveillance ofShigella
infections in Henan Province, the predominant serotypes fluc-tuated (Fig. 1). Serotype 2a was the predominant serotype (26%) in 2000 and 2001, declined to less than 10% in 2004, and rose again to dominance (42%) in 2007 (Fig. 1). In contrast, the serotype X variant first appeared in Henan Province in 2001 and was the most prevalent serotype between 2002 and 2006, accounting for 14%, 35%, 47%, 48%, and 27% of the isolations in the respective years (Fig. 1). However, it declined to only 15% in 2007, back to the 2002 level. To determine whether the serotype X variant has spread to other parts of China, a national survey was conducted from 2005 onward using two surveillance posts in each of 10 selected provinces/ cities. The serotype X variant was found to be the most prev-alent serotype in Shanxi Province in 2006 (67%) and 2007 (33%) and in Gansu Province, Anhui Province, and Shanghai in 2007 with 67%, 54%, and 35% of the isolations ofS. flexneri, respectively.
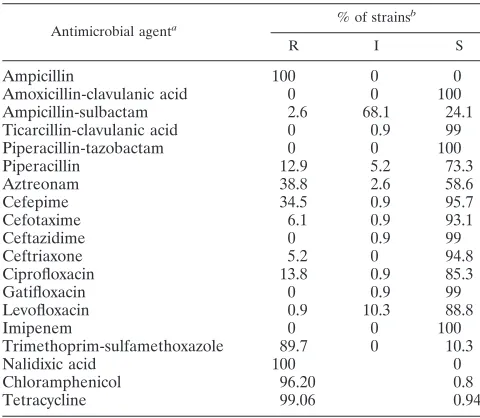
A total of 1,890S. flexneriisolates were tested for resistance to the commonly used antibiotics by the Henan Provincial CDC, and 116 serotype X variant isolates were selected for retesting and confirmation by the National Laboratory in Bei-jing. All of the 116 isolates were resistant to ampicillin and nalidixic acid, among which 96% were also resistant to chlor-amphenicol and tetracycline and 89.7% were resistant to tri-methoprim-sulfamethoxazole (Table 1). Resistance to ampicil-lin is significantly higher than previously reported levels of 53% in China and 84% in Asia (35, 37).
Genes gained, lost, or mutated in S. flexneri serotype X
variant isolate 2002017.To further elucidate genetic changes
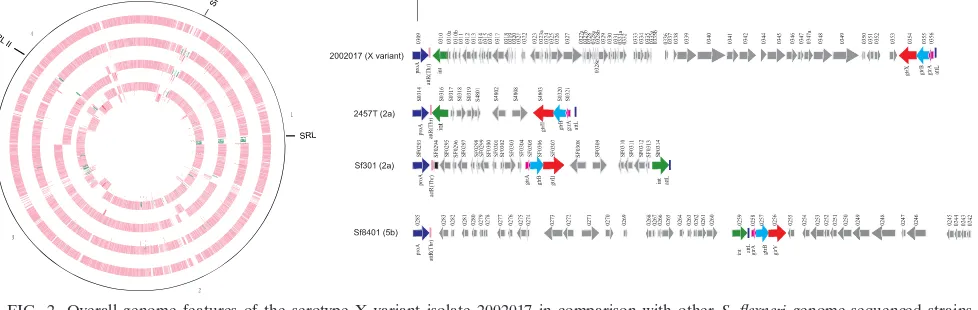
that occurred in the serotype X variant, an isolate, 2002017, obtained from a 2-year-old patient in 2002 in Henan Province, was sequenced. Its genome is composed of one chromosome and five plasmids including a large virulence plasmid and a drug resistance plasmid for sulfonamide and streptomycin re-sistance (Table 2; Fig. 2).
Compared to the published genome sequences ofS. flexneri
strains 2457T, Sf301, and Sf8401 (16, 21, 38), 2002017 gained three genomic islands. The first genomic island is a 37,006-bp
Shigellaserotype conversion island carrying genes for O-anti-gen modification and was named the serotype X variant SHI-O (Fig. 2; see also Fig. S1 in the supplemental material). The 2002017 SHI-O is located at the same site as the other S. flexneriSHI-Os reported previously (Fig. 2) (13). It contains thegtrgenes for serotype X conversion because the observed
gtrsequence is identical to the SfXgtrgenes published previ-ously (10, 34).
The second genomic island is a multiantibiotic resistance island encoding tetracycline, chloramphenicol, ampicillin, and streptomycin resistance (see Fig. S2 in the supplemental ma-terial). It is similar to theShigellaresistance locus (SRL) island initially discovered in anS. flexneri2a strain, YSH6000 (31, 32). The first 8 kb and the last 7 kb of the multiantibiotic resistance island are almost identical between 2002017 and YSH6000. However, the 2002017 island has an additional set of tetracy-cline resistance genes and an extra 22 genes of various func-tions but does not contain the iron acquisition system present in the YSH6000 SRL. This island was named serotype X vari-ant SRL.
[image:3.585.43.283.68.219.2]The third genomic island is a composite transposon contain-ing multiantibiotic resistance genes (20, 29) and was named
TABLE 1. Antibiotic resistance profiles ofS. flexneriserotype X variant isolates
Antimicrobial agenta % of strains
b
R I S
Ampicillin 100 0 0
Amoxicillin-clavulanic acid 0 0 100
Ampicillin-sulbactam 2.6 68.1 24.1
Ticarcillin-clavulanic acid 0 0.9 99
Piperacillin-tazobactam 0 0 100
Piperacillin 12.9 5.2 73.3
Aztreonam 38.8 2.6 58.6
Cefepime 34.5 0.9 95.7
Cefotaxime 6.1 0.9 93.1
Ceftazidime 0 0.9 99
Ceftriaxone 5.2 0 94.8
Ciprofloxacin 13.8 0.9 85.3
Gatifloxacin 0 0.9 99
Levofloxacin 0.9 10.3 88.8
Imipenem 0 0 100
Trimethoprim-sulfamethoxazole 89.7 0 10.3
Nalidixic acid 100 0
Chloramphenicol 96.20 0.8
Tetracycline 99.06 0.94
a
Results for the first 16 antibiotics were based on tests of 116 serotype X variant isolates using the Microscan Gram-negative panel Neg Combo 31 (Sie-mens Healthcare Diagnostics, United Kingdom). The remaining antibiotics were tested using the calibrated dichotomous sensitivity test (CDS) method, as the Microscan panel does not contain these antibiotics.
b
[image:3.585.300.540.451.660.2]R, resistance; I, intermediate sensitivity; S, sensitivity. Resistance was deter-mined according to the manufacturer’s instructions for Microscan or by the annular radius using the CDS method.
FIG. 1. Temporal trends ofS. flexneriserotypes in Henan Province, China. Only the top four predominant serotypes for each year are shown. The frequencies were based on 1,890 isolates from Henan Province, and the number of isolates per year varies.
on May 16, 2020 by guest
http://jcm.asm.org/
serotype X variant SRLII. It is 15,360 bp in length and carries gene cassettes for dihydrofolate reductase (dfrA1), streptothri-cin acetyltransferase (sat1), and aminoglycoside adenyltrans-ferase (aadA1) conferring resistance to trimethoprim, strepto-thricin, and streptomycin/spectinomycin, respectively. In addition to these islands, 2002017 gained 13 other genes, of which 11 are single-gene gains compared with the three pub-lished S. flexneri genomes (Table 3). The majority of these genes are of unknown function.
Shigellaas a host-adapted pathogen has undergone consid-erable genome decay (16, 38). The loss of gene functions seems to be continuing with 37 new pseudogenes in 2002017 in ad-dition to the 194 pseudogenes shared with 2457T (see Table S1 in the supplemental material). Thirty-seven percent of the new pseudogenes are genes of bacteriophage or insertion sequence (IS) origin or of unknown functions.
Comparison with the other threeS. flexnerigenomes, 2457T, Sf301, and Sf8401 (16, 21, 38), showed that 2002017 is closest to 2457T (Fig. 3). With the use of otherE. coligenomes as an outgroup, Sf8401 was shown to have diverged first. We were able to allocate most of the sequence differences between 2002017 and 2457T to specific lineages by comparison with the genomes of Sf301 and Sf8401 using the approach described previously (8). The base differences, commonly referred to as single-nucleotide polymorphisms (SNPs), can be classified as recombinational or mutational, based on the difference in the
distribution of SNPs introduced by recombination and muta-tion, as recombinant segments have a higher frequency of SNPs (8). The differences between 2002017 and 2457T are largely due to mutational changes, almost all of which can be attributed to either the 2002017 or 2457T genome (Fig. 3; see also Fig. S3 in the supplemental material). There are 221 SNPs in 2002017, 33% more than in 2457T (166 SNPs). A higher proportion of the SNPs are nonsynonymous (62%, Fig. 3). We looked at the functional categories of the 186 genes with one or more nonsynonymous SNPs in 2002017 using clusters of or-thologous groups (COG) and found that only one category, “replication and repair,” was significantly overrepresented (Z
test,P⬍0.0001; see Fig. S4 in the supplemental material). All except for five genes in this category are IS encoded. The changes in two (gyrAandparC) of the five non-IS-related genes may be associated with quinolone resistance, as 2002017 is resistant to nalidixic acid (11, 30, 41). The genomes are also affected by recombination although to a much lesser extent than by mutational changes, with 18 recombinational events affecting 10 genes in 2002017 (see Table S2 in the supplemen-tal material). Only two of the recombination genes have known functions: dacA, encoding D-alanyl-D-alanine
[image:4.585.44.541.81.189.2]carboxypepti-dase, andmtlD, encoding mannitol-1-phosphate 5-dehydroge-nase; the significance of which is unknown. The data suggest that the level of recombination inS. flexneriis low.
TABLE 2. Features of theS. flexneriserotype X variant 2002017 genome
Featurea
Chromosome
Plasmid
pSFxv_1 pSFxv_2 pSFxv_3 pSFxv_4 pSFxv_5
Total length (bp) 4,650,865 223,364 6,850 6,200 4,042 3,180
No. of ORFs 4,380 302 11 8 4 6
Percentage of CDS 83.3 79.3 64.2 71.5 58.9 61.8
G⫹C content (%) 50.85 45.9 44.03 52.58 52.62 45.03
IS elements (%) 491 (6) 157 (32) 1 0 0 0
No. of pseudogenes 249
No. of rRNAs (16S/23S/5S) 7/7/8
No. of tRNAs 101
a
ORFs, open reading frames; CDS, coding sequence.
FIG. 2. Overall genome features of the serotype X variant isolate 2002017 in comparison with otherS. flexnerigenome-sequenced strains (2457T, Sf301, and Sf8401). The locations of SRLs and SHI-O are indicated. From the outer ring to the innermost in order are 2002017, 2457T, Sf301, and Sf8401. The inset depicts the structures of the SHI-Os in the 4 genomes. For details of 2002017 SHI-O see Fig. S1 in the supplemental material.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:4.585.47.536.529.684.2]The serotype X variant emerged from a novel sequence
type ofS. flexnericirculating in China for years.To
deter-mine the phylogenetic relationship ofS. flexneriserotype X variant, an extended MLST scheme of 15 genes as described by Lacher et al. (17) was used to type 37 serotype X variant and 74 other serotype isolates from Henan and other prov-inces. We found that all 37 serotype X variant isolates be-long to a new sequence type (ST), ST91. ST91 differs from ST18 (represented by Sf301) and ST86 (represented by 2457T) by only a single nucleotide inlysPandrpoS, respec-tively, and thus forms a clonal complex with these STs. All except 5 of the 74 isolates from other serotypes (1a, 2a, 2b, 3a, 4a, 5b, X, and Y) were also typed as ST91. For the remaining five isolates, one belongs to ST18 while the other four were new sequence types, ST92, ST96, ST97, and ST98. These results suggested that a predominant sequence type, ST91, ofS. flexnerihas been persistently circulating in China for many years. The presence of multiple serotypes in ST91 FIG. 3. The evolutionary relationship of the four
[image:5.585.42.539.79.472.2]genome-se-quenced strains ofS. flexneri. The root of the tree was determined using genome sequences of E. coli K-12 (GenBank accession no. NC_000913) and EDL933 (GenBank accession no. NC_002655) as an outgroup. The number of genes gained (⫹) or lost (⫺) was marked on each branch. The number of mutational and recombinational changes was marked on 2002017 and 2457T only. rec, recombination; sSNP, synonymous SNPs; nsSNP, nonsynonymous SNPs. Sequence types are shown in parentheses beside the strain names. For a detailed compar-ison, see Fig. S3 in the supplemental material.
TABLE 3. Gained and lost genes ofS. flexneriserotype X variant 2002017
Locus taga Gene name Function Categoryb
Gained genes
SFxv_1259 Hypothetical protein EcHS_A1225 Unknown
SFxv_1369 ycgY Hypothetical protein SFV_1210 Unknown
SFxv_1537 Putative bacteriophage protein Unknown
SFxv_1540 COG3646: uncharacterized phage-encoded protein Unknown
SFxv_1938 Putative IS1-encoded protein Unknown
SFxv_2305 Phage integrase family protein Replication and repair
SFxv_2368 yegE Putative sensor-type protein Signal transduction
SFxv_2400 yehC Uncharacterized fimbrial chaperoneyehCprecursor Cell motility
SFxv_2807 Hypothetical protein Unknown
SFxv_2909 IS4ORFc Replication and repair
SFxv_3441 Putative pilus biogenesis initiator protein Cell motility
SFxv_3833 Hypothetical protein Unknown
SFxv_4762 Conserved hypothetical protein Unknown
Lost genes
SF0654 tatE Twin arginine translocase protein E Signal transduction
SF0721 Putative tail component of prophage CP-933K Phage related
SF1039 csgC Putative curli production protein Cell communication
SF1137 Hypothetical bacteriophage protein Phage related
SF1494 celF Cryptic phospho-beta-glucosidase Carbohydrate metabolism
SF1495 ydjC Hypothetical protein Unknown
SF1496 katE Hydroperoxidase II Energy metabolism
SF1497 Cell division modulator Cell growth and death
SF1498 ydjO Hypothetical protein Unknown
SF1881 Putative tail component of prophage CP-933K Phage related
SF2066 Hypothetical protein Unknown
SF2067 yeeX Hypothetical protein Unknown
SF2133 alkA 3-Methyl-adenine DNA glycosylase II Replication and repair
SF2135 Putative chaperonin Unknown
SF2136 Hypothetical protein Unknown
SF2137 Hypothetical protein Unknown
SF2463 flxA Hypothetical protein Unknown
SF2611 Putative tail component of prophage CP-933K Phage related
SF2718 Hypothetical protein Unknown
SF3737 yieC Putative receptor protein Unknown
SF3738 yieL Putative xylanase Unknown
SF3739 yieK Putative 6-phosphogluconolactonase Unknown
SF3744 yieI Predicted inner membrane protein Unknown
SF3745 yieH Predicted hydrolase Unknown
SF4175 yjbA Phosphate-starvation-inducible protein PsiE Unknown
a
Locus tags for lost genes are from 2457T.
b
Based on clusters of orthologous groups of proteins (COG) database.
c
ORF, open reading frame.
on May 16, 2020 by guest
http://jcm.asm.org/
indicates that serotype switching within the clone has oc-curred many times and that the serotype X variant emerged recently.
Serotype switching and spreading of the epidemic clone.To
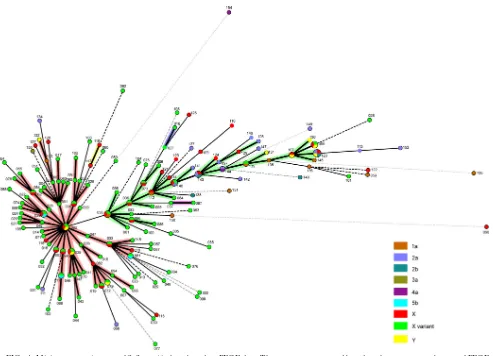
investigate serotype switching further and the spread of the epidemic clone ST91, S. flexneri isolates were analyzed by PFGE, which is a more discriminatory method than MLST. A total of 655 isolates obtained from Henan and 10 other prov-inces of China were analyzed and divided into 154 pulse types. Pulse type CN002 (199 isolates) was the most frequent pulse type, accounting for 30.38% of the isolates. Ninety-six pulse types were represented by a single isolate. We found that 24 pulse types contain more than one serotype, suggesting that serotype switching occurred within a pulse type. Pulse type CN120 contains the largest number of serotypes with four serotypes. Seven pulse types (CN002, CN038, CN039, CN058, CN069, CN140, and CN143) contain three serotypes, and the remaining 16 contain two (Fig. 4). Assuming that only one of the serotypes in each pulse type is the founder, 33 serotype switching events occurred in these 24 pulse types.
To determine the relationships of the pulse types and asso-ciated serotype diversity, we constructed a minimum spanning
tree (MST) for all 154 pulse types (Fig. 4). We assigned pulse types differing by one band to the same clonal complex using the terminology of multilocus sequence typing. The 154 pulse types were grouped into three clonal complexes with clonal complex 1 being the largest. The MST allowed further identi-fication of serotype conversion events as shown in Fig. 4 when one pulse type gives rise to another accompanied by a change of its serotype. At least 57 serotype switching events can be identified. The founder pulse types for two of the three clonal complexes (CN002 and CN058) contain serotype X variant isolates, indicating that the serotype X variant arose consider-ably earlier. Among the 655 isolates, 189 were from other parts of China and the majority shared the same pulse types with isolates from Henan Province, suggesting that the same pulse types were spreading concurrently to other parts of China.
DISCUSSION
The predominantShigellaspecies in resource-poor countries isS. flexneri, and the distribution ofS. flexneriserotypes varies geographically and temporally. Statistically significant shifts of
[image:6.585.44.537.63.419.2]S. flexneriserotypes between years were observed in several FIG. 4. Minimum spanning tree ofS. flexneriisolates based on PFGE data. The tree was constructed based on the presence or absence of PFGE bands. Pulse types with a band difference of one are connected by a thick line while those with a difference of two are connected by a thin line and those with a difference greater than two are connected by a dashed line. Clonal complexes are shaded. Each pulse type is color coded by serotype(s), and the pulse type is labeled alongside the circle. The circle size is proportional to the number of serotypes in the given pulse type.
on May 16, 2020 by guest
http://jcm.asm.org/
developing countries including Indonesia, Bangladesh, Paki-stan, and China (35–37). These epidemiological observations were unexplained, but it was generally assumed that the sero-type shifts were due to different clones replacing each other over time with each serotype being treated as a clone. In this study we combined epidemiological surveillance and genetic analysis to find a single epidemic clone causing the majority of theS. flexneriinfections in China and found that switching of serotypes was occurring frequently within the epidemic clone. Serotype switching was also reflected in the shifts of serotypes over time (Fig. 1). Therefore, we uncovered a novel mecha-nism of epidemic persistence ofS. flexneri over long periods through serotype switching to escape infection-induced immu-nity. This finding has major implications for prevention. As immunity toS. flexneri is serotype specific (15), infection-in-duced protection against one serotype will provide protection only against reinfection by the homologous serotype. Frequent serotype switching in S. flexneri may partly explain why the incidence of shigellosis in China and other developing coun-tries remains high despite the fact that safe water supplies, sanitation, hygiene, and the economy have greatly improved in recent years (26, 35). The easy conversion among theS. flexneri
serotypes through modification of the O antigen and rapid emergence of new serotypes render the population susceptible to serotypes to which they have not been exposed previously. The emergence of the serotype X variant and frequent se-rotype switching in an epidemic sequence type require updat-ing of the current vaccine development and other prevention and control strategies against shigellosis. Major efforts have been directed to develop vaccines against the predominantS. flexneriserotype 2a. Our mice experiment data show no cross-protection between serotype X variant and serotype 2a (data not shown), and thus, the only vaccine currently in use against
S. flexneriin China, a live serotype 2a vaccine, is expected to offer no protection against the X variant serotype. Since a novel serotype can appear and increase to a high frequency in a very short time and serotypes can interconvert frequently, a rapid vaccine development cycle would be most beneficial to provide timely protection against newly emerged serotypes such as the serotype X variant (18, 19). Our results also high-light that for a vaccine to be successful in controllingS. flexneri
infections, the vaccine must cover multiple serotypes. A num-ber of strategies have been tried to develop such vaccines (14, 22). Noriega et al. (22) used twoS. flexneriserotypes (2a and 3a) to cover all “type” and “group” antigenic factors for S. flexneri serotypes 1 to 5, but the vaccine offered protection against only four of the sixS.flexneriserotypes tested (22). Finally, the choice of a live vaccine candidate should be from among the currently circulating epidemic clones, in this case ST91.
The acquisition of multiantibiotic resistance by the genome-sequenced serotype X variant isolate and likely the majority of the isolates of the ST91 clone presents new challenges in clin-ical therapy for shigellosis (31). The genomic sequence data showed that the serotype X variant isolate, 2002017, gained two multiantibiotic resistance genomic islands encoding resis-tance to six antibiotics including tetracycline, streptomycin, streptothricin, chloramphenicol, ampicillin, and trimethoprim, of which the latter three are among the first-line antibiotics recommended for treating shigellosis in China (Fig. 2 and 3) (36, 37). The uncontrolled use of antibiotics in many Asian
countries including China is likely to provoke a major crisis as the multidrug-resistant clone can and may have already spread to other parts of the world (26).
In conclusion, this study has identified a new serotype, the serotype X variant, ofS. flexneriwhich was prevalent in several provinces in China. Genome sequencing of a serotype X vari-ant isolate revealed the genetic basis of serotype conversion by the acquisition of an SfX bacteriophage, although the “variant” factor remains to be identified. The genome sequencing also revealed the presence of multidrug resistance islands and a plasmid. Analyses of MLST and PFGE data showed that epi-demics in China in recent years were caused by a single se-quence type, ST91. The clone evades infection-induced immu-nity through serotype switching. Its epidemic capability was further enhanced by multidrug resistance including that to all first-line therapeutic drugs for treating shigellosis. These find-ings underscore the challenges to the current vaccine develop-ment and control strategies for shigellosis.
ACKNOWLEDGMENTS
This work was supported by grants (2005CB522904 and 2008DFA31830 to J.X.) from the Ministry of Science and Technol-ogy and by a grant from the State Key Laboratory for Infectious Disease Prevention and Control, People’s Republic of China.
REFERENCES
1.Adams, M. M., G. E. Allison, and N. K. Verma.2001. Type IV O antigen modification genes in the genome of Shigella flexneri NCTC 8296.
Micro-biology147:851–860.
2.Ahmed, A. M., K. Furuta, K. Shimomura, Y. Kasama, and T. Shimamoto.
2006. Genetic characterization of multidrug resistance in Shigella spp. from
Japan. J. Med. Microbiol.55:1685–1691.
3.Allison, G. E., and N. K. Verma.2000. Serotype-converting bacteriophages
and O-antigen modification in Shigella flexneri. Trends Microbiol.8:17–23.
4.Buchrieser, C., P. Glaser, C. Rusniok, H. Nedjari, H. D’Hauteville, F. Kunst, P. Sansonetti, and C. Parsot.2000. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella
flexneri. Mol. Microbiol.38:760–771.
5.Carlin, N. I., and A. A. Lindberg.1987. Monoclonal antibodies specific for Shigella flexneri lipopolysaccharides: clones binding to type IV, V, and VI antigens, group 3,4 antigen, and an epitope common to all Shigella flexneri
and Shigella dysenteriae type 1 strains. Infect. Immun.55:1412–1420.
6.Clemens, J. D., K. Kotloff, and B. A. Kay.1999. Generic protocol to estimate the burden of Shigella diarrhoea and dysenteric mortality, WHO/V&B/ 99.26. World Health Organization, Geneva, Switzerland.
7.Darling, A. C., B. Mau, F. R. Blattner, and N. T. Perna.2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements.
Genome Res.14:1394–1403.
8.Feng, L., P. R. Reeves, R. Lan, Y. Ren, C. Gao, Z. Zhou, Y. Ren, J. Cheng, W. Wang, J. Wang, W. Qian, D. Li, and L. Wang.2008. A recalibrated molecular clock and independent origins for the cholera pandemic clones.
PLoS One3:e4053.
9.Gordon, D., C. Abajian, and P. Green.1998. Consed: a graphical tool for
sequence finishing. Genome Res.8:195–202.
10.Guan, S., D. A. Bastin, and N. K. Verma.1999. Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX.
Microbiology145:1263–1273.
11.Heisig, P.1996. Genetic evidence for a role of parC mutations in develop-ment of high-level fluoroquinolone resistance in Escherichia coli.
Antimi-crob. Agents Chemother.40:879–885.
12.Hitchcock, P. J., and T. M. Brown. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained
poly-acrylamide gels. J. Bacteriol.154:269–277.
13.Ingersoll, M., E. A. Groisman, and A. Zychlinsky.2002. Pathogenicity islands
of Shigella. Curr. Top. Microbiol. Immunol.264:49–65.
14.Jennison, A. V., F. Roberts, and N. K. Verma. 2006. Construction of a
multivalent vaccine strain ofShigella flexneriand evaluation of
serotype-specific immunity. FEMS Immunol. Med. Microbiol.46:444–451.
15.Jennison, A. V., and N. K. Verma.2004.Shigella flexneriinfection:
patho-genesis and vaccine development. FEMS Microbiol. Rev.28:43–58.
16.Jin, Q., Z. Yuan, J. Xu, Y. Wang, Y. Shen, W. Lu, J. Wang, H. Liu, J. Yang, F. Yang, X. Zhang, J. Zhang, G. Yang, H. Wu, D. Qu, J. Dong, L. Sun, Y. Xue, A. Zhao, Y. Gao, J. Zhu, B. Kan, K. Ding, S. Chen, H. Cheng, Z. Yao,
on May 16, 2020 by guest
http://jcm.asm.org/
B. He, R. Chen, D. Ma, B. Qiang, Y. Wen, Y. Hou, and J. Yu.2002. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through com-parison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res.
30:4432–4441.
17.Lacher, D. W., H. Steinsland, T. E. Blank, M. S. Donnenberg, and T. S. Whittam.2007. Molecular evolution of typical enteropathogenic Escherichia coli: clonal analysis by multilocus sequence typing and virulence gene allelic
profiling. J. Bacteriol.189:342–350.
18.Levine, M. M.2006. Enteric infections and the vaccines to counter them:
future directions. Vaccine24:3865–3873.
19.Levine, M. M., K. L. Kotloff, E. M. Barry, M. F. Pasetti, and M. B. Sztein.
2007. Clinical trials of Shigella vaccines: two steps forward and one step back
on a long, hard road. Nat. Rev. Microbiol.5:540–553.
20.Lichtenstein, C., and S. Brenner.1982. Unique insertion site of Tn7 in the
E. coli chromosome. Nature297:601–603.
21.Nie, H., F. Yang, X. Zhang, J. Yang, L. Chen, J. Wang, Z. Xiong, J. Peng, L. Sun, J. Dong, Y. Xue, X. Xu, S. Chen, Z. Yao, Y. Shen, and Q. Jin.2006. Complete genome sequence of Shigella flexneri 5b and comparison with
Shigella flexneri 2a. BMC Genomics7:173.
22.Noriega, F. R., F. M. Liao, D. R. Maneval, S. Ren, S. B. Formal, and M. M. Levine.1999. Strategy for cross-protection amongShigella flexneriserotypes.
Infect. Immun.67:782–788.
23.Pryamukhina, N. S., and N. A. Khomenko.1988. Suggestion to supplement Shigella flexneri classification scheme with the subserovar Shigella flexneri
4c: phenotypic characteristics of strains. J. Clin. Microbiol.26:1147–1149.
24.Pupo, G. M., R. Lan, and P. R. Reeves.2000. Multiple independent origins
of Shigella clones ofEscherichia coliand convergent evolution of many of
their characteristics. Proc. Natl. Acad. Sci. U. S. A.97:10567–10572.
25.Ribot, E. M., M. A. Fair, R. Gautom, D. N. Cameron, S. B. Hunter, B. Swaminathan, and T. J. Barrett.2006. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7,
Salmonella, and Shigella for PulseNet. Foodborne Pathog. Dis.3:59–67.
26.Sansonetti, P. J.2006. Shigellosis: an old disease in new clothes? PLoS Med.
3:e354.
27.Simmons, D. A., and E. Romanowska.1987. Structure and biology of Shigella
flexneri O antigens. J. Med. Microbiol.23:289–302.
28.Stagg, R. M., P. D. Cam, and N. K. Verma.2008. Identification of newly recognized serotype 1c as the most prevalent Shigella flexneri serotype in
northern rural Vietnam. Epidemiol. Infect.136:1134–1140.
29.Sundstrom, L., and O. Skold.1990. The dhfrI trimethoprim resistance gene of Tn7 can be found at specific sites in other genetic surroundings.
Antimi-crob. Agents Chemother.34:642–650.
30.Tatusov, R. L., N. D. Fedorova, J. D. Jackson, A. R. Jacobs, B. Kiryutin, E. V. Koonin, D. M. Krylov, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, B. S. Rao, S. Smirnov, A. V. Sverdlov, S. Vasudevan, Y. I. Wolf, J. J. Yin, and D. A. Natale.2003. The COG database: an updated version includes
eu-karyotes. BMC Bioinformatics4:41.
31.Turner, S. A., S. N. Luck, H. Sakellaris, K. Rajakumar, and B. Adler.2003. Molecular epidemiology of the SRL pathogenicity island. Antimicrob.
Agents Chemother.47:727–734.
32.Turner, S. A., S. N. Luck, H. Sakellaris, K. Rajakumar, and B. Adler.2001. Nested deletions of the SRL pathogenicity island of Shigella flexneri 2a. J.
Bacteriol.183:5535–5543.
33.Venkatesan, M. M., M. B. Goldberg, D. J. Rose, E. J. Grotbeck, V. Burland, and F. R. Blattner.2001. Complete DNA sequence and analysis of the large
virulence plasmid of Shigella flexneri. Infect. Immun.69:3271–3285.
34.Verma, N. K., D. J. Verma, P. T. Huan, and A. A. Lindberg.1993. Cloning and sequencing of the glucosyl transferase-encoding gene from converting
bacteriophage X (SFX) of Shigella flexneri. Gene129:99–101.
35.von Seidlein, L., D. R. Kim, M. Ali, H. Lee, X. Wang, V. D. Thiem, D. G. Canh, W. Chaicumpa, M. D. Agtini, A. Hossain, Z. A. Bhutta, C. Mason, O. Sethabutr, K. Talukder, G. B. Nair, J. L. Deen, K. Kotloff, and J. Clemens.
2006. A multicentre study of Shigella diarrhoea in six Asian countries:
dis-ease burden, clinical manifestations, and microbiology. PLoS Med.3:e353.
36.Wang, X. Y., L. Du, L. Von Seidlein, Z. Y. Xu, Y. L. Zhang, Z. Y. Hao, O. P. Han, J. C. Ma, H. J. Lee, M. Ali, C. Q. Han, Z. C. Xing, J. C. Chen, and J. Clemens.2005. Occurrence of shigellosis in the young and elderly in rural China: results of a 12-month population-based surveillance study. Am. J.
Trop. Med. Hyg.73:416–422.
37.Wang, X. Y., F. Tao, D. Xiao, H. Lee, J. Deen, J. Gong, Y. Zhao, W. Zhou, W. Li, B. Shen, Y. Song, J. Ma, Z. M. Li, Z. Wang, P. Y. Su, N. Chang, J. H. Xu, P. Y. Ouyang, L. von Seidlein, Z. Y. Xu, and J. D. Clemens.2006. Trend and disease burden of bacillary dysentery in China (1991–2000). Bull. World
Health Organ.84:561–568.
38.Wei, J., M. B. Goldberg, V. Burland, M. M. Venkatesan, W. Deng, G. Fournier, G. F. Mayhew, G. Plunkett III, D. J. Rose, A. Darling, B. Mau, N. T. Perna, S. M. Payne, L. J. Runyen-Janecky, S. Zhou, D. C. Schwartz, and F. R. Blattner.2003. Complete genome sequence and comparative
genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun.71:
2775–2786.
39.West, N. P., P. Sansonetti, J. Mounier, R. M. Exley, C. Parsot, S. Guadag-nini, M. C. Prevost, A. Prochnicka-Chalufour, M. Delepierre, M. Tanguy, and C. M. Tang.2005. Optimization of virulence functions through
gluco-sylation ofShigellaLPS. Science307:1313–1317.
40.Yang, F., J. Yang, X. Zhang, L. Chen, Y. Jiang, Y. Yan, X. Tang, J. Wang, Z. Xiong, J. Dong, Y. Xue, Y. Zhu, X. Xu, L. Sun, S. Chen, H. Nie, J. Peng, J. Xu, Y. Wang, Z. Yuan, Y. Wen, Z. Yao, Y. Shen, B. Qiang, Y. Hou, J. Yu, and Q. Jin.2005. Genome dynamics and diversity of Shigella species, the
etio-logic agents of bacillary dysentery. Nucleic Acids Res.33:6445–6458.
41.Yoshida, H., M. Bogaki, M. Nakamura, and S. Nakamura.1990. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia
coli. Antimicrob. Agents Chemother.34:1271–1272.