Amnah H Alyaqoub
School of Biological Sciences
A structure/function analysis of
Runx transcription factor complex
interactions
A thesis submitted to the University of Manchester
for the degree of Doctor of Philosophy in the Faculty
of Biology, Medicine and Health
2 Table of Contents Table of Contents……….………..……...………....2 List of Figures……….….……….……….……….6 List of Tables………..………..………..……9 List of Abbreviations ……….………....10 List of Equations…………..……….……….…………...…12 Abstract.………...…13 Declaration.………14 Copyright statement.……….….14 Acknowledgements……….…………...…15 Chapter 1 Introduction...16
1.1 The Runx family of Transcription factors...16
1.1.1 Runx2 in skeletal development...16
1.1.2 Runx2 in the skeletal disorder Cleidocranial Dysplasia...17
1.1.3 Runx2 domains...18
1.1.3.1 NMTS...20
1.1.4 Runx1...20
1.1.4.1 Runx/CBFβ in Acute myeloid leukaemia...20
1.1.4.2 Runx/CBFβ in Breast cancer...21
1.1.5 Role of CBFβ...21
1.1.6 The structure of Runt and in complex with DNA...22
1.1.7 CBFβ structure... 24
1.1.8 CBFβ/Runt structure... 25
1.1.9 CBFβ/Runt/DNA structure... 26
1.1.10 CBFβ and Runt Mutagenesis...28
1.2 Filamin A (FLNA) function and structure...29
1.2.1 FLNA function... 29
1.2.2 FLNA structure...30
3
1.2.3 Runx2/CBFβ and FLNA... 33
1.2.3.1 Runx2...33
1.2.3.2 CBFβ... 33
1.3 Project Aims...35
Chapter 2 Materials and Methods...36
2.1 Materials...36
2.1.1 Chemicals and reagents...36
2.1.2 Kits used...36
2.2 Methods...40
2.2.1 Molecular biology techniques...40
2.2.1.1 Polymerase chain reaction PCR...40
2.2.1.2 Restriction enzyme digestion analysis... 42
2.2.1.3 DNA gel extraction...43
2.2.1.4 DNA Ligations and bacterial transformation...43
2.2.1.5 DNA digestion...44
2.2.1.6 Agarose gel electrophoresis...44
2.2.1.7 Mini/Maxi prep plasmid DNA purification...44
2.2.1.8 DNA concentration...45
2.2.1.9 DNA sequencing...45
2.2.1.10 Site-directed mutagenesis (SDM)...45
2.2.2 Proteins techniques...47
2.2.2.1 Cell culture and transient transfection...47
2.2.2.2 Whole-cell lysis...47
2.2.2.3 Cytoplasmic and nuclear extraction...48
2.2.2.4 Transformation of competent bacterial cells...48
2.2.2.5 LB and minimal media preparation...48
2.2.2.6 Protein overexpression (Luria broth/Minimal media)...49
2.2.2.7 Cell lysis……….49
4
2.2.3.1 Tagged protein purification………50
2.2.3.2 Affinity protein purification………50
2.2.3.2.1 Gravity polyprep column………..50
2.2.3.2.2 Glutathione pre-packed column (10ml)………..51
2.2.3.3 Size exclusion chromatography………51
2.2.4 Protein detection……….…52
2.2.4.1 SDS polyacrylamide gel electrophoresis……….52
2.2.4.2 Immunoblotting/western blotting procedure………52
2.2.5 Protein quantification………..53 2.2.5.1 Nanodrop spectrophotometer……….53 2.2.5.2 Protein assay……….53 2.2.5.3 280nm UV spectrometer………..53 2.2.6 Protein-binding assays……….…53 2.2.6.1 Pulldown assay……….53
2.2.6.2 Electrophoretic mobility shift assays (EMSA)……….54
2.2.7 Thermal ramp experiments (fluorescence, SLS and DLS)……….55
2.2.8 NMR spectroscopy………..…55
2.2.8.1 Sample preparation………..55
2.2.8.2 NMR Experiments………..56
2.2.8.3 Data process………..56
2.2.8.4 Figures generation……….56
Chapter 3 Investigating the interaction between Runx2 and FLNA...57
3.1 Introduction...57
3.2 Mapping FLNA interaction with CBFβ………..58
3.2 CBFβ interacts with only FLNA 16-24………62
3.3 Runx2 interacts with FLNA 16-24 through NMTS domain……….66
3.4 Runx2 interacts with specific domains in FLNA……….69
3.5 Summery and discussion………71
5
4.1 Introduction...72
4.2 Purification of FLNA domain 23...76
4.3 15N-HSQC spectrum of FLNA domain 23...78
4.4 Purification of Runx2 NMTS 391-438...86
4.5 15N-HSQC spectrum of Runx2 NMTS 391-438...88
4.6 Characterisation of two time points effect of Runx2 NMTS 391-438 on FLNA domain 23 in complex……….………91
4.6.1 15N-HSQC spectrum of FLNA domain 23/Runx2 GST-NMTS 391-438 complex...91
4.6.2 15N-HSQC spectrum of FLNA domain 23/Runx2 NMTS 391-438 complex..95
4.6.3 15N-HSQC spectrum of FLNA domain 23/Runx2 NMTS 391-438 (with P.I) complex...99
4.7 Purification of Runx2 NMTS 391-424...104
4.8 15N-HSQC of Runx2 GST-NMTS 391-424 compared to NMTS 391-424...106
4.9 15N-based sequential assignments for Runx2 NMTS 391-424...109
4.10 Complex of Runx2 GST-NMTS 391-424/FLNA domain 23...112
4.11 Complex of Runx2 NMTS ATFTYT/FLNA domain 23...116
Chapter 5 Characterisation of CBFβ mutations in breast cancer patients...120
5.1 Introduction...120
5.2 Choice of mutations...121
5.3 CBFβ missense mutations affect the formation of a heterodimerisation complex and DNA/Runt/CBFβ complex...124
5.4 Characterising the thermal stability of CBFβ mutants...129
5.5 Production of CBFβ WT and L21V...136
5.6 NMR spectra of L21V mutant...140
5.7 Backbone resonance assignment for CBFβ WT...142
5.8 L21V fold compared to WT...148
Chapter 6 Discussion and future directions...150
6.1 FLNA domain 23 bound specifically to full-length Runx2 NMTS...150
6.2 CBFβ mutations reduce the formation of CBFβ/Runt and CBFβ/Runt/DNA complexes………152
6
Word count (35966)
List of Figures
Figure 1.1 The structure of Runx shared domains 19
Figure 1.2 Ribbon diagram of the solution structure of Runt domain 23
Figure 1.3 Ribbon representation of the CBFβ structure 24
Figure 1.4 A ribbon diagram of the Runt domain/CBFβ heterodimer 25
Figure 1.5 Surface representation of the ternary complex CBFβ/Runt/DNA 27
Figure 1.6 A schematic representation of FLNA protein structure 31
Figure 1.7 crystal structure of FLNA domain 32
Figure 3.1 Purification of CBFβ and Runt GST fusion proteins used in the
pulldown assay
60
Figure 3.2 Purification of GST fusion FLNA 61
Figure 3.3 CBFβ purified from GST did not interact with GST-FLNA 24 63
Figure 3.4 Optimising the GST-based pulldown assay of FLNA 24 and CBFβ 64
Figure 3.5 GST pulldown interaction assays between the various fragments of
FLNA and CBFβ are not sufficiently robust
65
Figure 3.6 Purification of GST-fusion Runx2 fragments 67
Figure 3.7 Mapping of interaction domains in C-terminus of Runx2 with FLNA
16-24
68
Figure 3.8 Full-length of Runx2 interacts specifically with 22-23 and 23
domains of FLNA
70
Figure 4.1 Domain 23 of FLNA crystal structure 74
Figure 4.2 NMTS motif from Runx1 75
Figure 4.3 Purification of 15N-labelled domain 23 77
Figure 4.4 15N-HSQC spectrum of domain 23 at pH 7.3 80
Figure 4.5 Domain 23 spectrum pH comparisons 81
Figure 4.6 Backbone amide assignment for domain 23 on 15N-HSQC spectra at
pH 7.3 and 6.5
7
Figure 4.7 The result of amide backbone assignment for domain 23 (2428-2516
residues)
84
Figure 4.8 Overlay assignment categories on domain 23 crystal structure 85
Figure 4.9 Purification of 15N-labelled GST-NMTS/NMTS 391-438 87
Figure 4.10 GST-NMTS/NMTS 391- 438 constructs comparison 90
Figure 4.11 GST-NMTS 391-438 and GST-NMTS 391-438/domain 23 comparison 93
Figure 4.12 Domain 23 and domain 23/GST-NMTS 391-438 comparisons 94
Figure 4.13 NMTS 391-438 and NMTS 391-438/domain 23 comparison 96
Figure 4.14 Domain 23 and domain 23/NMTS 391-438 comparisons 98
Figure 4.15 NMTS 391-438 and NMTS 391-438/domain 23 comparison 101
Figure 4.16 Monitoring chemical shift perturbations of domain 23 in response to
NMTS 391-438
103
Figure 4.17 An elution profile of 15N-labelled NMTS 391-424 105
Figure 4.18 15N-HSQC spectrum of GST-NMTS 391-424 at pH 6.7 107
Figure 4.19 15N-HSQC spectrum of NMTS 391-424 at pH 5.7 108
Figure 4.20 HSQC-TOCSY spectrum of NMTS 391-424 (-2L-34H residues) 110
Figure 4.21 NMTS 391-424 NOESY spectra 111
Figure 4.22 15N-HSQC spectrum of GST-NMTS 391-424 with backbone assignment 112
Figure 4.23 GST-NMTS 391-424 and GST-NMTS 391-424/domain 23 comparison 115
Figure 4.24 Monitoring chemical shift changes in domain 23 upon addition of
ATFTYT peptide
119
Figure 5.1 Targeted point mutagenesis of the CBFβ 122
Figure 5.2 The production of CBFβ point mutants 123
Figure 5.3 Purification of CBFβ mutations following bacterial expression 125
Figure 5.4 Effect of All the missense CBFβ mutations on Runt in a GST pulldown
assay
126
Figure 5.5 DNA/Runt/CBFβ (WT) Electrophoretic mobility shift assays (EMSAs)
optimisation
8
Figure 5.6 Comparison of relative binding affinity of the CBFβ mutations and WT
for DNA/Runt/CBFβ heterocomplex formation
128
Figure 5.7 Initial DLS measurements for CBFβ mutants 131
Figure 5.8 Effect of thermal ramp experiments on CBFβ WT stability 132
Figure 5.9 5.9 Effect of thermal ramp experiments on CBFβ Mutants stability and
prone to aggregate
135
Figure 5.10 Development of L21V mutant purification 138
Figure 5.11 15N-labelled CBFβ WT and L21V purifications for NMR studies 139
Figure 5.12 CBFβ WT compared to L21V 141
Figure 5.13 Backbone resonance assignment for CBFβ WT spectrum 144
Figure 5.14 Final backbone amide assignment of CBFβ WT 147
9 List of Tables
Table 2.1 Reagents and chemicals used in this thesis 36
Table 2.2 Antibodies used in this thesis 37
Table 2.3 List of plasmids were previously generated in house or gifted 37
Table 2.4 General buffers and solutions that were generated for this thesis and
the technique they were used in
38
Table 2.5 (A) List of plasmids that generated for FLNA
(B) List of plasmids that generated for Runx2
40
Table 2.6 Typical PCR program 42
Table 2.7 Restriction enzymes used for generated plasmids. 43
Table 2.8 Vectors used for generated plasmids 44
Table 2.9 Primers generated for FLNA sequencing 45
Table 2.10 Oligonucleotides used to generate human CBFβ point mutants 46
Table 2.11 List of lysis buffers used for each protein 50
Table 2.12 Gel filtration buffers for proteins prepared for NMR study 52
Table 2.13 Buffers that used to each binding protein assay. 54
Table 2.14 EMSA reaction protocol 55
Table 4.1 Comprehensive backbone resonance assignment data of domain 23 83
10 List of Abbreviations
2D Two dimensional
3D Three dimensional
a.a. Amino acid
Amp Ampicillin
BCM Barycentric mean
BMRB Biological magnetic resonance data bank
BSA Bovine Serum Albumin
CBFβ Core binding factor beta
CCPN Collaborative computing project for NMR
CS or δ Chemical shift
CSP Chemical shift perturbation
DLS Dynamic light scattering
D2O Deuterated water
DNA Deoxyribonucleic Acid
DTT Dithiothreitol
EDTA Ethylenediaminetetraacetic acid
EMSA Electrophoretic mobility shift assay
FLNA Filamin A
GFP Green fluorescent protein
GST Glutathione S-transferase
HSQC Ig
Heteronuclear single quantum coherence
Immunoglobulin
IPTG Isopropyl β-D-1-thiogalactopyranoside
MW Molecular weight
11
NH Amide group
NMR Nuclear magnetic resonance
NMTS Nuclear matrix targeting sequence
NOE Nuclear overhauser effect
NOESY Nuclear overhauser effect spectroscopy
OD600 Optical density at 600nm
PAGE Polyacrylamide gel electrophoresis
PBS Phosphate buffered saline
PCR Polymerase chain reaction
PDB PSA
Protein data bank
Proline-serine-threonine
Runx Runt-related transcription factor
SDM Site-directed mutagenesis
SDS Sodium dodecyl sulphate
SDS-PAGE Sodium dodecyl sulphate polyacrylamide gel electrophoresis
Static light scattering Static light scattering
Tm Melting point
TOCSY Total correlation spectroscopy
TRIS Trisaminomethane
UV Ultra-violet
WT Wild-type
β-ME β-mercaptoethanol
12 List of Equations
Equation 2.1 Beer-Lambert law used to calculate the concentration of protein. 53
13 Abstract
Filamin A (FLNA), an actin-binding protein, interacts with a wide range of proteins with great functional diversity. Previous studies have demonstrated that FLNA can regulate transcription factor activity. The mammalian Runx family of transcription factors, Runx1, Runx2 and Runx3 have essential roles in development and disease. In order to regulate transcription the Runx proteins form obligate dimers with a co-regulator, CBFβ. Previous studies have shown that CBFβ interacts with FLNA. In addition, FLNA has been shown to interact directly with Runx2 to supress Runx2-mediated transcription. However, to date the precise nature of the interaction between Runx2 and FLNA has not been determined. The aim of this study was therefore to determine the minimal domains on Runx2 and FLNA that mediate the interaction and subsequently determine the structural basis of the interaction using Nuclear Magnetic Resonance (NMR) spectroscopy. Understanding the structural basis of this interaction may be important in the design of therapeutic reagents to modify Runx function in human disease.
A series of Runx2 and FLNA polypeptides were generated and the minimal regions required to interact with each other was determined using GST pulldown assays. It was established that the interaction between Runx2 and FLNA is mediated primarily by Ig domain 23 of FLNA and a short region on Runx2 known as the Nuclear Matrix Targeting Sequence (NMTS). NMR was subsequently used to investigate how FLNA interacts with Runx2, and confirmed that domain 23 of FLNA is responsible for the interaction with the NMTS domain of Runx2. A partial region of the NMTS was resolved using backbone NMR assignment. A two-mode interaction was discovered through chemical shift differences between domain 23 and NMTS. These findings reveal a novel interaction between domain 23 in FLNA and NMTS domain in Runx2.
Experiments were also performed on mutant forms of CBFβ found in patients with breast cancer. The impact of these mutations on CBFβ structure and function has not been previously characterised. Nine CBFβ missense mutations were expressed and purified and their ability to interact with the Runt domain of Runx1 was analysed by GST pulldown and electrophoretic mobility shift assays. The mutations affected the formation of the CBFβ/Runt complex either by disrupting the interface or by affecting the stability of the protein.
14 Declaration
No portion of the work referred to in the thesis has been submitted in support of an application for another degree or qualification of this or any other university or other institute of learning
Copyright Statement
i. The author of this thesis (including any appendices and/or schedules to this thesis) owns certain copyright or related rights in it (the “Copyright”) and s/he has given The University of Manchester certain rights to use such Copyright, including for administrative purposes. ii. Copies of this thesis, either in full or in extracts and whether in hard or electronic copy, may be made only in accordance with the Copyright, Designs and Patents Act 1988 (as amended) and regulations issued under it or, where appropriate, in accordance with licensing agreements which the University has from time to time. This page must form part of any such copies made.
iii. The ownership of certain Copyright, patents, designs, trademarks and other intellectual property (the “Intellectual Property”) and any reproductions of copyright works in the thesis, for example graphs and tables (“Reproductions”), which may be described in this thesis, may not be owned by the author and may be owned by third parties. Such Intellectual Property and Reproductions cannot and must not be made available for use without the prior written permission of the owner(s) of the relevant Intellectual Property and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and commercialisation of this thesis, the Copyright and any Intellectual Property and/or Reproductions described in it may take place is available in the University IP Policy (see http://documents.manchester.ac.uk/DocuInfo.aspx?DocID=2442 0), in any relevant Thesis restriction declarations deposited in the University Library, The University Library’s regulations (see http://www.library.manchester.ac.uk/about/regulations/) and in The University’s policy on Presentation of Theses
15 Acknowledgements
I would like to thank everyone who contributed to the work presented in this thesis. First, I would like to specifically thank my supervisor Paul Shore and co-supervisor Jon Waltho for their patient guidance, support and advice over the last four years. I would also like to express my gratitude to my lab mates in both the MIB and MS buildings for their help and support: Hannah, Henry, Mimi, Richard and Zhalgas. A special thanks goes to Matt Cliff for all his assistance and advice.
Finally, I would like to express my appreciation for my family, especially my parents, siblings and friends for their continued support and encouragement during my time here.
16 Chapter 1 Introduction
Gene transcription is controlled by myriad interactions between transcriptional regulators. The exquisite interplay between these proteins ultimately determines gene expression. Many developmental disorders and cancers are caused by mutations that disrupt the interactions between transcription factors. It is therefore essential that the molecular basis for transcription factor interactions is understood so that therapies can be developed to modulate transcription factor interactions in the diseased state.
The mammalian Runx family of transcription factors, Runx1, Runx2 and Runx3 have essential roles in development and disease. Previous work has shown that Filamin A (FLNA), an actin-binding protein, interacts with Runx transcription factor complexes to regulate transcription factor. FLNA has been shown to interact directly with Runx2 to supress Runx2-mediated transcription. However, to date the precise nature of the interaction between Runx2 and FLNA has not been determined. The overarching aim of this study was therefore to determine the minimal domains on Runx2 and FLNA that mediate the interaction and subsequently determine the structural basis of the interaction using Nuclear Magnetic Resonance (NMR) spectroscopy. Understanding the structural basis of this interaction could potentially be important in the design of therapeutic reagents to modify Runx function in human disease.
1.1 The Runx family of Transcription factors
1.1.1 Runx2 in skeletal development
Skeletal development is a complex process that involves bone growth and remodelling (Liu and Lee, 2013, Rahman et al., 2015, Huang et al., 2007, Zhang et al., 2012, Raggatt and Partridge, 2010). The cells that compose the skeleton such as osteoblasts, chondrocytes, fibroblasts, myoblasts, adipocytes and tendon cells are derived from mesenchymal stem cells (MSCs). In contrast, the osteoclasts are derived from hematopoietic stem cells (HSCs). In bone development, MSCs differentiate into osteoblasts to form bone directly. Then cartilage is formed by chondrocytes. Eventually, differentiated chondrocytes undergo
17
apoptosis, some mature and mineralizing osteoblasts become buried within the bone matrix and formed osteocytes. Cartilaginous structures are then substituted by bone. The transcription factor Runx2 is a master regulator of osteoblast differentiation (Ducy et al., 1997, Deng et al., 2008, Komori, 2005). Runx2 is a Runt-related transcription factor, which belongs to the Runx family. Runx family proteins share a conserved runt domain that directly binds to DNA. Runx2 forms an obligate heterodimer with core-binding factor beta (CBFβ) which confers high affinity DNA-binding.
The first evidence that Runx2 is essential for bone development was the complete lack of an ossified skeleton due to an absence of osteoblasts in Runx2-/- mice (Komori et al., 1997, Otto et al., 1997). Bone matrix protein genes that are expressed by osteoblast are also absent in Runx2-/- mice, such as Spp1, Ibsp, and Bglap2 (Komori et al., 1997, Inada et al., 1999). Expression of bone matrix genes at different levels, including Spp1, Ibsp, Bglap2, Col1a1, and Fn1, can be up-regulated by Runx2 as well as activated promoters of Spp1, Bglap2 and Col1a1/2 (Ducy et al., 1997, Harada et al., 1999, Lee et al., 2000, Sato et al., 1998, Banerjee et al., 1997, Jimenez et al., 1999, Kern et al., 2001). Runx2 is necessary for chondrocyte maturation. Chondrocyte maturation is also inhibited in Runx2-/- mice (Inada et al., 1999). The expression of genes normally observed in terminally differentiated chondrocytes is virtually suppressed in the Runx2-/- skeletons, such as Spp1, Ibsp and Mmp13. Runx2 directly regulated Spp1 and Mmp13 (Sato et al., 1998, Jimenez et al., 1999, Porte et al., 1999, Selvamurugan et al., 2000, Hess et al., 2001).
1.1.2 Runx2 in the skeletal disorder Cleidocranial Dysplasia
Mutation in the Runx2 gene is the main cause of cleidocranial dysplasia (CCD). CCD is characterised by defects in teeth, absence of clavicles, delayed closure of the skull, and a short stature (Otto et al., 1997, Lee et al., 1997, Mundlos, 1999). CCD is caused by a variety of mutations in Runx2, but no clear genotype-phenotype correlations have been identified (Otto et al., 2002). Most mutations arise in the region encoding the Runt domain of Runx2, pointing to the importance of this domain in Runx2 function (Otto et al., 2002). The types of mutations found include missense, nonsense, translocation, splicing and deletion mutations that are detected in the majority of CCD patient (Hoeijmakers, 2001). Many studies have been performed on missense Runx2 mutations in vitro. Mutations in
18
the Runt domain result in disruption of the DNA binding, whereas C-terminal mutations affect the ability of Runx2 to transactivate (Tang et al., 2007, Lee et al., 2015, Wang et al., 2010, Zhang et al., 2000a). In addition, mutations in the Runt domain can abrogate the interaction between the Runt domain and its obligate binding partner CBFβ (Zhou et al., 1999). Indeed, several patients with a deletion of the CBFβ gene also show CCD-like phenotype, indicating that the complex is essential for normal skeletal development (Goto et al., 2004).
1.1.3 Runx2 domains
The domain structures of Runx proteins are shown in Figure 1.1. The conserved domains in all these proteins are Runt domain (RD), a nuclear localisation signal (NLS), a nuclear matrix targeting signal (NMTS), and at the C-terminus there is sequence a VWRPY region (Wheeler et al., 2000). All three Runx genes can be expressed from two promoters; the distal P1 and proximal P2. These promoters give rise to two isoforms with different amino termini; the type I isoform starts at the N-terminus with amino acid sequence MASNS, and type II isoform starts with MRIPV (Ducy et al., 1997, Wheeler et al., 2000, Banerjee et al., 2001). In Runx2, the isoform I is commonly found in osteoblast, spleen and T-cells, while isoform II is expressed only in osseous cells (Ogawa et al., 1993b, Satake et al., 1995, Ducy et al., 1997, Banerjee et al., 2001, Prince et al., 2001).
Both Runx2 isoforms share the same structural domains (Stock and Otto, 2005). A unique domain is found in Runx2 compared to the other Runx proteins members: a glutamine-alanine rich (QA) domain at its N-terminal end. This sequence motif plays an important role in Runx2 transactivation; its deletion results in a decrease in Runx2 transcriptional activity by up to 75% (Thirunavukkarasu et al., 1998). The Runt domain is responsible for DNA binding as well as protein-protein interactions (Kamachi et al., 1990, Kanno et al., 1998). A proline-serine-threonine rich (PST) domain in Runx2 is the main activation domain in the transactivation function (Kanno et al., 1998). Complete deletion of this region causes a 4-5 fold drop in the Runx2 transactivation function (Thirunavukkarasu et al., 1998). The NLS and NMTS domains direct localisation of Runx2 proteins to the nucleus and to subnuclear foci (Zaidi et al., 2001). Deletion of this nine amino acid length NLS domain led to accumulation in cytoplasmic Runx2 (Thirunavukkarasu et al., 1998, Kanno et al., 1998). The C-terminal VWRPY motif involved in repression of Runx2, forms a co-repressor
19
complex with other proteins (Javed et al., 2000, Silverstein and Ekwall, 2005). Removal of VWRPY enhanced Runx2 transactivation and further deletion of C-terminal regions also increased transactivation (Thirunavukkarasu et al., 1998).
Figure 1.1 The structure of Runx shared domains. The
schematic diagram of the Runx proteins. RD: the conserved Runt domain. NLS: nuclear localisation signal peptide. NMTS: nuclear matrix-targeting signal. VWRPY: C-terminal repression motif. Non-conservative regions are in yellow.
N LS NMTS V W R P Y N RD C
20 1.1.3.1 NMTS
NMTS is a unique Runx protein domain that is sufficient to direct Runx proteins to matrix associated subnuclear positions to where gene expression occurs (Zeng et al., 1997, Zaidi et al., 2001). The NMTS is required for cell differentiation and tissue-specific development, as demonstrated by in vivo and genetic approaches (Yergeau et al., 1997, Choi et al., 2001). Mutations in Runx2 targeting subnuclear localisation are associated with skeletal disorders (Zhang et al., 2000b, Chen et al., 2017, Choi et al., 2001). Deletion of the C-terminal containing NMTS in Runx2 increases the intranuclear mobility of proteins (Harrington et al., 2002).
The NMTS is 31 amino acids long (Zeng et al., 1997, Zeng et al., 1998). The only resolved structure study for the NMTS domain is of Runx1 fused to GST (Tang et al., 1999). This X-ray crystal structure revealed that the NMTS forms two loops (I + II) connected by a flexible turn. Loop I and II through the tips of the finger-like structure were predicted to be involved in protein-protein interactions (Tang et al., 1999). Mutation studies at loop II in NMTS showed significantly decreases Runx2 nuclear matrix association (Zaidi et al., 2006). This supports the proposed structural model in which these loops interact with the nuclear matrix.
1.1.4 Runx1
Runx1 is a master regulator of haematopoiesis (Mikhail et al., 2006). The major functional role of Runx1 was first revealed after the generation of Runx1 null mice (Wang et al., 1996, Okuda et al., 1996). The mice died due to loss of foetal liver haematopoiesis. Runx1 is required for formation of haematopoietic stem cells (HSCs) (Chen et al., 2009b).
1.1.4.1 Runx/CBFβ in Acute myeloid leukaemia
Disruption of Runx/CBFβ interaction or CBFβ mutation leads to compromised transcriptional activities and stability. Acute myeloid leukaemia (AML) is an example associated with result of Runx1 mutations leading to loss of function, or the case of inversion mutation due to absence of CBFβ function (Liu et al., 1993, Shigesada et al., 2004, Goyama and Mulloy, 2011). An inversion mutation in CBFβ results in a protein bound in a complex in cytoplasm, preventing Runx1 from entering the nucleus to bind to DNA
21
(Yoshida et al., 2005, Richter et al., 2016). Most Runx1 point mutations found in clinical samples occurred in the Runt domain, in which mutated Runx1 maintains its binding to CBFβ but not to DNA (Osato, 2004, Ptasinska et al., 2012).
1.1.4.2 Runx/CBFβ in Breast cancer
CBFβ expression in breast cancer cells has been found to be mainly required for migration (Pratap et al., 2005, Mendoza-Villanueva et al., 2010). It demonstrated the role of Runx2/CBFβ in cancer cells’ invasion of bone. Also, some known targeted genes of Runx2/CBFβ require CBFβ for expression. Runx1 and CBFβ abnormalities have also been reported in patients with breast cancer (Banerji et al., 2012, Ellis et al., 2012). Missense mutations in the Runt domain of Runx1 and CBFβ suggests that their transcriptional activites influence breast cancer growth. However, Runx2 mutation has not been reported in cancer. Taken together, this suggests that Runx/CBFβ functions are lost in breast cancer.
1.1.5 Role of CBFβ
Death occurring during embryonic development in CBFβ knockout mice is thought to be due to a lack of definitive haematopoiesis and haemorrhage (Sasaki et al., 1996, Wang et al., 1996). The same detected phenotypes have been seen in Runx1 knockout mice (Wang et al., 1996). This indicates the essential role of CBFβ in haematopoiesis along with Runx1. Other models such as CBFβ knock-in mice showed normal haematopoietic function, yet the mice died soon after birth (Kundu et al., 2002). CBFβ knock-in transgenic mice showed rescue of haematopoiesis but no haemorrhaging, confirming the requirement of CBFβ for haematopoiesis (Miller et al., 2002, Yoshida et al., 2002) . The mice also showed several skeletal defects. These defects were similar to those reported in Runx2 knockout mice, but less severe. Rather than a lack of bone ossification in Runx2 deficiency, with transgenic CBFβ, mice experience delayed bone ossification (Kundu et al., 2002, Miller et al., 2002, Yoshida et al., 2002). This further suggested that CBFβ is essential for bone development and is required in Runx family proteins. Several patients with deletion of the CBFβ gene also showed CCD-like phenotypes, suggesting that the complex is essential for normal skeletal development (Goto et al., 2004).
22
A recent study by Wu et al (2014) shows further evidence of the critical role of CBFβ in cartilage and bone development; its deletion in mice in undifferentiated MSC stem cells, affects chondrocytes’ and osteoblasts’ development, leading to severe skeletal defects and death shortly after birth from respiratory distress. This study also suggested that during osteoblastogenesis, CBFβ is likely to interact mainly with Runx2, while it interacts with both Runx1 and Runx2 during chondrogenesis. This latter finding implies a regulatory role for Runx2/CBFβ in cartilage and bone development (Wu et al., 2014).
1.1.6 The structure of Runt and in complex with DNA
Although Runx proteins have specific individual roles, they share a highly conserved sequence region termed the Runt domain (Rennert et al., 2003, Coffman et al., 1996). The runt domain was first recognised in Drosophila melanogaster in the runt gene that is responsible for embryonic segmentation. This domain is located towards the N-terminus of each Runx protein. The Runt domain binds to DNA via a consensus sequence PyGPyGGTPy (Py is pyrimidine) (Kamachi et al., 1990). The runt domain structure is an S-type immunoglobulin (Ig) fold, consisting of seven anti-parallel strands to yield a β-sandwich arrangement (Figure 1.2) (Berardi et al., 1999, Nagata et al., 1999). This S-type Ig motif is observed among Ig folds found in DNA binding transcription factors such as p53, NFAT, NF-kB and STAT1 (Cho et al., 1994, Zhou et al., 1998, Chen et al., 1998). These proteins mainly differ in the connecting loop regions. The Runt domain differs from other Ig motif transcription factors in that it is at least 40 amino acids shorter with a short anti-parallel β-strand added at the N-terminus.
The Runt domain contacts the major and minor grooves of DNA mainly using two loops and a C-terminal tail surrounding the sugar-phosphate backbone. In the major groove, hydrogen bonds are formed between the three key guanines and three arginine residues, and in the minor groove a different loop section interacts with them. Several interactions between the side chains and the backbone of the Runt domain and the sugar-phosphate backbone of DNA are further maintained by this complex (Tahirov et al., 2001, Warren et al., 2000).
23
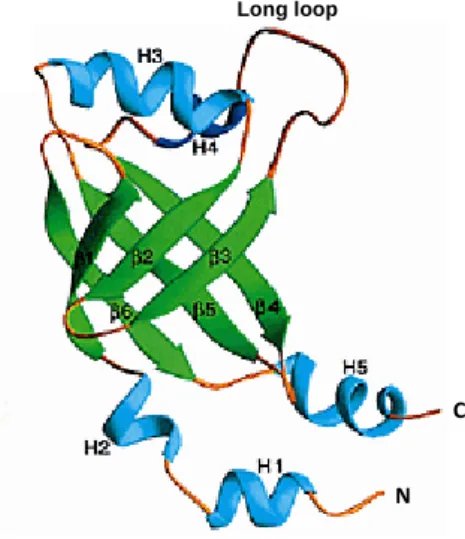
Figure 1.2 Ribbon diagram of the solution structure of Runt domain. The β-strands are labelled sequentially from β1 to β7.
The Runt domain shows an S-type Ig fold. Adapted from (Berardi et al., 1999)
24 1.1.7 CBFβ structure
The Runt domain also binds to the dimeric partner protein CBFβ. The first CBFβ structures were determined in solution by NMR analysis (Goger et al., 1999, Huang et al., 1999). The CBFβ structure adopts a unique arrangement of α and β folds, parted by loops into two folded regions and the N-terminus in close proximity with the C-terminal end of the protein (Figure 1.3). The initial studies established that an N-terminal 141 amino acid fragment of CBFβ is responsible for the heterodimerisation domain with Runt (Ogawa et al., 1993a, Huang et al., 1998). CBFβ (141) binds to Runt with the same affinity as a full-length isoform of CBFβ (187). CBFβ (141) essentially also has the same fold as CBFβ (187) when its heterodimerisation domain was isolated. For these reasons, apparently, most structural studies focused on CBFβ (141). In addition, a truncated CBFβ (135) can still form a stable heterodimer and is suggested to have fully functional protein activity; however, truncation beyond 135 leads to complete loss of heterodimerisation with Runt (Kagoshima et al., 1996).
Figure 1.3 Ribbon representation of the CBFβ structure. It
shows β-strands (green), α helices (cyan) and a long loop. (Goger et al., 1999)
Long loop
C
25 1.1.8 CBFβ/Runt structure
CBFβ increases the affinity of the Runt domain for DNA by approximately six- to tenfold, but does not itself make contact with DNA (Figure 1.4). This effect is increased to at least fortyfold in the full-length Runx proteins. This suggests that other regions within the Runx proteins mediate auto-inhibition of DNA binding (Kanno et al., 1998, Gu et al., 2000, Inman et al., 2005). CBFβ also stabilises the Runx proteins by protecting them from proteolytic degradation by the proteasomes (Nimmo and Woollard, 2008, Tahirov et al., 2001, Tang et al., 2000a, Tang et al., 2000b, Wang et al., 1993, Kamachi et al., 1990, Bravo et al., 2001).
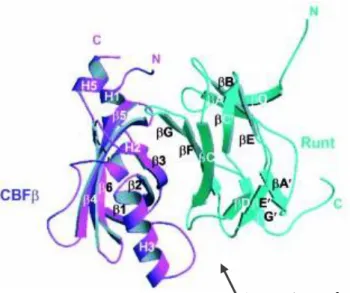
Figure 1.4 A ribbon diagram of the Runt domain/CBFβ heterodimer. It shows the large binding interface between the
Runt domain and CBFβ. Strands and helices are labelled. (Warren et al., 2000)
26 1.1.9 CBFβ/Runt/DNA structure
Several X-ray crystalllography studies were conducted to compare the binary complex CBFβ/Runt, Runt/DNA and ternary complexes of CBFβ/Runt/DNA in relation to free form proteins, to investigate the mechanism underlines these bindings. These studies suggested that the allosteric nature of CBFβ plays a role in the process of regulation of Runt/DNA binding (Bravo et al., 2001, Backstrom et al., 2002, Tahirov et al., 2001, Bartfeld et al., 2002). CBFβ was shown to allosterically enhance DNA binding in the Runt domain by stabilising the flexible regions of the Runt domain in contact with DNA binding via a specific hydrogen bonding arrangement (Bravo et al., 2001, Warren et al., 2000).
These studies also revealed that several regions of the Runt domain, especially the S- switch termed region, βG-G’ loop (which is involved in the conformational exchange of the Runt domain/DNA complex) are quenched upon introduction of CBFβ, suggesting this as the binding interface (Figure 1.5). The changes are in agreement with NMR studies that highlight the same region that experiences major chemical shift changes upon CBFβ binding (Tang et al., 2000a). Similar conformational changes in the Runt domain upon binding CBFβ and/or DNA indicate that CBFβ’s effect is to accommodate a favourable equilibrium in the Runt domain for the existing structure. NMR relaxation analysis provides further evidence that CBFβ quenches conformational exchange in the S-switch region upon binding to the binary complex Runt/DNA (Yan et al., 2004). This study supports the idea of free Runt that can take two forms, binding to DNA with either high or low affinity, and switch between them.
DNA binding has also been observed to quench the conformational exchange of Runt in a study showing a mutation study in the S-switch region (Li et al., 2006). These characteristics provide insights into CBFβ allosteric regulation mechanism upon DNA binding, in which CBFβ enhances DNA binding by the Runt domain through securing these dynamic areas in a favourable structure that interacts with DNA.
27
DNA
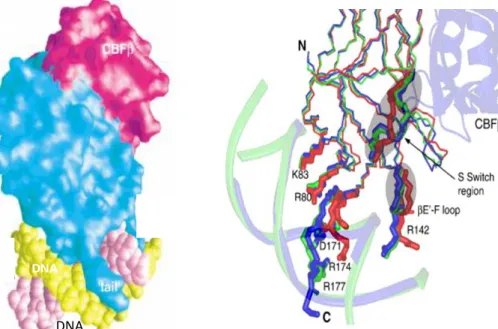
Figure 1.5 Surface representation of the ternary complex CBFβ/Runt/DNA. (A) The Runt domain makes contact with
CBFβ and contacts DNA through its C-terminal loop. (Bravo et al., 2001) (B) The S-switch region of the runt domain involved in the DNA interactions. Comparison of structures among the Runt domain (red), the Runt domain/DNA binary complex (green) and the CBFβ/Runt domain/DNA ternary complex (blue). (Li et al., 2006)
DNA
28 1.1.10 CBFβ and Runt Mutagenesis
Several mutagenesis studies on Runt and CBFβ have been carried out to characterise the effects of the mutations on the structural integrity and the energetic critical residues for Runt/DNA as well as CBFβ/Runt binding (Nagata and Werner, 2001, Bravo et al., 2001, Tahirov et al., 2001). This approach supported by NMR analyses when chemical shifts occurred upon binding of Runt or CBFβ, was used to map the contact surfaces and direct mutagenesis studies.
Mutagenesis studies on the residues at the interface of CBFβ with Runt/DNA showed that some mutants exhibit defective heterodimerisation, including N63K, M101Q, I102E, N104K and E135R (Nagata and Werner, 2001). These were residues observed to be side chains facing outward, towards the Runt domain, providing a rational explanation for protein-protein disruption. The Asn 104 residue in particular appeared to be the greatest contributor to heterodimerisation and was identified as a critical hotspot for binding energy to Runt (Tang et al., 2000b). Interestingly, Asn 63 substituted with alanine, whose side chain points toward the surface, contacts four amino acids in Runt, one of which mutated in a CCD patient (Thr 149); however, this did not affect Runt interaction (Tang et al., 2000b).
29 1.2 Filamin A (FLNA) function and structure
1.2.1 FLNA function
Filamins are large cytoplasmic proteins that stabilise the form of three-dimensional F-actin networks and connect them to cellular membranes via binding to transmembrane receptors or ion channels. Thus, they serve as scaffolding molecules and influence protein cellular localisation and facilitate protein-protein interactions (Popowicz et al., 2006, Stossel et al., 2001). FLNA is one of the three human filamin isoforms (along with filamin B and C). Their genes are highly conserved and share 70% sequence identity (van der Flier and Sonnenberg, 2001). All of them are widely expressed during development. FLNA is the most abundant isoform in adults and is the first identified that crosslinks actin in a non-muscle cell (Hartwig and Stossel, 1975).
FLNA has been shown to be essential in normal development. Hence, mutations in the respective genes cause developmental malformation in the bone, brain and heart (Feng and Walsh, 2004). FLNA mutation was first identified in human periventricular nodular heterotropia (PVNH), showed brain malformation due to abnormal neuronal migration as the neurons fail to migrate into the developing cerebral cortex (Fox et al., 1998). Other FLNA mutations studies have shown different congenital malformations such as frontometaphyseal dysplasia (FMD) (Robertson et al., 2003, Ithychanda et al., 2017). FLNA functions are associated with a wide range of interacting partners, including several transmembrane proteins: ion channels, integrins, intracellular signalling molecules, kinases, transcription factors and cytoskeletal proteins (Li et al., 2000, Stossel et al., 2001). These interactions are regulated through mechanical forces, competition, phosphorylation, proteolysis and multimerization (Li et al., 2000, Stossel et al., 2001). Although full-length FLNA is mainly localised in the cytoplasm, multiple studies showed that it localised to the nucleus both as a full-length protein and as a cleaved C-terminal fragment (Loy et al., 2003, Ozanne et al., 2000, Bedolla et al., 2009, Wang et al., 2007). The nuclear FLNA C-terminal fragment has been shown to inhibit transcription factor FOXC1 (Loy et al., 2003). It also acts as a repressor for androgen receptor transactivation through a competing co-activator (TIF2) (Loy et al., 2003). FLNA serves as the scaffolding crosslinking for signalling, and at the same time is associated with transcription factors, implying that FLNA plays a role involving the cytoskeleton in gene regulation expression.
30 1.2.2 FLNA structure
The molecular weight of FLNA is 280 kDa and consists of two subunits that self-assemble (Figure 1.6) (Nakamura et al., 2007, van der Flier and Sonnenberg, 2001). Each monomer is composed of an N-terminal region with an actin-binding domain (ABD) that consists of two calponin homology (CH) domains (CH1 and CH2). The ABD plays a key role in FLNA subunit attachment to F-actin thereby crosslinking actin filaments (Stossel and Hartwig, 1975, Janmey et al., 1990, Brotschi et al., 1978). The ABD is followed by 24 immunoglobulins Ig-like (IgFLN) repeats/domains of around 100 amino acids forming anti-parallel β sheets (Nakamura et al., 2011, Gorlin et al., 1990). FLNA is divided into rod 1 and 2. Two flexible hinge regions separate the rod 1 domain (repeats 1-15) and rod 2 domain (repeats 16-23) and repeat 24 (van der Flier and Sonnenberg, 2001, Gorlin et al., 1990). Besides the flexibility of hinges added to the structure, they are sites of proteolytic cleavage (Popowicz et al., 2006). Whereas the rod 1 domain has a secondary ABD with lower affinity, the rod 2 domain (C-terminal) mediates the majority of the known interacting partner proteins (Nakamura et al., 2007, Nakamura et al., 2011, Zhou et al., 2007). The repeat 24 is a self-association domain that mediates the homodimerisation of FLNA, permitting the formation of V-shaped flexible structures in the cross-linking of actin filamins that is essential for its function (Pudas et al., 2005). In addition, structural studies of FLNA have found that it is able to arrange linearly and also form multi-domain modules (Tossavainen et al., 2012, Ruskamo et al., 2012, Heikkinen et al., 2009, Lad et al., 2007, Sethi et al., 2014).
31
Figure 1.6 A schematic representation of FLNA protein structure. FLNA is a homodimer forming
a V shape. Each monomer is divided into three major domains: (ABDs); the N-terminal actin binding domains (yellow), FLNA (1–24); the Ig-like FLN domains, which consist of rod 1 and rod 2 (red and blue); and FLNA (24), which is the C-terminal domain that mediates the dimerization (green). Hinges 1 and 2 separate rod 1 from rod 2, and rod 2 from rod 24. Modified from (Pezeshgi and Mofrad, 2014)
32 1.2.2.1 FLNA domain structure
The FLNA domain structure consists of seven to eight β strands that are assembled into two β sheets forming β sandwiches (Figure 1.7) (van der Flier and Sonnenberg, 2001, Gorlin et al., 1990). C and D strands form a common ligand-binding interface referred to as the CD face. FLNA domains’ CD faces have been shown to interact with proteins such as FilGAP, platelet glycoprotein (GP) and integrin β-subunit cytoplasmic tails (Nakamura et al., 2009, Nakamura et al., 2006, Kiema et al., 2006, Takala et al., 2008). Some examples of mechanisms involving CD faces are a mechanism called β-sheet augmentation, when the peptides form a further antiparallel β strand next to the C strand, and the D strand participates in the IgFLN 24 dimerisation interface (van der Flier and Sonnenberg, 2001, Razinia et al., 2012, Seo et al., 2009, Sethi et al., 2014, Smith et al., 2010). Another mechanism involved is a mechanical regulation; FLNA domains 18-19 and 20-21 have shown a unique inter-domain interaction in which the A strand folds along the CD face of the next domain, resulting in auto-inhibited ligand binding (Lad et al., 2007, Razinia et al., 2012, Pentikainen and Ylanne, 2009, Chen et al., 2009a).
Figure 1.7 crystal structure of FLNA domain. An example of
FLNA Ig-like repeat fold (domain 10). The N-terminus in dark blue and C-terminus in dark red. (Page et al., 2011)
C
33 1.2.3 Runx2/CBFβ and FLNA
1.2.3.1 Runx2
It has been shown that both Runx2 and FLNA knockout mice display bone defects (Komori et al., 1997, Feng et al., 2006). Mutations of both Runx2 and FLNA genes have been shown to cause skeletal disorders in patients (Robertson et al., 2003, Feng et al., 2006, Mundlos, 1999). Direct interaction of FLNA and Runx2 has been characterised in vivo and in vitro in the nucleus (Tang, 2007, Camacho, 2011).
1.2.3.2 CBFβ
The cytoplasmic CBFβ is expressed in murine embryos, specifically in the skeletal myogenic cells (Chiba et al., 1997). It is colocalised with F-actin in the established skeletal muscle fibres and has an affinity with cytoskeletal structure (Tanaka et al., 1997). CBFβs also located where many actin-associated proteins are abundant, on or near the Z-line of muscle fibres (Chiba et al., 1997).
FLNA has also been found to interact with CBFβ in the cytoplasm (Yoshida et al., 2005). FLNA was shown to prevent CBFβ from enters the nucleus to bind to Runx1 and enhance its transcription. Multiple studies also showed the interaction between FLNA and other transcription factors, such as Smads (Sasaki et al., 2001), Androgen receptors (Loy et al., 2003), FOXC1 (Berry et al., 2005),and P73α (Kim et al., 2007), which indicates that it is involved in regulating gene expression.
Recent studies by Johnson (2012) were carried out to identify drugs with possible chondrogenic and chondroprotective capacity using a high-throughput, image-based screen (Johnson et al., 2012). They identified a molecule called Kartogenin (KGN) that was capable of stimulating chondrogenic differentiation of bone marrow MSCs in culture and showed cartilage repair in two joint mouse OA models. KGN functions through binding the FLNA; it directly targets the pathway of FLNA and CBFβ by binding to FLNA and inhibiting its interaction with CBFβ (Johnson et al., 2012). A more recent study by Decker, R (2014) has suggested KGN as a stimulator of limb skeletal growth and a facilitator of joint formation (Decker et al., 2014). Thus, formation of a regenerative and repair tool for unsolved joint pathologies, such as osteoarthritis (OA), congenital joint dysplasia and severe joint injuries receives substantial attention in current research (Umlauf et al., 2010,
34
Sandell, 2012, Onyekwelu et al., 2009); however, its mechanism of action on developing skeletal cells and its therapeutic potential are still unclear.
35 1.3. Project Aims
The aim of this study was to investigate how FLNA interacts with CBFβ and Runx2. This objective was to be carried out by mapping the interaction domains of FLNA/CBFβ and FLNA/Runx2. Then, the relation between them was assessed by finding the most suitable complex for the structural analysis using NMR spectroscopy.
Another aim was to characterise the impact of CBFβ missense mutations with structural evidence on its loss of function, including the impact on Runt and DNA binding. Thus, the objective was to investigate these breast cancer-associated mutations using in vitro assays and NMR spectroscopy.
Objectives:
1. To identify minimal region of Runx2 and CBFβ with FLNA that form a complex. 2. To determine the structure of the minimal Runx2/FLNA complex.
36 Chapter 2 Materials and methods
2.1 Materials
2.1.1 Chemicals and reagents
All chemicals were purchased from Sigma Aldrich or else stated (Table 2.1). Table 2.1 Reagents and chemicals used in this thesis
Supplier Item ThermoFisher Scientific DTT L-Glutamine Lipofectamine 2000 LightShift Poly (dI-dC) Opti-MEM Penicillin/Streptomycin Tween 20 Glycerol Ni-NTA resin GE Healthcare Thrombin
Bioline Isopropyl β-D-1-thiogalactopyranoside (IPTG) Mytaq-red
Bio-Rad Bromophenol blue
Pre-made 10/12/4-15/4-20% SDS-PAGE gels
Precision Plus ProteinTM Dual color standards/Xtra Prestained
protein ladder range from 250-10/ 250-2 kDa National
diagnostic
Protogel acrylamide Bio-West Fetal bovine serum
Expedeon Amintra glutathione Resin Instant Blue
Roche EDTA-free protease inhibitor
Lonsa agarose
2.1.2 Kits used
37 Table 2.2 Antibodies used in this thesis
Antibody Supplier Dilution in milk blocking solution (µl)
CBFβ Abcam 1:500
FLAG Sigma 1:1000
Histone H3 Cell signaling technology 1.5:3000
Runx2 Santa Cruz 1:5000
GST GE lifesciences 1:5000
GFP Abcam 1:5000
Goat IgG Abcam 1:5000
Rabbit IgG Cell signaling technology 1:5000 Mouse IgG Cell signaling technology 1:3000
Table 2.3 List of plasmids were previously generated in house or gifted
Plasmid Description Origin
pRK5- Runx2-Flag
Full-length Runx2 (528 a.a.) with C-terminal flag tag
[Mus musculus, P1 promotor the type II isoform]
In house
GFP-FLNA
Full-length FLNA with N-terminal GFP tag In house 16-24 FLNA 16-24 domain FLNA [Human (ABP-280)] In house GST-CBFβ
Full-length CBFβ (187 a.a) in pGEX-2TK [Human, isoform 2]
In house
His-CBFβ Full-length CBFβ in pET14b In house
GST-Runt 60-181a.a. from 450 a.a Runx1 in pGEX-2TK [Homo sapiens]
38
GST-2TK pGEX-2TK vector GE Healthcare
GST-6p-1 pGEX-6p-1 vector Gifted from
Sharrock laboratory (University of Manchester)
Table 2.4 General buffers and solutions that were generated for this thesis and the technique they were used in. All buffers made up using water produced by a MiLLi-Q system.
Name Components Use
BufferX 20mM Tris, 150/300mM NaCl, 0.2mM EDTA, 0.2mM EGTA (~pH8)
General Protein lysis and purification 5xSDS sample buffer 10mM Tris-HCl (pH 6.8), 10mM SDS, 45% Glycerol, 12% 2-mercaptoethanol, 0.01% Bromophenol blue Gel electrophoresis 12%Separating gel 12%Acrylamide, 140mM SDS, 125mM Trizma (pH 6.8), 0.02% TEMED, 0.05% APS SDS-PAGE
4%Stacking gel 4%Acrylamide, 140mM SDS, 375mM Trizma (pH 8.8), 0.02% TEMED, 0.05% APS SDS-PAGE 10xBinding buffer 100mM Tris, 500mM KCl, 10mM DTT (pH 7.5) EMSA Extraction buffer A 10mM HEPES (pH 7.9), 10mM KCl, 1mM DTT, 0.5mM PMSF, 0.1mM EGTA, 0.1mM EDTA Cytoplasmic extraction Extraction buffer B 20mM HEPES (pH 7.9), 400mM NaCl, 1mM DTT, 1mM PMSF, 0.1mM EGTA, 0.1mM EDTA Nuclear extraction
39
Imidazole buffer
30/250mM Imidazole in bufferX His-tag purification
5x TBE For 1L: 108g Tris-base, 55g Boric Acid (ortho), 40ml 0.5M EDTA pH8
EMSA
1x TBS-Tween 100mM Trizma, 300mM NaCl, 0.1% (v/v) Tween 20 (pH 8)
Western blot
1xTransfer buffer
400mM Glycine, 50mM Trizma, 20% methanol, 0.1% (w/v) SDS
Western blot
1% TAE 2M Tris-HCl, (pH 8), 0.05mM EDTA, 6% glacial acetic acid
Molecular cloning
Trace elements
For 100ml:
CaCl2 .2H2O 550mg, ZnSO4 .7H2O 220mg MnSO4 .H2O 140mg, CoCl2 .6H2O 45mg H3Bo4 40mg, CuSO 4 .5H2O 40mg Na2MoO4 .2H2O 26mg, KI 26mg EDTA 500mg, FeSO4 .7H2O 375mg
Minimal media component
40 2.2 Methods
2.2.1 Molecular biology techniques
2.2.1.1 Polymerase chain reaction PCR
PCR reactions were set up using 200ng of template plasmid, 1µl from each primer (20μM), 25µl of MyTaq Red Reaction buffer and up to 50µl of sterilised water. The complete lists of plasmids generated for this thesis are shown in Table 2.5 A typical program for DNA amplification is shown below in Table 2.6 Five µl of the PCR products were then analysed on a 1%agarose gel to check if the reaction was successful. The rest of the product was purified using a PCR purification kit according to manufacturer’s instructions.
Table 2.5 (A) List of plasmids that generated for FLNA
Plasmids Primers Domain 24 (2552-2650) F 5' gatcggatcccctgggcctgctgacgccag R 5' gatcgaattctcagggcaccacaacgcggtag Domain 23 (2425-2520) F 5' gatcggatccgagcctgggcatggaggggaccca R 5' gatcgaattctcatgtgactttggccttgaaggggct Domain 22 (2329-2425) F 5' gatcggatccgcttctccgtctggcgacgc R 5' gatcgaattctcactccccaactcggatcttgaaggggc Domain H2-24 (2520-2650) F 5' gatcggatccacaggcccccgtctcgtcag R 5' gatcgaattctcagggcaccacaacgcggtag Domain 23-24 (2425-2650) F 5' gatcggatccgagcctgggcatggaggggaccca R 5' gatcgaattctcagggcaccacaacgcggtag Domain 22-23 (2329-2520) F 5' gatcggatccgcttctccgtctggcgacgc R 5' gatcgaattctcatgtgactttggccttgaaggggct
41 Domain 22-24 (2329-2650) F 5' gatcggatccgcttctccgtctggcgacgc R 5' gatcgaattctcagggcaccacaacgcggtag Domain 16-21 (1779 – 2325) F 5' gatcggatccggtgtcaatgggctggatgt R 5’ gatcgtcgactcacacaggcaccacgaaggggc Domain 16-24 (1779 – 2647) F 5' gatcggatccggtgtcaatgggctggatgt R 5' gatcgtcgactcagggcaccacaacgcggt
Table 2.5 (B) List of plasmids that generated for Runx2
Plasmids Primers 235-528 F 5' gatcggatccagaaggcacagacagaagct R 5' gatcgtcgactcaatatggccgccaaacagact 22-233 F 5' gatcggatccagcaccagccggcgcttca R 5' gatcgtcgactcattcccggggaccgtccac 235-438 F 5' gatcggatccagaaggcacagacagaagct R 5' gatcgtcgactcattgggaagagccggggtagggt 235-429 F 5' gatcggatccagaaggcacagacagaagct R 5' gatcgtcgactcacaggtacgtgtggtagtgag 233-391 F 5' gatcggatccgaaccaagaaggcacagac R 5' gatcgtcgactcactcagtgagggatgaaatgc 391-528 F 5' gatcggatccgagagccgcttctccaacccac R 5' gatcgtcgactcaatatggccgccaaacagact 391-438 F 5' gatcggatccgagagccgcttctccaacccac R 5' gatcgtcgactcattgggaagagccggggtagggt
42 391-424 F 5' gatcggatccgagagccgcttctccaacccac R 5' gatcgtcgactcagtgagtggtggcggacatg 411-438 F 5' gatcggatccgtcacgtcaggcatgtccct R 5' gatcgtcgactcattgggaagagccggggtagggt 511-528 F 5' gatcggatccccaactgttttgaattctagc R 5' gatcgtcgactcaatatggccgccaaacagact
Table 2.6 Typical PCR program
Steps Temperature (°C) Time (s)
Initial denaturation (1cycle)
95°C 300
Denaturing 95°C 50
Primer annealing Subject to primers used (used online oligocalculater)
50
Primer extension 72°C 25
No. of cycles 35
2.2.1.2 Restriction enzyme digestion analysis
The restriction enzyme’s (NEB) digestion of plasmids occurred in a total reaction volume of 50µl. Two µg of plasmid were added to a mix containing 1µl from each enzyme, 5µl of the reaction buffer and topped off with free DNase/RNase H2O. Reactions were then
incubated for 1–2 hours at 37°C. The reaction was performed for the insert and the vector from the same master mix reaction. Both were purified using a PCR purification kit before ligation. The pGEX-2TK vector was not empty and contained another protein, so it was subjected to DNA gel extraction prior to ligation.
43 Table 2.7 Restriction enzymes used for generated plasmids.
Plasmids Runx2 domains
FLNA 16-21 FLNA 16-24
FLNA domains (others)
Restriction enzymes BamH1, Sal1 BamH1, EcoR1
2.2.1.3 DNA gel extraction
The digested pGEX-2TK vector was run on a 1%agarose gel. The agarose gel was visualised under UV, and the desired DNA fragments were cut out. The gel slice was then weighted and further purified using the QIAGEN Gel Extraction Kit, according to the manufacturer’s protocol. The collected DNA was then quantified and subjected to ligation.
2.2.1.4 DNA Ligations and bacterial transformation
Ligations were carried out using a 1:3ng molar ratio of vector fragment (Table 2.8) to insert plasmid, for a total reaction volume of 20µl. The reaction carried out contained the calculated vector to insert ratio, 1 unit of T4 DNA ligase, 2µl of ligation buffer and enough
sterile ddH2O to reach the final volume. A control reaction with no insert plasmid was also
set up. The reactions were then incubated for 2–3 hours at 37°C. Next, the ligation mixtures were transformed.
44 Table 2.8 Vectors used for generated plasmids.
Plasmids FLNA domains Runx2 domains
FLNA 16-21 FLNA 16-24
Vectors description
Domains cloned downstream of GST in pGEX-2TK containing Ampicillin resistance and a thrombin cleavage site.
Domains cloned downstream of GST in pGEX-6p-1 containing Ampicillin resistance and a PreScission Protease cleavage site.
2.2.1.5 DNA digestion
The Miniprep plasmid DNA for the various constructs was incubated with the constructs’ restriction enzymes and with the specific buffer to determine if the ligation was successful. A single cut or a double cut was performed to cleave one side or both sides of the DNA’s inserted fragments from their corresponding vector. These digestion samples were run on 1% agarose gel and the presence of the correctly sized cloned insert was confirmed. These plasmids were then quantified and sequenced.
2.2.1.6 Agarose gel electrophoresis
The DNA was resolved and its size identified via agarose gel electrophoresis. The DNA was tested on 1% agarose gels that contained 10µl SYBR Safe DNA gel stain (Thermo fisher scientific) for each 100ml of agarose gel. DNA samples were mixed with 6xDNA loading dye (NEB) prior to loading into the gel and were run alongside an appropriately sized DNA ladder. The gels were then immersed in a 1xTAE running buffer and run at 80V for around 30 minutes. The DNA was then visualised under UV light (Biorad).
2.2.1.7 Mini/Maxi prep plasmid DNA purification
For a small- or large-scale DNA preparation, a mini prep was employed. The transformed colonies were used to inoculate 5/100ml of LB-Amp via shaking at 200rpm and were incubated overnight at 37°C. The DNA was then extracted using the QIAGEN Mini/Maxi prep kits as instructed by the manufacturer. The purified plasmid DNA was eluted in H2O,
45 2.2.1.8 DNA concentration
DNA was quantified using Nanodrops and measuring the absorbance at 260nm of a diluted DNA sample.
2.2.1.9 DNA sequencing
DNA sequencing was performed to verify the cloning and mutagenesis. Sequencing reactions consisted of 150–300ng of plasmid DNA, 2µl of 100μM pGEX primer stock (forward or reverse) and enough ddH2O to reach a final volume of 10µl. The Stopford
in-house DNA sequencing facility carried it out.
The case with FLNA 16-24 and 21-24, primers were designed to check the long length of these two domains in addition to the pGEX primers (Table 2.9).
Table 2.9 Primers generated for FLNA sequencing
FLNA 16-24 FLNA 16-21 primerA: acctaaaggtcggctctgct primerB: gcagccccttctctgtga primerC: tggcttatgtggtccaggag primerD: gtctgcttacggagcaggtc primerA: cttcactgctcgggtcacag primerB: cctactgccccacagagc 2.2.1.10 Site-directed mutagenesis (SDM)
The mutagenic oligonucleotides used to generate the human CBFβ point mutants are shown in Table 2.10. The primers were designed using the web-based QuikChange Primer Design Program, available online and accessed in 2017. Primers were named using single-letter amino acid code of the mutation they were designed to produce. The template used for the PCR reaction was a 2TK-pGEX vector containing full-length human CBFβ (187 a.a.). Human CBFβ point mutants were produced using the QuikChange II XL Site-Directed Mutagenesis Kit, following the manufacturer’s instructions. The primer sequences used were designed using the online QuickChange Primer design program. XL10-Gold ultracompetent cells were transformed with 2µl of the Dpn I-treated DNA sample reaction. Colonies were selected and sent for DNA sequencing.
46 Table 2.10 Oligonucleotides used to generate human CBFβ point mutants.
Primer Name
Primer Sequence (5' to 3') From To
R9G a25g F’ 5'-tcgtgcccgaccagggaagcaagttcgag-3' R’ 5'-ctcgaacttgcttccctggtcgggcacga-3' AGA GGA F12L c36a F’ 5'ccgaccagagaagcaagttagagaacgaggagtt-3' R’ 5'-aactcctcgttctctaacttgcttctctggtcgg-3' TTC TTA R83L g248t F’ 5'-gcgacaaacacctagcctagagtatgtcgacttag-3' R’ 5'-ctaagtcgacatactctaggctaggtgtttgtcgc-3' CGA CTA V95E t284a F’ 5'-aagagaagcaggcaaggaatatttgaaggctccca-3' R’ 5'-tgggagccttcaaatattccttgcctgcttctctt-3' GTA GAA A99V c296t F’ 5'-ggcaaggtatatttgaaggttcccatgattctgaatgga-3' R’ 5'-tccattcagaatcatgggaaccttcaaatataccttgcc-3' GCT GTT p100A c298g F’ 5'-gcaaggtatatttgaaggctgccatgattctgaatgga-3' R’ 5'-tccattcagaatcatggcagccttcaaatataccttgc-3' CCC GCC
47 G121D g362a F’ 5'-ctccaaagactggatgatatgggctgtctggag-3' R’ 5'-ctccagacagcccatatcatccagtctttggag-3' GGT GAT G123C g367t F’ 5'-caaagactggatggtatgtgctgtctggagtttgatg-3' R’ 5'-catcaaactccagacagcacataccatccagtctttg-3' GGC TGC A132T g394a F’ 5'-ggagtttgatgaggagcgaacccagcagga-3' R’ 5'-tcctgctgggttcgctcctcatcaaactcc-3' GCC ACC 2.2.2 Protein techniques
2.2.2.1 Cell culture and transient transfection
A HeLa cell line was used and maintained in ‘DMEM complete media’ that contained 10%FBS, 2mM L-Glutamine and 50μg/ml of penicillin/streptomycin. Prior to experiments, cells were grown for two days.
The cells were initially counted and plated overnight and left to reach 80–90% confluence at the time of transfection. The media was then changed to ‘serum-free media’, and the cells were transfected using a 3:1 ratio of Lipofectamine 2000 to DNA plasmid (12μg of 16– 24 GFP-FLNA or Runx2-Flag). The cells were then incubated for 48 hours prior to harvest. 2.2.2.2 Whole-cell lysis
Cell media were discarded, and cells were washed with cold PBS. An RIPA buffer containing a 1xprotease inhibitor cocktail was added to the plate that was placed on ice (15μl/cm2 of
the growth area). After a 15 minute incubation, cells were dislodged via scraping. The cell suspension was then centrifuged for 15 minutes at 13,200rpm and a temperature of 4°C. The supernatant containing soluble protein from the whole cell extraction was transferred to a new tube.
48 2.2.2.3 Cytoplasmic and nuclear extraction
Cells were washed in PBS and lysed on ice for 15 minutes in ice-cold extraction buffer A (5μl/cm2 of growth area). A 1xprotease inhibitor cocktail, DTT and PMSF were freshly
added into the working buffer. The cells were then left to swell. Next, 10%NP-40 (0.5μl/cm2 of the growth area) was added and were vortexed for 10s prior to centrifuge at
4°C for 1 minute at 13,200rpm. The supernatant, cytoplasmic protein, was collected into a fresh tube. The nuclear pellet left in the tube was then washed twice in extraction buffer A (5μl/cm2 of growth area) and lysed in ice-cold extraction buffer B (0.5μl/cm2 of the
growth area). A 1xprotease inhibitor cocktail, DTT and PMSF were freshly added to the working buffer. The cell suspension was vortexed for 1 hour to shear the nuclear membrane in the cold room. The lysate was then centrifuged at 4°C for 10 minutes at 13,200rpm, and the soluble protein of the nuclear extract was transferred into a fresh tube.
2.2.2.4 Transformation of competent bacterial cells
BL21 gold (DE3) (Agilent) bacterial cells were transformed with pGEX-2TK CBFβ (WT or mutant), Runt, FLNA fragments and Runx2 domains. Rosetta™ 2(DE3) pLysS Competent Cells (Novagen) were also used for CBFβ WT/L21V, 23th Ig repeats and NMTS constructs. The transformation process was performed following the manufacturer’s protocol. The cells were then plated on appropriate antibiotic resistance LB agar plates and incubated at 37°C overnight.
2.2.2.5 LB and minimal media preparation
LB-Amp media were prepared using 1 tablet for every 50ml of ddH2O. The media were then autoclaved, and the ampicillin was added to give a final concentration of 100µg/ml. The LB agar was prepared in the same way but with LB-agar tablets.
Minimal media preparation included two parts. First was the weighting 3g of Na2HPO4 ,
1.5g of KH2PO4 and 0.25g of NaCl for 500ml of a medium, which was then autoclaved. The other components were freshly added on the day the 500 ml of a medium was used. These components were 325μl of trace elements, 7.5ml of 20%w/v glucose, 50μl of 10mg/ml thiamine stock, 0.5g/ml of 15NH
49
CaCl2; all were autoclaved or filter sterilized. This medium was used when labelling
proteins with 15N for NMR studies, as required.
2.2.2.6 Protein overexpression (Luria broth/Minimal media)
A single colony from LB plates was used to inoculate the Ampicillin-medium culture overnight at 37°C. This culture was then added to 100 dilutions of fresh media-Ampicillin media. Ampicillin with a final concentration of 100μg/ml was always added. In the case of uniformly labelled 15N-proteins, the overnight culture was added into minimal media until
the OD600 absorbance was around 0.07.
Growth was then continued until OD600 readings were around 0.4 for CBFβ, Runt and
Runx2 domains and around 0.8 for FLNA fragments. Then, IPTG was added to a final concentration of 200–400µM to induce protein expression. Further growth at 37°C occurred for 2 hours for the Runt and Runx2 domains and for 3 hours for CBFβ, the 23 domain was incubated at 25°C overnight. All the incubations were carried out shaking at 200rpm. Cells were harvested via centrifugation (Beckman) at 5000–5500rpm for 20–30 minutes at 4°C. Bacteria pellets were either stored at -80°C or subjected to lysis.
2.2.2.7 Cell lysis
For every 500ml culture, the bacteria pellet was resuspended in 20ml of cold lysis buffer (Table 2.11) containing a complete protease inhibitor cocktail. The cells were then lysed via 4–5 minutes of sonication on ice—10s on and 30s off—at 15Amp. The lysis suspension was then centrifuged either for 40–50 minutes at 4100rpm or for 30–40 minutes at 13,000rpm and at 4°C to pellet insoluble fraction. The supernatant was transferred to a fresh tube and subjected to further protein analysis. For purifying NMR proteins, about 10ml of fresh, cold lysis buffer containing a protease cocktail was added after the sonication and spinning step to control the proteolysis. All samples subjected to purification were immediately or occasionally kept in the cold room for no longer than overnight. CBFβ WT/L21V could be stored at -20°C.