UNIT 2.5
Production of Monoclonal Antibodies
Highly specific antibodies can be obtained by fusing immune B cells from the spleen with tumor cells to produce hybridomas, each of which will then secrete a single antibody. The desired antibody-producing hybridoma can be identified by a screening process. If this hybridoma is subjected to a cloning process in which clones are selected, such that all progeny are derived from a single cloned parental cell, a monoclonal antibody is obtained. Monoclonal antibodies have high specificity and can be produced in large quantities. Thus, these biological reagents have been used extensively as probes in a wide range of systems including the characterization of novel cell-surface and soluble proteins and carbohydrates, as enzyme catalysts, and for targeting in immunotherapy (see commentary).
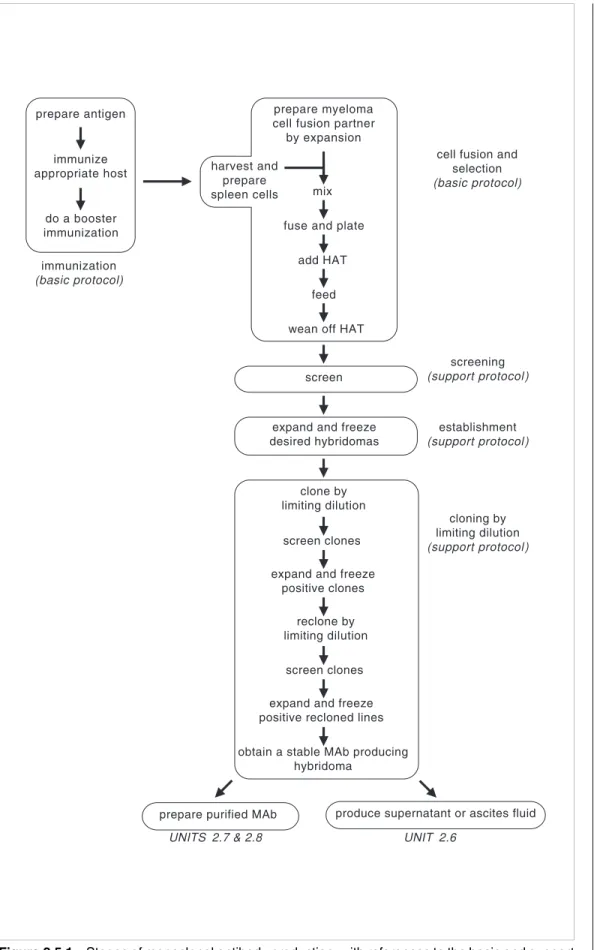
This unit describes the production of monoclonal antibodies beginning with basic proto-cols for immunization and cell fusion and selection.Support protoproto-cols are provided for screening primary hybridoma supernatants for antibodies of desired specificity, estab-lishment of stable hybridoma lines, cloning of these B cell lines by limiting dilution to obtain monoclonal lines, and preparation of cloning/expansion medium (thymocyte-con-ditioned medium). Figure 2.5.1 summarizes these stages and notes the protocols in this and subsequent units in which they are detailed. A major commitment of time and labor is necessary but, if successful, the monoclonal antibody may be an extremely valuable reagent that will be available in large quantities.
Submission of monoclonal antibodies to the American Type Culture Collection (ATCC) for distribution to the scientific community is encouraged. Moreover, the ATCC serves as a repository for cell lines should the line be lost in the investigator’s laboratory due to unforeseen circumstances.
NOTE: Sterile technique must be used in all procedures that utilize tissue culture cells.
BASIC PROTOCOL
IMMUNIZATION TO PRODUCE MONOCLONAL ANTIBODIES
A wide variety of antigen preparations have been used successfully to produce mono-clonal antibodies (see critical parameters discussing antigen preparation). The following protocol provides an immunization schedule for the production of most antibodies, although several different schedules can be used. In this protocol, emulsified antigen is injected intraperitoneally into the species of choice. A booster injection is administered 10 to 14 days after the primary immunization. Three days after the booster injection, the animals’ spleens are ready for cell fusion (second basic protocol).
Materials
Antigen
Complete Freunds adjuvant (CFA; Sigma)
Animal: pathogen-free mouse, hamster, or rat (Armenian hamsters from Cytogen Research are recommended; see critical parameters for discussion of animal choice and UNIT 1.1)
Incomplete Freunds adjuvant (IFA; Sigma), optional 1- to 2-ml glass syringes with Luer-Lok tips, sterile 3-way stopcock
20- and 22-G needles, sterile
Additional reagents and equipment for handling and restraint of animals (UNIT 1.3) and intraperitoneal injection (UNIT 1.6)
Contributed by Wayne M. Yokoyama Current Protocols in Immunology (1995) 2.5.1-2.5.17 Copyright © 1995 by John Wiley & Sonc, Inc. Supplement 13
2.5.1
Production of Monoclonal Antibodies
immunization (basic protocol) prepare antigen immunize appropriate host do a booster immunization harvest and prepare spleen cells screen
cell fusion and selection (basic protocol) screening (support protocol) establishment (support protocol) cloning by limiting dilution (support protocol) prepare myeloma cell fusion partner by expansion
mix fuse and plate
add HAT feed wean off HAT
expand and freeze desired hybridomas
clone by limiting dilution
screen clones expand and freeze
positive clones reclone by limiting dilution
screen clones expand and freeze positive recloned lines obtain a stable MAb producing
hybridoma
prepare purified MAb produce supernatant or ascites fluid
UNITS 2.7 & 2.8 UNIT 2.6
Figure 2.5.1 Stages of monoclonal antibody production, with references to the basic and support
protocols in this unit (as well as subsequent units) that describe the steps.
Current Protocols in Immunology
2.5.2
Induction of Immune Responses
CAUTION: CFA is an extremely potent inflammatory agent, particularly if introduced intradermally or into the eyes. Profound sloughing of skin or loss of sight may occur. Self-injection can cause a positive TB skin test and lead to a granulomatous reaction. Use gloves and protective eyewear when handling CFA.
1. Prepare antigen using 2 × 106 to 5 × 107 cells or 1 to 50 µg protein or peptide per animal to be immunized in normal saline.
The antigen may be in several different forms depending on the desired property of the MAb and the method of screening (see critical parameters for discussion of antigen preparation and screening assays). If cells are the immunogen, wash three times in serum-free medium before immunization. Plan the immunization of several animals (enough for several fusions) so that primed and boosted animals will be ready 3 days before fusion (see second basic protocol).
To minimize the risk of introducing a pathogen into the rodent colony, screen cells for pathogens by antibody-production assay (UNIT 1.1).
2. Draw up antigen into a sterile 1- to 2-ml glass syringe with a Luer-Lok tip. Connect syringe to a 3-way stopcock.
3. Completely resuspend CFA to disperse the Mycobacterium tuberculosis bacilli which settle to the bottom of the container with time. Draw up a volume of CFA equal to the antigen volume in a syringe and connect to the antigen-containing syringe. 4. Emulsify antigen and CFA by discharging antigen into CFA, then discharging back
and forth until a thickened mixture results. Test whether the emulsion is stable—a stable emulsion will not disperse when a drop of it is placed in water.
See UNIT 2.4 for further discussion of immunization. Figure 2.4.1 illustrates the double-syr-inge device.
5. Transfer all of the CFA/antigen emulsion to one syringe and remove the other syringe and stopcock. Attach a sterile 20-G needle to the syringe containing the emulsion. 6. Inject emulsion intraperitoneally into the animal using <0.2 ml/mouse, 0.5 to 1 ml/rat,
or 0.2 to 0.4 ml/hamster.
Be careful not to force the syringe plunger since excessive pressure may dislodge the needle and spray the emulsion. Introduce the needle through the skin and tunnel the needle between the skin and peritoneal wall before entering the peritoneal cavity at a site distant from the dermal puncture site. Twirl needle before withdrawal to minimize leakage.
Rats are generally anesthetized (UNIT 1.4) whereas mice and hamsters can be manipulated with one hand and do not require anesthetic.
7. Boost animal after 10 to 14 days with approximately the same dose of antigen as in step 5. If cell fusion is planned for 3 days after boosting, immunize with antigen alone in aqueous solution, or intact cells in suspension. If a fusion is not immediately planned, boost the animal with antigen emulsified in IFA (which does not contain Mycobacterium tuberculosis bacilli).
Do not use CFA for the booster immunizations as this will cause intense inflammation and increased anti-TB antibody response.
If desired, antibody titers can be assayed by ELISA (UNIT 2.1) or immunoprecipitation (UNIT 8.3), 7 to 10 days after the primary and booster immunizations.
Current Protocols in Immunology
2.5.3
Production of Monoclonal Antibodies
BASIC PROTOCOL CELL FUSION AND SELECTION OF HYBRIDOMAS
While animals should be immunized as soon as the decision has been made to produce a monoclonal antibody and the antigen prepared, do not perform cell fusion until the screening assay (first support protocol) has been perfected. Artifactual results that may arise from conditioned media must be identified before cell fusion, because after a fusion there is only a finite amount of time available to assay for the desired monoclonal antibody. Prior to cell fusion, the partner (myeloma) cell line is expanded and a booster injection of antigen is administered to the primed animals. On the day of fusion, the spleens are harvested. Spleen cells and partner cells are washed, harvested, and mixed. Cell fusion is performed at 37°C in the presence of polyethylene glycol (PEG). The resulting pellet is harvested and plated into tissue culture plates. After incubation with hypoxanthine, aminopterin, and thymidine (HAT) medium and feeding over ∼2 weeks, the hybridomas are ready for screening (first support protocol).
Materials
SP2/0-Ag14 myeloma cell line (drug-marked, nonsecretory; ATCC #CRL 1581) Complete DMEM-10 and -20 media (APPENDIX 2) with 10 mM HEPES and
1 mM sodium pyruvate
Primed animal; mouse, hamster, or rat (10 to 14 days after primary immunization; see first basic protocol)
Complete DMEM medium (APPENDIX 2), serum-free 50% polyethylene glycol (PEG), sterile
Ammonium chloride solution
Complete DMEM-20 (APPENDIX 2) with 10 mM HEPES, 1 mM sodium pyruvate, and 1× HAT or 1× HT supplement
175-cm2 flasks
Fine-mesh metal screen
50-ml conical polypropylene centrifuge tubes Beckman TH-4 rotor or equivalent
96-well flat-bottom microtiter plates
Additional reagents and equipment for animal euthanasia (UNIT 1.8), spleen removal (UNIT 1.9), and counting cells and assessing cell viability by Trypan blue exclusion (APPENDIX 3)
Prepare myeloma cells (one week before fusion)
1. One week before fusion, begin expansion of SP2/0-Ag14 myeloma cell line (the fusion partner cell line) in complete DMEM-10/HEPES/pyruvate (see critical pa-rameters). By the day cell fusion is to be performed, the following total number of myeloma cells must be available (in multiple 175-cm2 flasks containing 100 ml each), depending upon the source of the primed animal: mouse spleen, 1 × 108 cells in two or three flasks; hamster spleen, 2 × 108 cells in three or four flasks; and rat spleen, 5-10 × 108 cells in ten flasks.
Two mouse or hamster spleens, or one rat spleen, will provide enough cells for the fusion (see step 7).
Boost primed animal (three days before fusion)
2. Three days before fusion, boost primed animal(s) according to step 7 of the first basic protocol.
Prepare reagents and split myeloma cells (one day before fusion)
3. One day before fusion, prepare all reagents and media, particularly 50% PEG.
Current Protocols in Immunology Supplement 3
2.5.4
Induction of Immune Responses
4. One day before fusion, split SP2/0-Ag14 myeloma cells (from step 1) into fresh complete DMEM-10/HEPES/pyruvate medium.
Vigorous growth of the SP2/0-Ag14 cells is generally required for good fusion.
Check myeloma cells and prewarm reagents (day of fusion)
5. Use an inverted microscope to check the SP2/0-Ag14 myeloma cells to make sure they are growing vigorously (refractile and not pyknotic), they are not contaminated (no obvious bacteria or fungi), and there are enough cells for the fusion.
It is better to postpone the fusion than to perform an ill-advised fusion, since the entire selection and screening effort will take ∼3 weeks.
6. Prewarm the following in a 37°C water bath:
Three 400- and three 600-ml beakers, each containing ∼100 ml H2O 20 ml sterile complete serum-free DMEM
5 ml sterile 50% PEG solution.
Harvest spleen and prepare cells
7. Sacrifice boosted animal(s) and aseptically harvest spleen(s).
Do not use anesthetics for sacrifice. Instead, use cervical dislocation for mouse, or CO2 asphyxiation for mouse, hamster, or rat to avoid introducing an anesthetic into the blood stream and therefore, into the cultures.
8. Transfer spleen to a sterile 100-mm-diameter petri dish filled with 10 ml sterile complete serum-free DMEM.
Perform all subsequent steps in a laminar flow hood.
9. Tease spleen into a single-cell suspension by squeezing with angled forceps or by chopping with fine-tipped dissecting scissors. Remove debris and disperse cells further by passage through a fine-mesh metal screen.
10. Transfer spleen cell suspension to a sterile 50-ml conical centrifuge tube and fill with sterile complete serum-free DMEM.
Do not use protein- or HEPES-containing medium because the PEG will precipitate proteins and HEPES can be toxic to cells during fusion.
11. Centrifuge 5 min in TH-4 rotor at 1500 rpm (500 ×g), room temperature, and discard supernatant.
12. Lyse red blood cells (RBC) by resuspending pellet in 5 ml ammonium chloride solution. Let stand 5 min at room temperature.
13. Add 45 ml sterile complete serum-free DMEM, and centrifuge as in step 11. 14. Resuspend pellet in 50 ml sterile complete serum-free DMEM. Centrifuge as in step
11. Repeat DMEM addition and centrifuging once (each repeat is a wash).
15. While spleen cells are being washed, separately harvest the SP2/0-Ag14 myeloma cells (from step 5) by transferring the cells to 50-ml conical centrifuge tubes. Centrifuge as in step 11. Resuspend myeloma cells in DMEM and pool all cells into one 50-ml conical centrifuge tube. Wash myeloma cells three times as in step 14. 16. Separately resuspend the spleen and myeloma cells in 10 ml complete serum-free
DMEM. Count cells and assess viability in each cell suspension using a hemacytome-ter and Trypan blue exclusion; there should be nearly 100% viability of both suspensions.
Supplement 3 Current Protocols in Immunology
2.5.5
Production of Monoclonal Antibodies
17. On basis of cell counts (from step 16), calculate the amount of complete DMEM-20/HEPES/pyruvate needed to plate cells at ∼2.5 × 106 total cells/ml. Prewarm this amount of complete DMEM-20/HEPES/pyruvate in 37°C water bath. Prepare 96-well flat-bottom plates by labeling them sequentially: one plate is required for each 10 ml of final cell suspension.
Perform cell fusion
18. Mix SP2/0-Ag14 myeloma and spleen cells at a 1:1 ratio in a 50-ml conical centrifuge tube. Fill the tube with complete serum-free DMEM.
Other cell ratios work. Successful fusions have been performed with a ratio of myeloma/spleen cells as low as 1:20.
19. Centrifuge cell mixture 5 min at 500 ×g, room temperature.
20. While cells are in the centrifuge, prepare three 37°C double-beaker water baths in the laminar flow hood by placing a 400-ml beaker (from step 6) containing 100 ml of 37°C water into 600-ml beaker containing 75 to 100 ml of 37°C water. Place the tubes of prewarmed 50% PEG solution and complete serum-free DMEM (from step 6) into two of the 37°C water baths in the hood.
21. Aspirate and discard supernatant from the mixed-cell pellet (from step 19).
22. Perform the cell fusion at 37°C by placing the tube containing the mixed-cell pellet in one of the double-beaker water baths in the laminar flow hood.
23. Using a 1-ml pipet, add 1 ml prewarmed 50% PEG to the mixed-cell pellet drop-by-drop over 1 min, stirring the cells with the pipet tip after each drop-by-drop. Stir for an additional minute.
24. Using a clean pipet, add 1 ml prewarmed complete serum-free DMEM to the cell mixture drop-by-drop over 1 min, stirring after each drop. Repeat once with an additional 1 ml of prewarmed complete serum-free DMEM.
25. With a 10-ml pipet, add 7 ml prewarmed complete serum-free DMEM drop-by-drop over 2 to 3 min.
Macroscopic clumps of cells should be obvious at this point. 26. Centrifuge 5 min at 500 ×g, room temperature.
27. While the cells are in the centrifuge, rewarm the beaker water baths to 37°C and place in the hood. Place prewarmed complete DMEM-20/HEPES/pyruvate (from step 17) in the beaker water bath.
28. Discard the supernatant (from step 26). Place tube in the beaker water bath.
29. With a clean 10-ml pipet, forcefully discharge 10 ml prewarmed complete DMEM-20/HEPES/pyruvate to the cell pellet.
30. Repeat step 29 until the total volume of prewarmed complete DMEM-20/HEPES (calculated in step 17) is added. If necessary, allow clumps to settle and disrupt with the pipet tip. Further warming of cell suspension is no longer required.
If the total volume exceeds 50 ml, gently aspirate and transfer to another sterile container such as a tissue culture flask.
31. Gently aspirate 10 ml of cell suspension with a 10-ml pipet. Add 2 drops (100 to 125 µl) of suspension to each well of a 96-well flat-bottom plate (continue until entire suspension is plated). Incubate overnight in a humidified 37°C, 5% CO2 incubator.
Current Protocols in Immunology
2.5.6
Induction of Immune Responses
Vigorous pipetting of the cell suspension should be avoided at this point, as the newly formed hybrids are unstable. Moreover, the vigorous addition of cells to the wells with repeating micropipettor is not advised. Use a pipet aid and hold the 10-ml pipet at a 45° angle with the tip 1 to 2 cm above the well, bracing the pipet with a finger from the opposite hand. To avoid introducing contaminants, do not hold hands above the plate. A steady, even flow of drops from the pipet will allow the most efficient delivery of cell suspension or medium to the wells. Use a fresh pipet to withdraw additional cell suspension.
As an optional step to minimize fibroblast overgrowth, permit the fibroblasts in bulk-fused cell suspension to adhere overnight to tissue culture flasks before seeding the 96-well plates.
Many investigators select their hybridomas under bulk conditions—i.e., they incubate large numbers of cells per well in larger plates or flasks. This makes feeding easier, but allows fast-growing hybridomas to overgrow the others. Since nonproducing hybridomas tend to grow faster, especially in the hamster-mouse fusions, hybridomas are isolated initially in multiple small wells in this protocol. The primary hybridomas tend to be monoclonal. This is especially important when screening procedures are used that require differential reactivities, e.g., to different cell lines by flow cytometry analysis or to different antigen preparations. In those cases, multiple hybridomas per well will obscure the reactivity of the MAb of interest.
Monitor and feed cells
32. After one day of incubation, check wells under an inverted microscope. If seeded with the appropriate number of cells, there should be a nearly confluent monolayer of highly viable cells on the bottom and obvious clumps of cells.
33. Add 2 drops complete DMEM-20/HEPES/pyruvate/HAT to each well with a 10-ml pipet (see step 31). Place in humidified 37°C, 5% CO2 incubator.
Use a separate pipet for each microtiter plate and keep the same covers with each plate to ensure that each plate remains a separate unit and to avoid spreading contamination. It cannot be overemphasized that it takes practice and meticulous attention to possible sources of contaminants to keep these plates sterile during the subsequent 2-to 3-week feeding and monitoring schedule.
If plates become contaminated, discarding them is advised. Alternatively, contamination in one or two wells may be treated by aspirating the contents of the contaminated well with a sterile Pasteur pipet attached to a vacuum flask, rinsing the well with 70% ethanol, and wiping with a sterile cotton swab. Wash twice with ethanol. Finally, blot the well dry with the sterile cotton swab and blot the appropriate area of the cover with a sterile cotton swab soaked in 70% ethanol. Do not open contaminated plates while other plates are in the hood. 34. On days 2, 3, 4, 5, 7, 9, and 11, aspirate half the volume of each well using a sterile, short Pasteur pipet attached to a vacuum flask, holding pipet at a 45° angle and touching tip to surface of supernatant at the point where the liquid meets the opposite wall of the well. Feed the cells by adding 2 drops complete DMEM-20/HEPES/pyru-vate/HAT from a 10-ml pipet (see steps 31 and 33) to each well, and return to humidified 37°C, 5% CO2 incubator.
Use a separate Pasteur pipet for each plate to minimize spreading contamination. Since the frequency of successful viable hybridoma formation is ≤10−5, when HAT is added, profound cell death should be apparent at days 2 and 3 and the remaining viable cells should not be readily apparent until they have expanded. By day 7 to 9 for mouse-mouse fusions, day 11 for rat-mouse fusions, and day 14 for hamster-mouse fusions, clusters of hybridoma cells should become visible under the inverted microscope. If profound cell death is not apparent on days 2 and 3, check the medium containing HAT and the parental cell line by incubating an aliquot of the parental myeloma line with the medium containing HAT.
Current Protocols in Immunology
2.5.7
Production of Monoclonal Antibodies
The feeding schedule is not rigid except for the first 4 days, when it is necessary to remove the toxic products of cell death. Thereafter, feedings will depend on the actual number of cells deposited in the wells, efficiency of fusion, and appearance and growth of hybridomas. Do not allow wells to become yellow (acidic) for more than a day. Examine plates daily, even if cells are not scheduled to be fed, and feed plates if acidic wells are noted. 35. On day 14, repeat feeding protocol outlined in step 34 except use complete
DMEM-20/HEPES/pyruvate/HT to feed cells. Return to 37°C, 5% CO2 incubator.
Cells do not require more than one change of complete DMEM-20/HEPES/pyruvate/HT. After this change, the aminopterin (from prior addition of HAT medium) is apparently diluted out enough so that the cells can survive without additional HT.
36. On day 15 and subsequently, feed wells as noted using complete DMEM-20/HEPES/pyruvate without HAT or HT. The hybridomas are ready for screening when most of the wells containing growing cells demonstrate 10% to 25% confluence and when those with denser populations turn yellow within 2 days after feeding (see first support protocol).
If the screening assay requires a [3H]thymidine incorporation assay (APPENDIX 3), be aware
that the large amount of thymidine in complete DMEM-20/HEPES/pyruvate with HAT and HT will serve as a cold-label inhibitor of [3H]thymidine incorporation. At least 3 to 4
changes of complete DMEM-20/HEPES/pyruvate without HT are required to dilute out excess thymidine.
SUPPORT PROTOCOL SCREENING PRIMARY HYBRIDOMA SUPERNATANTS
The vast majority of wells will not contain the desired antibody or may contain nonpro-ducing hybridomas. The purpose of screening is to discover which wells (<1% to 5%) contain hybridomas that secrete the antibody of desired specificity (note that the antibody is not yet monoclonal.) Screening should be performed when most of the growing wells demonstrate 10% to 25% confluence when viewed with an inverted microscope or when some of the denser wells begin to turn yellow within 2 days after feeding. This point may be reached 10 to 14 days after a mouse-mouse or rat-mouse fusion, and 14 to 21 days after a hamster-mouse fusion. Although the most dense wells can be screened first and the less dense wells when they become more dense, this duplicates the effort required and is not recommended. Thus, the wells are fed and 2 days later, aliquots of the supernatants are tested in the screening assay for the presence of the desired antibody.
Additional Materials
Growing hybridomas (second basic protocol)
Additional reagents and equipment for ELISA (UNIT 2.1) and indirect immunofluorescence (UNIT 5.3)
1. Estimate the number of wells with growing hybridomas using an inverted microscope. Determine whether it will be more efficient to screen all wells or only the wells that contain hybridomas.
2. Allow hybridomas in the viable wells to grow in a humidified 37°C, 5% CO2 incubator without feeding for ≥2 days.
This is usually enough time to build a saturating titer of antibody in the culture supernatant. 3. Remove 100 µl from each well to be tested and use in a screening assay, such as an
ELISA or indirect immunofluorescence (see critical parameters).
Current Protocols in Immunology
2.5.8
Induction of Immune Responses
A micropipettor with disposable sterile tips is convenient. Use a new pipet tip with each well. If all wells are screened, a multichannel pipet is convenient for transfer to another 96-well assay plate.
Keep track of the plate number and well by its row letter and column number for each sample. This is frequently the origin of the MAb’s name.
4. After harvesting one entire plate, feed wells with fresh complete DMEM-20/HEPES before harvesting the next plate (see step 33 of the second basic protocol).
SUPPORT PROTOCOL
ESTABLISHMENT OF HYBRIDOMA LINES
Once candidate hybridomas are identified (see previous support protocol), they are expanded and fed, then the cells are both frozen and cloned by limiting dilution (see following support protocol). Unfortunately, this must be done for all candidate lines before the monoclonal antibody specificity is fully characterized. This additional work ensures that viable antibody-producing hybridomas are available after the screening process. To limit the amount of extra work involved, select the twenty best candidate wells. All twenty should be frozen and limiting-dilution plates set up while the supernatants are checked.
Additional Materials
Growing hybridomas (second support protocol) Cloning/expansion medium (fourth support protocol) 24-well microtiter plates
Additional reagents and equipment for cryopreservation of cells (APPENDIX 3) 1. When the growing hybridoma is 25% to 50% confluent in the 96-well plate (master
well), expand the candidate hybridoma to a well in a 24-well plate by resuspending the cells in the master well with a sterile pipet (a micropipettor set at 100 µl is convenient). Transfer the entire contents of the master well to a well in the 24-well plate.
Sufficient numbers of cells will remain to serve as backup to the expanded cells.
If a small number of cells are transferred, they may not expand. However, if there are fibroblasts in the master well, the hybridoma cells should be transferred as soon as possible to avoid fibroblast overgrowth in the well (if necessary, transfer to another 96-well plate). When transferring cells from a well with fibroblasts, be careful not to scrape the bottom of the well (which will loosen the fibroblasts).
2. Feed cells in the master well with 3 drops complete DMEM-20/HEPES/pyruvate from a 1-ml pipet (see step 33 of the second basic protocol). Incubate cells in humidified 37°C, 5% CO2 incubator.
3. Use a fresh pipet and feed cells in 24-well plate with 1 to 1.5 ml cloning/expansion medium. Incubate 2 to 3 days in humidified 37°C, 5% CO2 incubator.
Remember that there are only two wells in the world that contain this hybridoma and that each is a backup to the other in case of contamination or other undesirable circumstance. Therefore, once established, each is treated as an individual entity and fed with media from different bottles and different pipets.
4. When cells in the 24-well plate are 25% to 50% confluent (2 to 3 days), they are ready to be used in the third support protocol (cloning by limiting dilution).
Repeat steps 1 to 3 as necessary when cells are 25% to 50% confluent in master well. 5. After taking cells for limiting-dilution cloning, transfer remainder of cells in the
24-well plate to a 4-ml sterile capped tube and feed the cells in the 24-well plate with
Current Protocols in Immunology
2.5.9
Production of Monoclonal Antibodies
complete DMEM-20/HEPES/pyruvate.
6. Centrifuge the cells in the 4-ml tube 5 min at 500 × g, room temperature. Keep supernatant for further characterization of the antibody and as a control, and freeze cell pellet (APPENDIX 3).
The supernatant usually contains sufficient antibody that could be reassayed in the original screening test and/or in any confirmatory test. The cell line is not established until stable clones can be identified, frozen, and successfully thawed. If the limiting-dilution plate does not yield an antibody-producing line, the original cells from the 24-well plate can be thawed, seeded back into a 24-well plate, grown overnight, used to seed another series of limiting-dilution plates, and refrozen for safekeeping.
SUPPORT PROTOCOL CLONING BY LIMITING DILUTION
Monoclonal antibodies are secreted by the progeny of a single cell that can produce only a single antibody (assuming a nonsecretory fusion-partner line). Cloning is required to ensure that the problems of polyspecificity are avoided and the risk of overgrowth by nonproducing cells minimized. Although cloning can also be performed by the soft-agar technique, clones derived by this technique must be adapted to liquid culture before the supernatants can be tested (Coffino et al., 1972). Since cloning by limiting dilution allows direct testing of the supernatants, this method is much more advantageous (hence, the soft-agar technique will not be discussed here).
Additional Materials
Candidate hybridoma line (second support protocol)
1. Resuspend the candidate hybridoma line from step 4 of the second support protocol in their wells and count and assess viability of a small aliquot (50 µl) of cells using a hemacytometer and Trypan blue (APPENDIX 3).
2. Prepare 10 ml of cells at 50 viable cells/ml and 10 ml at 5 viable cells/ml in cloning/expansion medium.
The degree of dilution is usually very large and thus serial dilution may be needed. 3. Seed a 96-well plate with the cell suspensions at 200 µl/well. Incubate 7 to 10 days
in a humidified 37°C, 5% CO2 incubator.
There will be enough to seed half of the wells in the plate with each dilution at final concentrations of 10 cells/well and 1 cell/well.
4. Determine which dilution was optimal for monoclonal growth by determining the number of wells that show growing hybridomas.
Usually hybridoma growth is obvious by macroscopic visualization of the well bottoms. A microplate reading mirror (Flow Laboratories or Dynatech) is useful to avoid the necessity of holding the plate above one’s head.
Poisson statistics indicate that if <22% of the wells have growing cells (the proportion expected if the cells were seeded at 0.3 per well and had a cloning efficiency of 100%), then 88% of these wells have only one clone. However, primary hybridomas generally have a poor “plating efficiency” and thus, more cells need to be seeded in each well to derive a reasonable number of growing clones. Use the wells seeded with 1 cell/well if there are “growing” wells and, if necessary, the wells seeded with 10 cells/well. The frequency of antibody-producing clones is dependent on the time after the initial testing that the cloning plates were set up and the MAb species. Generally, the longer the wait before plating the primary hybridomas and the more phylogenetically distant from the mouse the source of spleen cells, the less frequent the positive clones.
Current Protocols in Immunology
2.5.10
Induction of Immune Responses
5. Inspect wells for monoclonality with an inverted microscope before feeding cells, looking for tight single clusters of cells as evidence of monoclonal growth. Polyclonal growth is evidenced by more than one cluster of cells; if possible, do not use these wells.
6. Use the screening assay that was used in the initial identification of the master well (see critical parameters) to test the monoclonal wells for desired antibody activity on day 7 to 14. Use an aliquot of the original hybridoma supernatant (from step 6, second support protocol) as a positive control.
Mouse-mouse hybridomas could be checked as early as 7 days after plating, whereas hamster-mouse hybridomas may require up to 14 days for growth. If any of the wells begin to turn yellow, they should be tested. Since the wells were not fed, any well with growing cells should have readily detectable antibody if the cells produce the desired MAb. Wait 2 days before testing if the wells were fed.
7. When the desired clone is identified, expand and freeze the well as for the primary hybridoma in the second support protocol.
8. Reclone one of the positive hybridoma clones as in steps 1 to 3; seed two new 96-well plates at 0.3 cells/well (60 viable cells in 40 ml cloning medium).
9. Repeat steps 4 to 6.
10. Expand and freeze the recloned hybridoma as in the second support protocol. 11. Wean the recloned cells to complete DMEM-10/HEPES/pyruvate by splitting the
cells 1:2 every day (for 3 days) with complete DMEM-10/HEPES/pyruvate. At this point, the desired hybridoma should be stable as a cell line.
Freeze multiple vials on different days with different aliquots of freezing medium (APPENDIX 3). Thaw representative vials and check cells for growth and supernatant for MAb activity in appropriate assay. The hybridoma can then be used for ascites production and for large-scale production of hybridoma supernatants (UNIT 2.6). The isotype of the MAb can now be determined (UNIT 2.2).
Even recloned hybridomas have an unstable phenotype, especially some hamster-mouse hybridomas, which may require additional recloning. Prolonged cultures in vitro may result in loss of MAb production. To minimize this problem, frozen aliquots of cells known to produce the MAb are necessary and should be verified as sources of viable cells.
An occasional cloned hybridoma will not tolerate complete DMEM-10/HEPES/pyruvate and will require higher concentrations of FCS. It may be necessary to wean cells first to DMEM-15 before weaning to DMEM-10. Mycoplasma contamination should be consid-ered (see APPENDIX 3).
SUPPORT PROTOCOL
PREPARATION OF CLONING/EXPANSION MEDIUM
Many investigators add feeder cells (i.e., peritoneal washout cells, splenocytes, or thymo-cytes) to produce conditioned media that seem to enhance hybridoma growth and cloning. The direct addition of irradiated, freshly isolated cells to wells, however, sometimes results in contamination. Therefore, cell-free, sterile-filtered supernatants of all suspen-sions are recommended to enhance hybridoma cloning efficiencies. The following pro-cedure describes the preparation of a cell-free thymocyte-conditioned medium from mice. After sacrificing several mice and removing each thymus, a single cell suspension is prepared and grown for several days. The supernatant is harvested, filter sterilized, and stored at −20°C.
Current Protocols in Immunology
2.5.11
Production of Monoclonal Antibodies
1. Sacrifice 5 or 6 mice, avoiding use of anesthetics (see annotation to step 7 of the second basic protocol).
Mice (e.g, BALB/c) should be 4 to 6 weeks old. Obtain pathogen-free mice from a reliable supplier that screens for mycoplasma contamination.
2. Aseptically remove the thymus (UNIT 1.9). Tease thymus into a single-cell suspension as in steps 8 to 11 of the second basic protocol. Resuspend cells in 20 ml of complete DMEM-20/HEPES/pyruvate.
3. Add 10 ml thymus cells to a 75-cm2 flask. Add complete DMEM-20/HEPES/pyruvate medium to final amount of ∼20 ml medium per thymus (maximum 60 ml cell suspension/flask). Incubate flask 4 to 5 days in upright position in a humidified 37°C, 5% CO2 incubator.
4. Transfer suspension to a sterile 50-ml conical centrifuge tube. Centrifuge suspension 5 min at 1000 ×g, room temperature, and harvest supernatant.
5. Filter sterilize supernatant through 0.45-µm filter. Freeze at −20°C in 10-ml aliquots. 6. Thaw and use at 10% to 20% final concentration in desired medium.
An alternative to feeder cells and thymocyte-conditioned medium is the use of a source of IL-6 (plasmacytoma growth factor), such as liposaccharide-stimulated P388D1 super-natant (UNIT 6.6).
REAGENTS AND SOLUTIONS Ammonium chloride solution
0.02 M Tris⋅Cl, pH 7.2 0.14 M NH4Cl
Complete DMEM-20/HEPES/pyruvate/HAT (or HT)
To complete DMEM-20 medium (APPENDIX 2) containing 10 mM HEPES and 1 mM pyruvate, add 100× HAT (hypoxanthine/aminopterin/thymidine) or 100× HT sup-plement to 1× final. Store at 4°C for up to 1 month.
100× HAT and 100× HT supplement are available commercially (e.g., Sigma).
50% polyethylene glycol (PEG)
Autoclave 10 g PEG 4000 (Merck or ATCC) in a Wheaton glass bottle and cool. Before it solidifies (at ∼55°C), add 10 ml complete serum-free DMEM. This makes enough for ∼20 fusions. The solution may be kept at room temperature for several months; it will become alkaline but this does not affect its performance.
Be sure not to use protein-containing medium because PEG precipitates proteins.
COMMENTARY
Background Information
Since the original description of a technique to produce monoclonal antibodies (MAb) of defined specificity (Köhler and Milstein, 1975), MAbs have proven to be powerful tools to analyze a myriad of biological systems. As a testimony to the broad utility of this tech-nique, the original authors noted above re-ceived the Nobel Prize for medicine in 1984. Furthermore, a search of Medline (computer-ized medical literature data) for 1988 to 1989
shows that more than 8700 citations referred to MAb.
To produce MAb, a suitable host is im-munized with an antigen and antibody-secret-ing B cells are immortalized by the fusion of the host immune B cells with a nonsecretory, drug-marked myeloma cell line. Since the un-fused normal B cells cannot survive long in an in vitro culture, they derive immortality by fusion to a partner tumor cell line. The tumor line is resistant to the purine analogue
6-Current Protocols in Immunology Supplement 3
2.5.12
Induction of Immune Responses
thioguanine because of deficiency of hypoxan-thine-guanine phosphoribosyl transferase (HGPRT). This deficiency results in lethal sen-sitivity to aminopterin, which blocks de novo synthesis of purines. The normal B cell is not sensitive to aminopterin when hypoxanthine and thymidine are supplied; salvage pathways utilizing HGPRT are necessary for survival. Thus, only hybridomas (normal B cells fused to tumor cell) will survive selection in HAT. If a hybridoma that produces the antibody of interest is identified and subjected to a cloning procedure that results in monoclonality (i.e., derivation of progeny cells from a single cell), a MAb of desired specificity is produced.
Although monoclonal antibodies are pow-erful tools, it cannot be overemphasized that a major commitment is necessary to identify a MAb of interest. The work is tedious and labor intensive. Moreover, in some cases, specific polyclonal antisera, which are generally easier to produce, may be as suitable as—and in some cases superior to—MAb. For example, specific antisera against defined synthetic peptides that will immunoprecipitate cell-surface antigens of interest can be made. Such antisera frequently will be useful in immunoblots whereas MAb, unless directly screened for such a purpose, may not recognize determinants on denatured antigens. Nevertheless, if the desired MAb is produced, this reagent has a wide variety of uses, particularly for the characterization of novel molecules, and as a specific antagonist or agonist of ligand-receptor interactions. More-over, large quantities of the MAb can be readily purified.
There is a large body of literature on MAb production. More specific elaboration of the methods described here are discussed by Goding (1986) and Harlow and Lane (1988). Furthermore, many reports describing refine-ments of the basic technique are frequently published in the Journal of Immunological Methods. With general basic skills in animal handling, tissue culture, and screening as-says, the novice should be able to produce MAb using the protocols in this unit. The major obstacle, as with any screening effort, is identification of the MAb of desired speci-ficity. Because the technique produces many MAb– secreting hybridomas, the temptation exists to keep all or selected hybridomas even though they are not producing the MAb of initial interest. A large number of hybridomas secreting antibodies to unique antigens may be isolated, but the extra effort required is considerable. Nevertheless, it is a strategy
that may prove useful while the attempt to identify the desired MAb proceeds. Many an-tibodies described in the literature are such by-products.
Critical Parameters
There are several major factors, other than technical, that should be considered before any MAb production project is begun.
While several fusion partners are available, the SP2/0-Ag14 myeloma is a good general-purpose cell line. The best sources of the cell line are ATCC or a laboratory actively produc-ing hybridomas. Some sublines do not fuse well, perhaps due to some genetic variation or to undetected contaminants such as myco-plasma.
The SP2/0-Ag14 cells should grow in sus-pension with minimal adherence to the tissue culture flask. The cells should not be over-grown and the medium should not become acidic (yellow). Healthy cells will be refrac-tile and none of the cells should be pyknotic when viewed with an inverted microscope. In general, the cells should be diluted (split) into fresh medium (≥1:5 if the cells will be used on the following day) when the cells in an undisturbed flask form a monolayer on the bottom of the flask (1-2 × 106 cells/ml). If the cells do not appear healthy even when grown at lower densities, mycoplasma contamination should be suspected.
Freeze multiple large aliquots of cells ( AP-PENDIX 3) so that only a few days of expansion
will be necessary for future fusions. If planned correctly, a primed animal can be boosted, cells thawed on the same day, and a fusion performed 3 days later.
Choice of animal for immunization
The first basic protocol on immunization outlines the production of MAb by in vivo immunization and concentrates on the use of rodents for immunization. Specialized work may require the use of splenic injections, in vitro immunizations, or human monoclonals; these techniques are much more difficult to implement successfully and are not recom-mended for the novice. Other protocols de-signed to eliminate unwanted MAb against particularly immunogenic epitopes, involve the use of cytotoxic agents immediately after the initial immunization with the unwanted anti-gens and should be considered only after some experience has been gained (Sharpe et al., 1985).
Four animal species (mouse, rat, hamster,
Supplement 3 Current Protocols in Immunology
2.5.13
Production of Monoclonal Antibodies
and rabbit) can be used for MAb production. The spleen cells from a given species must be able to produce stable MAb-producing hybri-domas with a rodent myeloma cell line. Al-though several rat and mouse tumor cell part-ners exist and all have been successfully used, the SP2/0-Ag14 mouse cell line from ATCC is recommended because it is a drug-marked, nonsecretory myeloma that does not constitu-tively produce either light or heavy chains. Therefore, hybridomas derived from fusion to SP2/0-Ag14 will not make chimeric MAb. This cell line also forms stable hybridomas with mouse, rat, and hamster B cells. Rabbit MAb have recently been described (Raybould and Takahashi, 1988) but little collective experi-ence has accumulated.
It is technically more difficult to produce stable MAb-producing hybridomas from ani-mals that are phylogenetically distant from the mouse. First attempts at producing MAb should use either the mouse or rat. Of the two, the mouse is the best choice for most xenogeneic antigens, such as human antigens, because many more antibodies have been produced in the mouse and thus, MAb to defined antigens are available commercially. Isotype matched MAb are obtained easily, which can be used as controls in the assays of interest. Moreover, the mouse is easier to handle, anti-Ig reagents spe-cific for each mouse Ig isotype are more com-monly available, and generally mouse MAb are easier than rat MAb to purify. If a mouse is to be immunized, the best choice is a BALB/c mouse, because the hybridomas resulting from fusion to SP2/0-Ag14 will be entirely of BALB/c origin and thus should grow in a BALB/c host for ascites fluid production. Moreover, BALB/c spleens are generally larger than spleens from other mouse strains.
In contrast, immunization of mice would not be appropriate for most mouse antigens unless an allotypic difference is known. Fortunately, the rat is a reasonable choice for many mouse antigens because rat MAb will frequently rec-ognize framework determinants on mouse pro-teins. Most of the commonly available rat strains can be used, although this laboratory usually employs Lewis rats. If rat MAb are produced, the mouse anti-rat κ MAb, MAR 18.5 (ATCC) is useful, as this MAb recognizes only rat κ light chains and binds protein A, and thus is easy to purify. Moreover, the hybridoma produces high titers of MAb in culture super-natants.
Although there are mouse MAb that recog-nize many major, functionally important
hu-man cell-surface and soluble antigens, it has proven difficult, despite considerable effort, to produce rat MAb to several of the homologous mouse antigens. It is possible that these mouse molecules are not antigenic in rat. Recently, it has been shown that some of these mouse mole-cules may be more antigenic in hamsters, pre-sumably due to the phylogenetic distance in-volved (Schreiber et al., 1985). However, ham-ster MAb are more difficult to produce because of fibroblast overgrowth and instability of hy-bridomas. Thus, hamsters are not an ideal choice for the novice until experience with several fusions is acquired. The Armenian ham-ster strain is the best suited for hybridoma production because the hybridomas are more stable and fibroblast overgrowth is less than with other available hamster strains. Unfortu-nately some Armenian hamster MAb are non-reactive with standard, commercially available anti-hamster immunoglobulin antibodies. However, many hamster MAb are reactive with protein A and/or with the mouse anti-rat κ MAb, RG7/7.6, which cross-reacts with ham-ster κ chains (Sanchez-Madrid et al., 1983). Finally, there are few serologic reagents avail-able for hamster pathogens and thus it is wise to quarantine hamsters—and the hybridomas derived from them—in case there are occult infections that could be spread to other rodents (UNIT 1.1).
The sex of the host animal does not appear to be important. It is usually advisable to munize young adult animals because the im-munization schedule may be prolonged. Fu-sions of spleens from older hamsters (>6 months old) tend to have more fibroblasts.
Antigen preparation and immunization
Many types of antigen preparations have been used successfully including whole cells, partially purified lymphokines and cytokines, solubilized cell membranes and protein bands isolated from SDS-polyacrylamide gels (UNIT 8.4). The nature of the antigen preparation to be
used is dependent on several factors, particu-larly the ease in preparing the antigen, the screening assay, and the desired specificity and property of the MAb.
Although it is desirable to immunize with a purified antigen to increase the frequency of hybridomas secreting the desired antibodies, in contrast to polyclonal antisera production, this is not a major requirement.
It is important to note that MAb are quite specific. It is possible to immunize with im-pure antigens or with multiple antigens, and
Current Protocols in Immunology
2.5.14
Induction of Immune Responses
with a highly specific screening assay, pick the MAb that identifies a specific antigen. In many instances, this is exactly the protocol used. For example, animals are usually immunized with whole cells to derive MAb that recognize a specific cell-surface antigen. The major advan-tage to whole-cell immunization is that the proteins will be in their native conformation and thus, the MAb produced will recognize these antigens in their native form. The major disadvantage to whole-cell immunization is the production of MAb to many other antigens, particularly those that have been previously produced (since they tend to be the most com-monly made); immunization of rats with whole mouse T cell suspensions will produce ≥25% anti-Thy-1 MAb (W. M. Y., unpub. observ.). Nevertheless, collective experience with whole-cell immunizations suggest that al-though the antigen preparation may be impure, it is important to consider the nature of the antigen relative to its native form.
Because protein purification frequently de-natures molecules and synthetic peptides usu-ally do not achieve native conformations, im-munization with synthetic peptides and gel-pu-rified proteins generally has resulted in the production of MAb that recognize the antigen in its denatured form. While such MAb may be useful for immunoprecipitation and im-munoblot studies, often they are not useful for flow cytometry analysis of cell-surface anti-gens (UNITS 5.3 & 5.4) or functional assays that
require binding of the antigen in its native conformation.
There are many immunization protocols used to produce MAb. The major requirements appear to be a primary immunization with an adjuvant and fusion 3 to 4 days after the boost. However, there are notable exceptions. For ex-ample, a single primary immunization and fu-sion 4 to 5 days later have been successfully used to produce MAb against cell-surface an-tigens (Logdberg et al., 1985). It is generally agreed that a successful fusion requires the presence of activated B cells.
Most injections can be given intraperi-toneally with good results. Many investigators prefer to boost intravenously with antigen with-out adjuvant. While this can be done via tail vein in the mouse, this vein is inaccessible in the rat or Armenian hamster and intravenous injections are generally not used in these ani-mals. Be aware that intravenous injections may occasionally result in fatal systemic reactions.
Many investigators screen their immunized animals for serum antibody titers before a final boost and fusion. This depends on the purity of the antigen and an assay that is not influenced by serum. However, many MAb have been successfully produced against cell-surface an-tigens without screening the sera before fusion even if whole cells are used to immunize.
Screening assays
The cell-fusion protocol outlined in this unit should yield hybridomas in ∼50% of the wells. The purpose of the screening assay (first sup-port protocol) is to exclude all hybridomas that are unlikely to produce the MAb of interest and yet include all likely candidates. In that regard, a reliable assay that will detect a few false positives but no false negatives will help de-crease the number of hybridomas from several hundred to ∼20. Depending on the difficulty of the assay and the availability of the antigen, all wells can be screened for reactivity or—espe-cially if most wells do not contain hybrid-omas—the wells can be screened first for visual evidence of growing hybridomas. Visual screen-ing can be problematic since it requires inspec-tion with an inverted microscope and a signifi-cant amount of bookkeeping to record which wells contain hybridomas (and which do not).
Screening assays should be perfected before the fusion is done. The assay should be rapid, with results available within 1 to 2 days, reli-able, sensitive, and simple to perform in large numbers (hundreds to thousands of wells). In the protocol described here, the assay must be performed with 100-µl volumes. Alternatively, the supernatants from several wells could be pooled to screen fewer samples. One clever screening approach is to pool 50 µl from each well in each horizontal row and then separately pool 50 µl from wells in each vertical column of a single plate. The well containing the de-sired hybridoma can be pinpointed by the posi-tive row and column. Because each supernatant is tested twice, the percentage of false positives is diminished. This technique should not be used for low-sensitivity assays as the super-natant is diluted significantly. Many different screening assays and techniques have been de-scribed. ELISA (UNIT 2.1) and
radioimmunoas-say (RIA) have been popular because of the above considerations.
Because MAb are exquisitely specific, those which recognize the same antigen may bind to slightly different epitopes and thus have
dif-Current Protocols in Immunology
2.5.15
Production of Monoclonal Antibodies
ferent functional properties. For example, if a MAb to a cell-surface antigen that inhibits ligand-receptor interaction is required, flow cytometry analysis (UNITS 5.3 & 5.4) may not be
the best choice for the initial screening assay because a MAb that binds well by flow cy-tometry analysis may not be functionally active and vice versa. However, if the desired property of the MAb could be improved by a change of the heavy chain, it is possible to isolate isotype-switch variants that may improve the useful-ness of a particular MAb. Such studies have been performed with isotype-switch variants of anti-CD3 MAb (van Lier et al., 1985). Never-theless, the ideal screening assay should iden-tify MAb with the desired property, not just the desired specificity.
Troubleshooting
The production of MAb is a prolonged pro-cedure. Success requires the optimization of several steps. The antigen preparation and im-munization protocol must be adequate; this can be checked by serum titers after immunization. If no hybridomas are produced, then the immu-nization protocol may not have produced acti-vated B cells, an unlikely situation if an adju-vant was used. Alternatively, hybridoma forma-t ion and growforma-th may be inadequaforma-te, particularly if the cells (spleen or fusion part-ner) were contaminated with mycoplasma or if the lot of fetal calf serum (FCS) does not sup-port hybridoma growth. Obtain the fusion part-ner from a reliable source, either a lab that is successfully producing hybridomas or from ATCC. Freeze aliquots of these cells as soon as possible. Obtain animals from specific patho-gen-free suppliers and cage them in uncontami-nated rooms. The FCS lot can be tested by assaying the cloning efficiency of stable hybri-doma lines and using a lot that supports high-efficiency cloning.
If hybridomas are produced at the antici-pated level but the desired MAb is not found, assay the master wells for other antibodies that should react with the immunogen. For example, if whole cells were used as an immunogen and the original assay was a functional one (e.g., inhibition of proliferation), several wells should be screened for antibodies that recog-nize cell-surface antigens by flow cytometry (UNITS 5.3 & 5.4); under these conditions, 25% to
50% of growing wells will produce antibodies that recognize cell-surface antigens. This result would prove that MAb were produced but that the desired specificity was not found. Unfortu-nately, this is a typical result and the source of
the major commitment in time and labor that MAb production requires. The solution to the problem is to perform more fusions and screen-ing. It is useful to consider alternatives to the immunization, choice of animals, and screen-ing procedure.
During the cloning procedures, there are several problems that may be encountered. If none of the cloned hybridomas secrete the desired MAb, either the hybridoma was not producing the MAb initially or the clones have lost the ability to produce the MAb. An aliquot of supernatant from an expanded master well can be tested in confirmatory assays. If none of the clones produce the MAb of interest, the frozen master-well cells should be carefully thawed, reseeded in multiple cloning plates, and then refrozen. Beware that mycoplasma contamination will affect cloning efficiency adversely.
Anticipated Results
With the cell-fusion protocol outlined in this unit (second basic protocol), hybridoma growth should be apparent on the days men-tioned and ≥50% of the master wells will con-tain hybridomas. Depending on the purity of the antigen used and the immunization proto-col, ≤1% to 5% of the wells will contain the desired hybridoma. If the screening assay dem-onstrates many positive wells, it is likely that these are false positives.
Cloning by limiting dilution of the primary hybridomas (third support protocol) should yield ≥10 to 50 “growing” wells/plate when seeded as described. The second cloning under more stringent conditions should yield ≥10 wells when seeded at 0.3 cells/well. Remember that the cloning efficiency of the primary hy-bridomas is relatively poor. If no viable clones are derived, seed additional wells at higher density and/or use a source of IL-6 (plasmacy-toma growth factor; Nordan and Potter, 1986; Bazin and Lemieux, 1989).
Time Considerations
The initial immunization to the final cloning of the desired MAb requires ≥2 to 3 months if the MAb is produced and identified in the first fusion. Usually, several fusions are required and thus, a fusion every 3 weeks may be nec-essary. During much of this time, especially during the 3 weeks after fusion, daily or every-other-day tasks are required. An entire day’s work is required when the actual cell fusion and screening assays are performed. Feedings re-quire 5 to 10 min/plate.
Current Protocols in Immunology
2.5.16
Induction of Immune Responses
Literature Cited
Bazin, R. and Lemieux, R. 1989. Increased propor-tion of B cell hybridomas secreting monoclonal antibodies of desired specificity in cultures con-taining macrophage-derived hybridoma growth
factor (IL-6). J. Immunol. Methods 116:245-249.
Coffino, P., Baumal. R., Laskov, R., and Scharff, M.D. 1972. Cloning of mouse myeloma cells and
detection of rare variants. J. Cell. Physiol.
79:429-440.
Goding, J.W. 1986. Monoclonal Antibodies: Princi-ples and Practice. Academic Press, San Diego. Harlow, E. and Lane, D. 1988 Antibodies: A
Labo-ratory Manual. Cold Spring Harbor LaboLabo-ratory, Cold Spring Harbor, N.Y.
Köhler, G. and Milstein, C. 1975. Continuous cul-tures of fused cells secreting antibody of
prede-fined specificity. Nature (Lond.) 256:495-497.
Logdberg, L., Gunter, K.C., and Shevach, E.M. 1985. Rapid production of monoclonal
antibod-ies to T lymphocyte functional antigens. J.
Im-munol. Methods 79:239-249.
Nordan, R.P. and Potter, M. 1986. A macrophage-derived factor required by plasmacytomas for
survival and proliferation in vitro. Science
233:566-568.
Raybould, T.J.G. and Takahashi, M., 1988. Produc-tion of stable rabbit-mouse hybridomas that
se-crete rabbit MAb of defined specificity. Science
240:1788-1790.
Sanchez-Madrid, F., Szklut, P., and Springer, T.A. 1983. Stable hamster-mouse hybridomas pro-ducing IgG and IgM hamster monoclonal
anti-bodies of defined specificity. J. Immunol.
130:309-317.
Schreiber, R.D., Hicks, R.D., Celada, A., Buch-meier, N.A., and Gray, P.W. 1985. Monoclonal
antibodies to murine γ-interferon which
differ-entially moderate macrophage activation and
an-tiviral activity. J. Immunol. 134:1609-1618.
Sharpe, R.J., Schweizer, R.T., Krisiunas, L., Miha-lyo, M.A., and Poow, L.M. 1985. Efficient pro-duction of T cell-specific monoclonal antibodies through initial tolerance induction to nonspecific
antigens. Transplant. Proc. 17:2757-2759.
van Lier, R.A., Boot, J.H., Verhoeven, A.J., de Groot, E.R., Brouwer, M., and Aarden, L.A., 1987. Functional studies with anti-CD3 heavy chain isotype switch-variant monoclonal antibodies. Accessory cell-independent induction of inter-leukin-2 responsiveness in T cells by
epsilon-anti-CD3. J. Immunol. 139:2873-2879.
Key References
Goding, 1986. See above.
An in-depth discussion of MAb production. Köhler and Milstein, 1975. See above.
The first description of MAb, for which the authors were awarded the Nobel Prize.
Oi, V.T. and Herzenberg, L.A. 1980.
Immunoglob-ulin-producing hybrid cell lines. In Selected
Methods in Cellular Immunology (B.B. Mishell and S.M. Shiigi, eds.) pp. 351-372. W.H. Free-man, New York.
This reference is the basis for this protocol and for the development of many MAb in the literature.
Contributed by Wayne M. Yokoyama University of California School of Medicine San Francisco, California
Current Protocols in Immunology
2.5.17
Production of Monoclonal Antibodies