organic papers
Acta Cryst.(2005). E61, o3145–o3146 doi:10.1107/S1600536805027443 Xuet al. C
17H9N3O2
o3145
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
4-[(1,3-Dioxo-2,3-dihydro-1
H
-isoindol-2-yl)methyl]-phthalonitrile
Xiu-Zhi Xu, Mei-Jin Lin, Jun-Dong Wang,* Nai-Sheng Chen and Jin-Ling Huang
Department of Chemistry, University of Fuzhou, Fuzhou 350002, People’s Republic of China, and State Key Laboratory of Structural Chem-istry, Fujian Institute of Research on the Structure of Matter, Fuzhou 350002, People’s Republic of China
Correspondence e-mail: wangjd@fzu.edu.cn
Key indicators
Single-crystal X-ray study
T= 298 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.037
wRfactor = 0.117
Data-to-parameter ratio = 13.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, C17H9N3O2, the phthalonitrile group
and the isoindole-1,3-dione group lie in approximately orthogonal planes and exhibit a dihedral angle of 92.76 (8).
Partial face-to-face overlap is observed between the two groups. In addition, the crystal packing is stabilized by C— H O and C—H N interactions.
Comment
The title compound, (I), is a precursor for the synthesis of amphiphilic phthalocyanine, which is utilized in the photo-dynamic therapy (PDT) of tumors (Huanget al., 2000). After phthalocyanine is formed, the phthalimide is easily converted to an amine, which is believed to have a better interaction with cell tissues. In the structure of a similar precursor (Zhuet al., 2005), the two groups are connected by a flexible butoxy chain. The difference between the earlier structure and (I) is that there is no O atom linked to the phthalocyanine ring, and we are investigating if this O atom will or will not influence the yield of active1O2in PDT (Kobayashiet al., 2003).
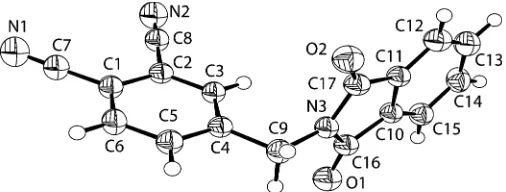
The molecular structure of (I) is shown in Fig. 1. The cyano-group N1—C7 [1.130 (3) A˚ ] and N2—C8 bonds [1.137 (3) A˚] are short enough to indicate their triple-bond character, and agree with the values reported by Zhuet al.(2005). The C1— C7 [1.436 (3) A˚ ] and C2—C8 [1.435 (3) A˚] bond distances are comparable to the mean value in phthalonitrile, 1.443 (8) A˚ , reported by Allen et al. (1987). The benzene ring of the phthalonitrile group and the isoindole ring system are essen-tially perpendicular to each other, with an angle of 92.76 (8).
In the crystal packing, the molecules are stacked along the short a axis with weak – interactions due to the partial overlap of the C1–C6 benzene rings and the isoindole ring systems, with perpendicular distances of 3.556 and 3.150 A˚ ,
respectively. In addition, the molecular packing is stabilized by C—H O and C—H N interactions (Table 1).
Experimental
4-Methylphthalic anhydride (50 g, 308 mmol) and urea (18.5 g, 308 mmol) were mixed and heated to 433 K; the mixture melted then solidified. The solid was washed with water and vacuum dried at 353 K to give 4-methylphthalimide (yield 87%, 43.5 g; m.p. 470.8– 471.4 K). All of the 4-methylphthalimide obtained was suspended in methanol (800 ml) and stirred for 5–7 days at room temperature while ammonia gas was pumped into the vessel. As the reaction progressed, the suspension dissolved and then a new suspension formed. After filtering and drying at 343 K, 4-methylphthalic diamide was obtained (yield 30%, 12 g; m.p. 455.4–455.6 K). 4-Methylphthalic diamide (10 g, 56.1 mmol) was suspended in pyridine (150 ml) and POCl3 (13 ml, 142 mmol) was added dropwise at 276–278 K. The
mixture was stirred at room temperature for 3 h and then poured into ice water; the precipitate was filtered off, washed with water and dried at 353 K, and 4-methylphthalonitrile was obtained (yield 60%, 4.6 g; m.p. 391.5–391.6 K). This was suspended in CCl4(70 ml), and thenN
-bromosuccimide (5.8 g, 32.6 mmol) and benzoyl peroxide (0.1–0.2 g) were added. The suspension was refluxed for 24 h, and then cooled to room temperature and filtered; the CCl4was evaporated and crude
bromomethylphthalonitrile (3.5 g) was obtained. The crude 4-bromomethylphthalonitrile (3.5 g, 15.8 mmol), phthalimide (2.4 g, 16.3 mmol) and K2CO3 (5 g, 36.2 mmol) were added to N,N
-dimethylformamide (60 ml), and then stirred at 353 K for 12 h. The reaction mixture was poured into ice water (800 ml), and the preci-pitate was separated by centrifugation, washed with water and vacuum dried at 353 K, resulting in crude (I) (1.7 g). The crude (I) was washed with CH3OH and recrystallized from tetrahydrofuran
and CH2Cl2repeatedly; colorless needle-shaped crystals of (I) were
obtained (m.p. 550–551 K). MS (m/z, %): 287 (M+, 100); IR (KBr, cm1): 2229 (C—N), 3069 (Ar—H), 1706 (C–O), 1776, 1420 (–CH2–),
1390, 1112 (N—H), 951 (C—H), 724; UV–vis: 227.41, 294.02 nm (CH2Cl2), 235.04, 284.18, 293.95 nm (THF);
1
H NMR (DMSO-d6):
4.919 (s, 2H), 7.910 (br, 5H), 8.115 (d, 1H), 8.183 (s, 1H).
Crystal data
C17H9N3O2 Mr= 287.27
Monoclinic,P21=c a= 4.9331 (3) A˚
b= 26.2490 (13) A˚
c= 10.4901 (6) A˚
= 93.0930 (13) V= 1356.37 (13) A˚3 Z= 4
Dx= 1.407 Mg m
3
MoKradiation Cell parameters from 6576
reflections
= 6.1–54.9
= 0.10 mm1 T= 298 (2) K Needle, colorless 0.700.100.08 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
!scans
11806 measured reflections 3107 independent reflections 1558 reflections withI> 2(I)
Rint= 0.044
max= 27.5
h= 0!6
k= 0!34
l=13!13
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.037 wR(F2) = 0.117
S= 1.00 3107 reflections 235 parameters
All H-atom parameters refined
w= 1/[2(F
o2) + (0.042P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.14 e A˚
3
min=0.23 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C3—H1 O2i
0.91 (2) 2.47 (2) 3.275 (3) 148 (2) C6—H3 N2ii
0.95 (2) 2.60 (2) 3.280 (3) 129 (2) C15—H9 O1iii 0.97 (2) 2.44 (2) 3.378 (3) 161 (2)
Symmetry codes: (i)x1;y;z; (ii)xþ1;yþ1 2;z
1
2; (iii)x;yþ1;zþ1.
All H atoms were located in a difference Fourier map and refined isotropically. The C—H distances lie in the range 0.89 (2)–1.01 (3) A˚ . Data collection:RAPID-AUTO (Rigaku, 1998); cell refinement:
RAPID-AUTO; data reduction:RAPID-AUTO; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
ORTEX (McArdle, 1995); software used to prepare material for publication:SHELXL97.
This work was supported by the Science Foundation of Fujian Province (No. E0310007) and the Science and Tech-nology Developing Foundation of Fuzhou University (No. 2004-XQ-10).
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2. pp. S1–19.
Huang, J. L., Chen, N. S., Huang, J. D., Liu, E. S., Xue, J. P., Yang, S. L., Huang, Z. Q. & Sun, J. C. (2000).Sci China Ser. B,30, 481–488.
Kobayashi, N., Ogata, H., Nonaka, N. & Luk’yanets, E. A. (2003).Chem. Eur. J.9, 5123–5134.
McArdle, P. (1995).J. Appl. Cryst.28, 65.
Rigaku (1998).RAPID-AUTO. Version 1.06. Rigaku Corporation, Tokyo, Japan.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
[image:2.610.314.567.71.167.2]Zhu, X.-L., Lin, M.-J., Wang, J.-D., Chen, N.-S. & Huang, J.-L. (2005).Acta Cryst.E61, o1214–o1215.
Figure 1
supporting information
sup-1
Acta Cryst. (2005). E61, o3145–o3146supporting information
Acta Cryst. (2005). E61, o3145–o3146 [doi:10.1107/S1600536805027443]
4-[(1,3-Dioxo-2,3-dihydro-1
H
-isoindol-2-yl)methyl]phthalonitrile
Xiu-Zhi Xu, Mei-Jin Lin, Jun-Dong Wang, Nai-Sheng Chen and Jin-Ling Huang
S1. Comment
The title compound, (I), is a precursor for the synthesis of amphiphilic phthalocyanine, which is utilized in the photodynamic therapy (PDT) of tumors (Huang et al., 2000). After phthalocyanine is formed, the phthalimide is easily converted to an amine, which is believed to have a better interaction with cell tissues. In the structure of a similar precursor (Zhu et al., 2005), the two groups are connected by a flexible butoxy chain. The difference between the earlier structure and (I) is that there is no O atom linked to the phthalocynine ring, and we are investigating if this O atom will or will not influence the yield of active 1O
2 in PDT (Kobayashi et al., 2003).
The molecular structure of (I) is shown in Fig. 1. The cyano-group N1—C7 [1.130 (3) Å] and N2—C8 bonds [1.137 (3) Å] are short enough to indicate their triple-bond character, and agree with the values reported by Zhu et al. (2005). The C1—C7 [1.436 (3) Å] and C2—C8 [1.435 (3) Å] bond distances are comparable to the mean value of phthalonitrile, 1.443 (8) Å, reported by Allen et al. (1987). The benzene ring of the phthalonitrile group and the isoindole ring system are perpendicular to each other, with an angle of 92.76 (8)°.
In the crystal packing, the molecules are stacked along the short a axis with weak π–π interactions due to the partial overlap of the C1–C6 benzene rings and the isoindole ring systems, with perpendicular distances of 3.556 and 3.150 Å, respectively. In addition, the molecular packing is stabilized by C—H···O and C—H···N interactions (Table 1).
S2. Experimental
4-Methylphthalic anhydride (50 g, 308 mmol) and urea (18.5 g, 308 mmol) were mixed and heated to 433 K; the mixture melted then solidified. The solid was washed with water and vacuum dried at 353 K to give 4-methylphthalimide (yield 87%, 43.5 g; m.p. 470.8–471.4 K). All of the 4-methylphthalimide obtained was suspended in methanol (800 ml) and stirred for 5–7 days at room temperature while ammonia gas was pumped into the vessel. As the reaction progressed, the suspension dissolved and then a new suspension formed. After filtering and drying at 343 K, 4-methylphthalic diamide was obtained (yield 30%, 12 g; m.p. 455.4–455.6 K). 4-Methylphthalic diamide (10 g, 56.1 mmol) was suspended in pyridine (150 ml) and POCl3 (13 ml, 142 mmol) was added dropwise at 276–278 K. The mixture was stirred at room
temperature for 3 h and then poured into ice water; the precipitate was filtered off, washed with water and dried at 353 K, and 4-methylphthalonitrile was obtained (yield 60%, 4.6 g; m.p. 391.5–391.6 K). This was suspended in CCl4 (70 ml),
and then N-bromosuccimide (5.8 g, 32.6 mmol) and benzoyl peroxide (0.1–0.2 g) were added. The suspension was refluxed for 24 h, and then cooled to room temperature and filtered; the CCl4 was evaporated and crude
4-bromomethyl-phthalonitrile (3.5 g) was obtained. The crude 4-bromomethyl4-bromomethyl-phthalonitrile (3.5 g, 15.8 mmol), phthalimide (2.4 g, 16.3 mmol) and K2CO3 (5 g, 36.2 mmol) were added to N,N-dimethylformamide (60 ml), and then stirred at 353 K for 12 h.
The reaction mixture was poured into ice water (800 ml), and the precipitate was separated by centrification, washed with water and vacuum dried at 353 K, resulting in crude (I) (1.7 g). The crude (I) was washed with CH3OH and recrystallized
(m/z, %): 287 (M+, 100); IR (KBr, cm−1): 2229 (C—N), 3069 (Ar—H), 1706 (C–O), 1776, 1420 (–CH
2–), 1390, 1112 (N
—H), 951 (C—H), 724; UV–vis: 227.41, 294.02 nm (CH2Cl2), 235.04, 284.18, 293.95 nm (THF); 1H NMR (DMSO-d6):
δ 4.919 (s, 2H), 7.910 (br, 5H), 8.115 (d, 1H), 8.183 (s, 1H).
S3. Refinement
[image:4.610.116.485.175.316.2]All H atoms were located in a difference Fourier map and refined isotropically. The C—H distances lie in the range 0.89 (2)–1.01 (3) Å.
Figure 1
The structure of (I), showing the atomic numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
4-[4-(1,3-Dioxo-2,3-dihydro-1H-isoindol-2-yl)methyl]phthalonitrile
Crystal data
C17H9N3O2
Mr = 287.27 Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 4.9331 (3) Å
b = 26.2490 (13) Å
c = 10.4901 (6) Å
β = 93.0930 (13)°
V = 1356.37 (13) Å3
Z = 4
F(000) = 592
Dx = 1.407 Mg m−3
Melting point: 550 K
Mo Kα radiation, λ = 0.71069 Å Cell parameters from 6576 reflections
θ = 6.1–54.9°
µ = 0.10 mm−1
T = 298 K Needle, colourless 0.70 × 0.10 × 0.08 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
Radiation source: Rotating anode
Graphite Monochromator monochromator
ω scans
11806 measured reflections 3107 independent reflections
1558 reflections with I > 2σ(I)
Rint = 0.044
θmax = 27.5°, θmin = 1.6°
h = 0→6
k = 0→34
l = −13→13
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.037
wR(F2) = 0.117
S = 1.00
3107 reflections 235 parameters 0 restraints
supporting information
sup-3
Acta Cryst. (2005). E61, o3145–o3146Secondary atom site location: difference Fourier map
Hydrogen site location: difference Fourier map All H-atom parameters refined
w = 1/[σ2(F
o2) + (0.042P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.14 e Å−3
Δρmin = −0.23 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C17 0.7488 (5) 0.45506 (8) 0.7767 (2) 0.0452 (5)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0690 (12) 0.0635 (10) 0.0591 (11) 0.0001 (9) −0.0150 (10) −0.0092 (8) N3 0.0472 (10) 0.0382 (9) 0.0425 (10) −0.0015 (9) 0.0045 (9) −0.0008 (8) O2 0.0583 (11) 0.0651 (10) 0.0642 (11) 0.0162 (9) −0.0093 (9) −0.0038 (9) N2 0.0887 (17) 0.0611 (13) 0.0777 (16) −0.0039 (12) 0.0414 (14) 0.0014 (11) N1 0.105 (2) 0.0569 (13) 0.0849 (18) −0.0153 (14) 0.0165 (15) 0.0083 (13) C1 0.0510 (13) 0.0399 (12) 0.0490 (13) −0.0011 (10) 0.0045 (11) −0.0004 (10) C2 0.0424 (12) 0.0428 (12) 0.0415 (13) −0.0004 (10) 0.0053 (10) 0.0008 (9) C3 0.0470 (13) 0.0386 (12) 0.0427 (13) 0.0048 (11) 0.0072 (10) −0.0027 (10) C4 0.0415 (12) 0.0402 (12) 0.0400 (12) 0.0000 (10) 0.0022 (10) −0.0026 (9) C5 0.0522 (14) 0.0462 (13) 0.0500 (14) 0.0037 (11) 0.0157 (12) −0.0058 (11) C6 0.0643 (16) 0.0392 (13) 0.0588 (16) 0.0045 (12) 0.0134 (13) −0.0065 (11) C7 0.0671 (17) 0.0448 (13) 0.0583 (15) −0.0030 (13) 0.0086 (13) 0.0006 (12) C8 0.0570 (15) 0.0434 (13) 0.0543 (15) −0.0022 (11) 0.0148 (13) 0.0017 (11) C9 0.0534 (16) 0.0426 (12) 0.0508 (15) −0.0008 (12) 0.0140 (12) −0.0033 (11) C10 0.0431 (13) 0.0399 (11) 0.0422 (13) −0.0052 (10) 0.0062 (10) 0.0025 (9) C11 0.0436 (13) 0.0408 (11) 0.0419 (13) −0.0042 (10) 0.0063 (11) 0.0001 (10) C12 0.0558 (16) 0.0591 (16) 0.0497 (15) −0.0042 (13) −0.0038 (13) −0.0066 (12) C13 0.0754 (18) 0.0484 (14) 0.0617 (17) −0.0015 (14) 0.0057 (15) −0.0138 (13) C14 0.0634 (17) 0.0422 (13) 0.0679 (17) 0.0046 (13) 0.0071 (14) −0.0020 (12) C15 0.0521 (14) 0.0475 (13) 0.0532 (15) 0.0026 (12) 0.0042 (12) 0.0059 (11) C16 0.0466 (13) 0.0457 (12) 0.0429 (13) −0.0063 (11) 0.0049 (11) 0.0035 (10) C17 0.0445 (13) 0.0484 (12) 0.0432 (13) −0.0016 (11) 0.0065 (11) 0.0032 (10)
Geometric parameters (Å, º)
supporting information
sup-5
Acta Cryst. (2005). E61, o3145–o3146C17—N3—C16 111.94 (17) C4—C9—H5 105.4 (14) C17—N3—C9 124.89 (19) H4—C9—H5 111 (2) C16—N3—C9 123.16 (19) C15—C10—C11 121.8 (2) C6—C1—C2 118.9 (2) C15—C10—C16 130.1 (2) C6—C1—C7 121.4 (2) C11—C10—C16 108.11 (18) C2—C1—C7 119.7 (2) C12—C11—C10 121.0 (2) C3—C2—C1 120.1 (2) C12—C11—C17 130.8 (2) C3—C2—C8 120.42 (19) C10—C11—C17 108.11 (18) C1—C2—C8 119.50 (19) C11—C12—C13 117.6 (2) C2—C3—C4 121.0 (2) C11—C12—H6 120.6 (15) C2—C3—H1 118.8 (13) C13—C12—H6 121.8 (15) C4—C3—H1 120.3 (13) C12—C13—C14 121.4 (2) C3—C4—C5 118.5 (2) C12—C13—H7 120.2 (15) C3—C4—C9 122.34 (19) C14—C13—H7 118.4 (14) C5—C4—C9 119.1 (2) C13—C14—C15 121.3 (2) C6—C5—C4 121.2 (2) C13—C14—H8 120.0 (15) C6—C5—H2 118.8 (13) C15—C14—H8 118.6 (15) C4—C5—H2 120.0 (13) C10—C15—C14 116.9 (2) C5—C6—C1 120.4 (2) C10—C15—H9 124.3 (13) C5—C6—H3 121.5 (13) C14—C15—H9 118.7 (13) C1—C6—H3 118.1 (13) O1—C16—N3 124.1 (2) N1—C7—C1 178.6 (3) O1—C16—C10 130.0 (2) N2—C8—C2 179.6 (3) N3—C16—C10 105.91 (18) N3—C9—C4 114.01 (18) O2—C17—N3 124.4 (2) N3—C9—H4 110.2 (13) O2—C17—C11 129.6 (2) C4—C9—H4 107.9 (13) N3—C17—C11 105.92 (19) N3—C9—H5 107.9 (14)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C3—H1···O2i 0.91 (2) 2.47 (2) 3.275 (3) 148.4 (17)
C6—H3···N2ii 0.95 (2) 2.60 (2) 3.280 (3) 128.6 (16)
C15—H9···O1iii 0.97 (2) 2.44 (2) 3.378 (3) 160.7 (17)