Published: May 10, 2011
pubs.acs.org/cm
Electrochemical Windows of Room-Temperature Ionic Liquids from
Molecular Dynamics and Density Functional Theory Calculations
Shyue Ping Ong,
†Oliviero Andreussi,
‡Yabi Wu,
†Nicola Marzari,
‡and Gerbrand Ceder*
,††Department of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge,
Massachusetts 02139, United States
‡Department of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, United Kingdom
b
S Supporting Information’INTRODUCTION
The electrochemical window of a solvent is an important design criteria for electrochemical applications. An electro chemical solvent, such as the electrolyte in a lithium-ion battery or supercapacitor, must support the voltage in which the electrochemical device operates. The electrochemical window is defined as the difference between the cathodic and anodic limits, which are the potentials at which reduction and oxidation of the solvent take place respectively. For certain applications (e.g., supercapacitors), it is the overall window that matters, while in other applications, the actual cathodic and anodic limits relative to some reference is the limiting factor. For example, in lithium-ion batteries where the anode potential is set by Li/Liþ, the cathodic limit relative to Li/Liþwould determine whether the solvent would be reduced by lithium metal, and the anodic limit would determine the voltage allowable for the cathode.
In the pursuit of higher energy density storage systems, the electrochemical windows of current electrolytes have become a limiting factor. For example, the typical organic carbonate electrolyte used in current lithium-ion batteries, a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) with LiPF6added to improve Liþconductivity, can only support a
voltage up to approximately 5 V.2Furthermore, this EC/DMC electrolyte is unstable against Li in the graphitic carbon anode and works only because of the formation of a passivating solid-electrolyte interphase (SEI) layer that prevents further reaction. Room-temperature ionic liquids (ILs) have garnered increas-ing interest as electrolytes in electrochemical applications.39As a class of materials, ILs generally exhibit low volatility, low
flammability, and high thermal stability, which provide signifi -cant safety advantages overflammable organic compounds. This is especially important in the application of Li-batteries beyond small-scale portable electronics to large-scale applications such as hybrid electric vehicles (HEVs) and electric vehicles. Many ILs also have wide electrochemical windows of approximately 56 V or more,10,11which are considerably larger than that of current organic electrolytes. A more electrochemically stable electrolyte could unlock the potential of high-voltage cathodes with higher power density. For example, LiNiPO4, which is predicted to have
a potential greater than 5 V,12 would have 50% higher energy
Received: March 7, 2011
Revised: April 26, 2011 ABSTRACT:We investigated the cathodic and anodic limits of six room-temperature ionic liquids (ILs) formed from a combination of two common cations, 1-butyl-3-methylimidazo-lium (BMIM) andN,N-propylmethylpyrrolidinium (P13), and three common anions, PF6,
BF4, and bis(trifluoromethylsulfonyl)imide (TFSI), using an approach that combines
molecular dynamics (MD) simulations and density functional theory (DFT) calculations. All interion interactions were taken into account by explicitly modeling the entire liquid structure using classical MD, followed by DFT computations of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies. The relative cathodic and anodic limits of BMIM PF6, BMIM BF4, BMIM TFSI, and P13
TFSI obtained from our approach are in fairly good agreement with existing experimental data. From our DFT calculations, we also obtained the cation- and anion-projected density of states (DOS), which provide information on the likely species contributing to reductive and
oxidative decomposition. Our predictions support Howlett et al.’s earlierfinding1that the TFSI anion is less stable than the P13 cation against reduction. In addition, our results provide surprising evidence of possible cation anodic instability; we predict the aromatic BMIM cation to be less stable against oxidation than the respective anions in BMIM PF6and BMIM BF4, and the P13
cation to be less stable against oxidation than the PF6anion in P13 PF6. We also present a comparison of the predictions of our
approach with that of simpler approximations based on in vacuo or polarizable continuum model calculations.
density than the Fe-based LiFePO4 system currently under
development for HEVs. Similarly, Li(Ni0.5Mn1.5)O4has higher
capacity, higher voltage, and better rate capability than the Mn-based spinel currently in use for large format batteries, but operates at a high voltage close to the breakdown of the carbonate electrolyte.13
In ILs, it has typically been assumed that the cathodic limit is set by the reduction of the cations and the anodic limit is set by the oxidation of the anions.14,15 However, recent experiments have demonstrated that this may not always be the case. For instance, experimental and theoretical investigations by Howlett et al.1 have provided evidence that the commonly used bis-(trifluoromethylsulfonyl)imide (TFSI) undergoes reductive de-composition at a potential that is more than 1 V above the potential at which the N,N-propylmethylpyrrolidinium (P13) cation decomposes.
In this work, we investigated the cathodic and anodic limits of six common ILs using an approach that combines molecular dynamics simulations and density functional theory calcu-lations.16In our approach, all interion interactions are taken into account by explicitly modeling the entire liquid structure, which is in contrast to previous approaches to calculating solvent electrochemical windows that focused primarily on isolated molecules or ion-pairs in vacuo or in a continuum solvent.1724 In addition, our approach also provides information on the species (cation or anion) contributing to the redox stability of the overall liquid. We show that the predictions from our approach support the previous finding that the TFSI anion is less stable than the P13 cation against reduction. Our results also provide surprising evidence that points to possible cation anodic instability, especially in the case of the aromatic 1-butyl-3-methylimidazolium (BMIM) cation. We compare the results obtained using this approach to the predictions of simpler approximations based on in vacuo and polarizable continuum model (PCM) calculations and discuss the limitations of each level of approximation. The approach used in this work can potentially be used to estimate the electrochemical windows of other solvents as well.
’METHODOLOGY
Estimating Cathodic and Anodic Limits of Common ILs.
We investigated all possible combinations of two common IL cations, 1-butyl-3-methylimidazolium (BMIM) and N,N-propylmethylpyrroli-dinium (P13), and three common IL anions, PF6, BF4, and
bis-(trifluoromethylsulfonyl)imide (TFSI), that is, a total of six different ILs (see Figure 1). These ions were chosen because of the widely available experimental data for BMIM PF6, BMIM BF4, BMIM TFSI,
and P13TFSI, as well as to study a diverse range of local environments, for example, the more charge-delocalized aromatic BMIM cation and TFSI anion versus the more charge-localized aliphatic P13 cation and PF6and BF4anions.
To our knowledge, there is no reported experimental data for P13 PF6
and P13 BF4in the literature. ILs based on the similar
tetraethylammo-nium cationþBF4or PF6have been reported to have fairly high melting
points above room temperature,25and it is unclear whether the P13 analogues are liquids at room temperature. We have included P13 PF6
and P13 BF4in our investigations only for the sake of completeness and
to ascertain general trends in the electrochemical windows with changes in cation or anion.
We assume that the cathodic limit is set by the lowest unoccupied molecular orbital (LUMO) and the anodic limit is set by the highest
occupied molecular orbital (HOMO). The potentials of the cathodic and anodic limits,VCLandVAL, for a one-electron transfer can then be
obtained from the respective orbital energies using the following expressions:
VCL ¼ εLUMO
e ð1Þ
VAL ¼ εHOMO
e ð2Þ
whereεHOMOandεLUMOare the energies of the HOMO and LUMO levels, respectively, andeis the electron charge.
For the six ILs chosen, we compared the cathodic and anodic limits predicted using three levels of approximations.
The simplest level of approximation is to calculate the electron affinities (EA) and ionization energies (IE) of each of the constituent ions individually in vacuo using theΔ-SCF approach, that is, by explicitly taking the difference between the calculated energy of an ion and the calculated energy of that ion with one additional or one less electron.26 The EA corresponds to the energy of the LUMO level, while the IE corresponds to the energy of the HOMO level. For an IL formed from a combination of a cation C and anion A, we assume that the cathodic limit is given by the maximum of the cathodic limits of the two ions, and the anodic limit is given by the minimum of the anodic limits of the two ions, that is:
VCLvacuum ¼maxðVCLvacuum,C ,VCLvacuum,A Þ ð3Þ
Vvacuum
AL ¼minðVALvacuum,C ,VALvacuum,A Þ ð4Þ
whereVCL,ivacuumandVAL,ivacuumare the in vacuo cathodic and anodic limits of
ion i, respectively.
The next level of approximation is to model each ion in solution using a continuum solvation model and to calculate the HOMO and LUMO levels in solution.27,28In this work, we have adopted the integral equation formalism variant of the polarizable continuum model (PCM).2946This model takes into account, in a mean-field manner, the stabilization of an ion from the dielectric response of its surroundings. Similar to the vacuum calculations, we assume the following:
VCLPCM ¼maxðVCLPCM,C,VCLPCM,AÞ ð5Þ
VPCM
AL ¼minðVALPCM,C,VALPCM,AÞ ð6Þ
The highest level of approximation is to calculate the HOMO and LUMO levels of the six ILs using a combination of molecular dynamics
Figure 1. IL ions investigated. Cations: (a) 1-butyl-3-methylimidazo-lium (BMIM); (b) N,N-propylmethylpyrrolidinium (P13). Anions: (c) Hexafluorophosphate (PF6); (d) tetrafluoroborate (BF4); (e)
(MD) simulations and periodic boundary condition (PBC) density functional theory (DFT) calculations on unit cells containing the IL cations and anions in their liquid configuration. These calculations were carried out in two steps:
(1) Each IL wasfirst equilibrated via MD simulations using well-established force-fields. From the MD simulations, we extracted 10 equilibrated snapshots spaced 1000 MD steps (∼2 ps of simulation time) apart for each IL system.
(2) The snapshots were then used as inputs for periodic boundary DFT calculations to determine the density of states (DOS) and the HOMO and LUMO energies at the MD-equilibrated geometry; that is, no further geometry optimization was per-formed. The HOMO and LUMO energies of each IL are then obtained as the average of the HOMO and LUMO energies of all 10 snapshots of each IL, and aligned to a common vacuum reference using its Hartree potential.
Computational Methods. Molecular Dynamics Simulations.
Our classical molecular dynamics (MD) simulations were carried out using the DLPOLY program.4749For the interaction potentials, the anions and the BMIM cation were modeled using the OPLS-AA50-based force field derived by Lopes et al.5153The force field for the P13 cation was derived from the standard OPLS parameters for amines,50,53while atomic charges were explicitly parametrized using the CHelpG procedure54on ab initio MP2/cc-pVTZ(-f) calculations. Equilibration of the systems in the NPT ensemble was enforced using the Berendsen thermostat and barostat.55 A simulation time step of 2 fs and an interaction cutoff radius of 15 Å were used throughout.
Initial configurations containing 128 ion pairs were generated starting from a face-centered cubic lattice with the ions occupying random lattice sites. Equilibration runs were performed at 500 K and zero pressure until convergence of the statistical average of the density was achieved. Following equilibration, configurations of clusters of 16 ion pairs were extracted from thefinal configuration of the trajectories. These smaller clusters were then equilibrated with periodic boundary conditions at 500 K and zero pressure, followed by quenching for a few nanoseconds at 300 K, to obtain the input structures for the DFT calculations.
Periodic Boundary Condition DFT Calculations.The DOS calcula-tions were performed using the PerdewBurkeErnzerhof (PBE) generalized gradient approximation functional (GGA) with an energy cutoff of 500 eV. Projected augmented wave (PAW)56pseudopotentials
were used, as implemented in the Vienna ab initio simulation package (VASP).57Because of the large size of the unit cells, only a singleΓk-point was used, and our convergence tests found that increasing k-point density to a largerΓ-centered 222 grid had a negligible effect on the orbital energies obtained. All calculations were nonspin-polarized.
To align the orbital energies and density of states obtained for all ILs to a common reference, we determined the Hartree potential for all ILs relative to a vacuum layer.5860A vacuum slab of equal size was attached to each IL unit cell in the [100] direction. Using this extended unit cell, we then calculated the Hartree potential using the same parameters as that used for the DOS computations. The average Hartree potential is then plotted as a function of
the cella-coordinate (see the Supporting Information) to determine the difference in the average Hartree potentials in the IL and vacuum regions.
For each IL, only one Hartree potential shift relative to vacuum was determined using the MD configuration that exhibited the smallest dipole between the two slab surfaces, that is, the ILþvacuum cell that had theflattest Hartree potential in the vacuum region was used. We kept a constant shift for each IL as the difference between the average Hartree potential in the IL region and in the vacuum region. The interfacial regions between the IL and vacuum where there are sharp changes in the Hartree potential were excluded in calculating the average Hartree potentials. The constant shift was then applied to the orbital energies and DOS for the IL. The calculated Hartree potential shifts for the six ILs are given in Table 1.
Vacuum and Polarizable Continuum Calculations. All vacuum and polarizable continuum model calculations in this Article were performed using the Gaussian 03 quantum chemistry package.61 Geometry optimizations were carried out at the B3LYP/6-31þG(d) level and were followed by single-point energy calculations at the B3LYP/6-311þG(2d,p) level. The hybrid B3LYP density functional based on Becke’s three-parameter exchange functional62and the corre-lation functional of Lee, Yang, and Parr63was chosen as it has been shown to provide good accuracy for EAs and IEs at a reasonable computation cost.64The inclusion of diffuse functions in the basis sets ensures an adequate description of the diffuse electron cloud of anions and the radicals formed from cation reduction. Closed-shell restricted wave function calculations were used in the treatment of the cations and anions, while unrestricted open-shell wave functions were used for the ions that have singly occupied orbitals. Frequency analysis was per-formed to ensure that structures obtained were minimum energy struc-tures rather than transition strucstruc-tures, and also to obtain the thermochemical corrections for the Gibbs free energy. The thermo-chemical corrections were then scaled using the factor of 0.9806 determined earlier by Scott et al.65for the B3LYP/6-31G(d) model chemistry.
All PCM calculations were performed using a dielectric constant of 12, which is typical of ILs.66
Table 1. Hartree Potential Shifts
ionic liquid shift (EHartreeIL EHartreevacuum)
BMIM PF6 5.07 eV
BMIM BF4 4.83 eV
BMIM TFSI 5.66 eV
P13 PF6 5.11 eV
P13 BF4 4.56 eV
P13 TFSI 5.42 eV
’RESULTS
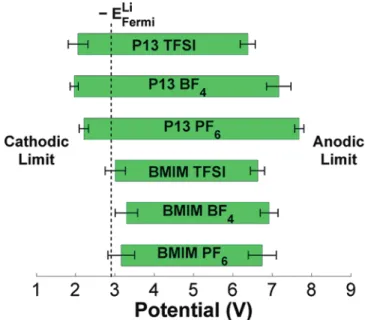
Electrochemical Windows from MDþDFT Calculations. Figure 2 summarizes the calculated electrochemical windows of the six ILs from our MDþDFT calculations. The solid green rectangular bars indicate the potential region for which a particular IL is stable. The minimum of the stable potential region is the cathodic limit (where reduction of the IL is expected to occur), while the maximum is the anodic limit (where oxidation of the IL is expected to occur). As the limits were determined as the average of 10 MD snapshots for each IL, the 95% confidence interval for each limit is indicated by the black dumbbells. In the figure, we also plotted the calculated Fermi level of lithium metal on the same potential scale using the same Hartree potential alignment approach. Our calculated value for EFermiLi of 2.91 eV (relative to vacuum) is in excellent agreement
with the experimental work function of lithium of 2.93 eV,67 which lends further support for our Hartree potential approach to aligning the orbital energies and DOS.
Before we outline the key observations from our results, we would like to note that comparing the predictions from our calculations with experiments is complicated by the fact that measured electrochemical windows depend heavily on the
measurement conditions.10,15 Typically, the electrochemical window is determined by performing a linear sweep voltammetry using inert electrodes (e.g., Pt or Au) and measuring the cathodic and/or anodic currents, which are indicative of electrolyte reduction or oxidation. However, reported electrochemical win-dows in the literature vary widely in the measurement conditions, differing in the type of electrodes, the arbitrary current cutoff used to determine the onset of redox processes (typically between 0.1 and 1.0 mA cm2), and the references used, some of which may not be strictly electrochemically defined. In the case of ILs, this difficulty is further compounded by their sensitivity to water, air, and other impurities.6870
Notwithstanding the above, a few consistent observations can be made from Figure 2 and experimental data from studies comparing several ILs:
(1) Relative cathodic stability of P13- and BMIM-based ILs: The P13-based ILs have significantly lower cathodic limits than the BMIM-based ILs, in agreement with the general experimental observation that aliphatic cations such as P13 are in general more stable than aromatic ones such as BMIM.10,14,15
ILs, which is in agreement with the relative anion stabilities calculated by Ue et al.18 and our previous work.23 On the other hand, all BMIM-based ILs have relatively similar anodic limits, regardless of the anion. As we shall demonstrate in the following subsection, we hypothesize that this is due to the relative instability of BMIM toward both reduction and oxidation.
(3) Underestimation of electrochemical windows: The pre-dicted electrochemical windows for the BMIM-based ILs are all around 3.6 V, which is somewhat smaller than the 4.24.6 V observed by Lewandowski et al.71
using Pt working electrodes in a Ag|Agþ in DMSO reference. However, it is well-known that DFT based on the local density approximation (LDA) and GGA underestimates the energy gap between occupied and unoccupied states by 30100%.7275Indeed, using the HeydScuseria
Ernzerhof hybrid functional (HSE06),76,77which is well-known to provide more accurate band gaps,75,78 we calculated the electrochemical windows of BMIM PF6,
BMIM BF4, BMIM TFSI, and P13 TFSI (the four
experi-mentally known ILs among the six we investigated) for a representative liquid configuration for each IL to be ap-proximately 4.9, 5.1, 5.5, and 6.1 V, respectively, signifi -cantly larger than the GGA electrochemical windows (see Figure 3). However, the HSE06 functional is far too expensive for the purposes of our investigations, which involve multiple calculations on unit cells containing more than 600 atoms in some cases. Despite this limita-tion of GGA, we observe that the relative trends in the limits are qualitatively the same in both HSE06 and GGA. In general, the electrochemical windows of all three BMIM-based ILs are fairly similar both in experi-ments and in our calculations.
(4) Electrochemical window of P13 TFSI: Our calculations predict the electrochemical window of P13 TFSI to be significantly wider than the BMIM-based ILs by approxi-mately 0.7 V. This prediction disagrees with Lewandowski et al.’s measurements,71which found the electrochemical window of P13 TFSI to be 3.7 V and somewhat smaller than that of the BMIM-based ILs. However, more recent work by Lewandowski et al.79found the electrochemical window (on glassy carbon) of 0.1 M LiTFSI dissolved in the similarN,N-butylmethylpyrrolidinium TFSI IL to be 5.1 V, slightly wider than the electrochemical window of 4.9 V measured for 0.1 M LiPF6 in BMIM PF6. This finding is in qualitative agreement with our predictions. Experiments by Barisci et al.80using Pt working electro-des and Ag wire reference also found the electrochemical window of P13 TFSI (5.7 V) to be larger than that of BMIM PF6(3.2 V). It should be noted, however, that
Barisci et al. measured P13 TFSI to have a higher anodic limit and lower cathodic limit than BMIM PF6, while our
calculations predict P13 TFSI to have both lower catho-dic and anocatho-dic limits than BMIM PF6.
(5) Stability versus lithium: The cathodic limits of the BMIM ILs are all above the Li potential, which indicates that BMIM is likely to be reduced by lithium metal. The P13 ILs, on the other hand, have cathodic limits that are below the Li potential, which suggest that they could be stable against lithium. This is consistent with thefindings of Fernicola et al.81who found that a mixture of the slightly differentN,N-butylethylpyrrolidnium TFSI with Li TFSI
was stable with lithium for at least a week of contact. However, there are conflicting reports on this matter, such as the work of Lewandowski et al.79who found that only piperidinium-based ILs have cathodic limits that are below the Li|SEI|Liþpotential.
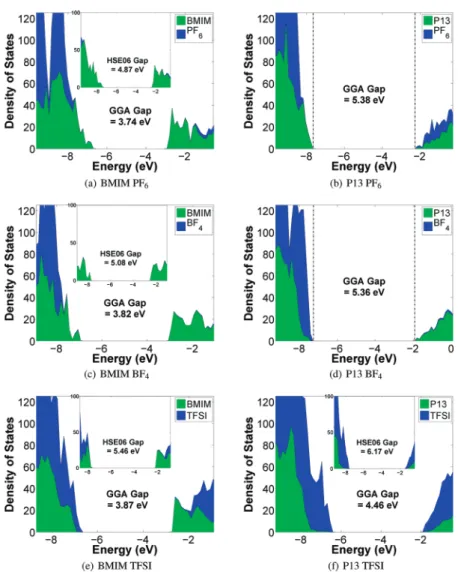
Calculated Density of States for ILs.Figure 3 shows repre-sentative plots of the projected DOS of the six ILs by cation and anion contributions. The cation- and anion-projected DOS allow us to determine the species in the IL that contributes to the HOMO (cathodic limit) and LUMO (anodic limit) levels. From the figures, we can make the following observations:
(1) Cathodic instability of TFSI in P13 TFSI: For P13 TFSI, the LUMO is dominated by the anion, which demon-strates that anion reductive stability could be an issue. This observation is in agreement with the experimental and theoretical work of Howlett et al.,1who showed that it is the TFSI anion that determines the cathodic limit in P13 TFSI.
(2) Possible cathodic instability of PF6in P13 PF6: For P13
PF6, our calculated DOS shows significant contributions
from both P13 and PF6 in the LUMO, which would
imply that PF6could contribute to the reductive stability
when paired with P13 as well. This is surprising, given than PF6is typically found to be the most stable anion
among the three studied.
(3) Anodic limits of TFSI-based ILs and P13 BF4: For
TFSI-based ILs and P13 BF4, the HOMOs are always
domi-nated by anion states, which implies that the anodic stability is set by the anion.
(4) Evidence of possible cation anodic instability: For BMIM BF4, BMIM PF6, and P13 PF6, the HOMO is dominated
by cation states, which implies that the oxidation stability is no longer limited by the anion, but rather by the stability of the cation. This observation challenges the prevailing assumption that it is always the anion that determines oxidative stability. To our knowledge, there is no experimental evidence pointing to possible cation oxidative instability in ILs thus far. The possible anodic in-stability of BMIM would also explain the earlier observation
that regardless of the variation in the anionic species, all BMIM-based ILs have relatively similar electrochemical windows. Because both the HOMO and the LUMO states of BMIM PF6and BMIM BF4are dominated by
cation states, we would expect the cation to set the electrochemical window. For BMIM TFSI, the LUMO states are dominated by the TFSI anion, but the cation states are only slightly lower in energy. We note that the HSE06 functional leads to results largely similar to those of GGA, but predicts the LUMO states to be dominated by BMIM instead of TFSI for BMIM TFSI (see insets in Figure 3).
Vacuum and PCM Electrochemical Windows.Figures 4 and 5 show the calculated cathodic and anodic limits for all ions investigated using vacuum and PCM calculations, respectively. For each IL formed from a particular cationanion pair, the overall cathodic limit is set by the maximum of cathodic limits of its constituent ions, and the overall anodic limit is set by the minimum of the anodic limit of its constituent ions. To take BMIM PF6as an example, the cathodic and anodic limits in vacuo
are set by the BMIM cation and PF6anion, respectively, while
the PCM cathodic and anodic limits are both set by the BMIM cation.
From Figure 4, we may observe that the vacuum calculations predict limits that are effectively set by respective ions, that is,
cathodic limits by cations and anodic limits by anions. On the other hand, the PCM calculations predict limits (cathodic and anodic) that are mostly set by the cations. Even P13 is predicted to have a lower anodic stability than the BF4and PF6 anions.
The significant stabilization from the isotropic dielectric re-sponse is evident from the significantly wider electrochemical windows (anodic limitcathodic limit) in the PCM calculations as compared to the vacuum calculations. Electrochemical windows in excess of 6 V are obtained, which are much wider than the typical 46 V observed experimentally for these ILs. This observation indicates that the PCM model may have over-stabilized the ions.
We note that recent solvatochromonic measurements82 as well as computer simulations8385have found that the effective solvent polarity of ILs is much higher than suggested by their dielectric constant. In particular, Reichardt et al.82have demon-strated that the effective solvent polarity of ILs, as measured by the solvatochromic absorption of the zwitterionic betaine dye 2,6-diphenyl-4-(2,4,6-triphenylpyridinium-1-yl)phenolate dyeET(30),
is comparable to that of molecular dipolar non-hydrogen bond donor (HBD) and dipolar HBD solvents. Hence, even though there is no simple mapping of theET(30) solvent polarity scale to
the necessary dielectric constant parameter required for PCM calculations, we performed PCM calculations using a higher dielectric constant of 30 to ascertain the effect of the dielectric constant on the predicted cathodic and anodic limits (see the Supporting Information). We found that some of the calculated electrochemical windows are slightly smaller (e.g., the electro-chemical window of BMIM PF6decreased from 6.51 to 6.34 V)
when a higher dielectric constant of 30 is used, due to the greater dielectric stabilization effect for more highly charged ions. None-theless, the general observation that PCM calculations signifi -cantly overestimate the electrochemical windows still holds.
’DISCUSSION
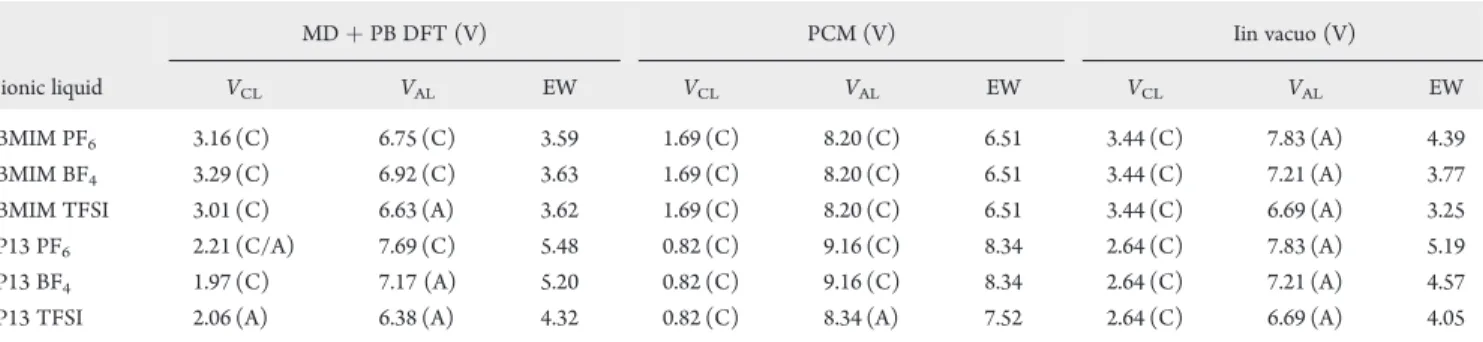
Table 2 summarizes the cathodic and anodic limits of the six ILs obtained from the three levels of approximations. We found that in vacuo calculations, which do not take into account any effect of the local environment in solution on the ion stability, result in limits that are effectively set by respective ions, that is, cathodic limits by cations and anodic limits by anions. While such calculations may be used to ascertain broad general trends in the relative stability of different ions (e.g., P13 versus BMIM or PF6
versus BF4versus TFSI), they are insufficient to provide
quanti-tative accuracy.
Table 2. Summary of the Electrochemical Windows (EW), Cathodic Limit (VCL), and Anodic Limit (VAL) for the Six ILs
Determined Using the Three Levels of Approximationsa
MDþPB DFT (V) PCM (V) Iin vacuo (V)
ionic liquid VCL VAL EW VCL VAL EW VCL VAL EW
BMIM PF6 3.16 (C) 6.75 (C) 3.59 1.69 (C) 8.20 (C) 6.51 3.44 (C) 7.83 (A) 4.39
BMIM BF4 3.29 (C) 6.92 (C) 3.63 1.69 (C) 8.20 (C) 6.51 3.44 (C) 7.21 (A) 3.77
BMIM TFSI 3.01 (C) 6.63 (A) 3.62 1.69 (C) 8.20 (C) 6.51 3.44 (C) 6.69 (A) 3.25
P13 PF6 2.21 (C/A) 7.69 (C) 5.48 0.82 (C) 9.16 (C) 8.34 2.64 (C) 7.83 (A) 5.19
P13 BF4 1.97 (C) 7.17 (A) 5.20 0.82 (C) 9.16 (C) 8.34 2.64 (C) 7.21 (A) 4.57
P13 TFSI 2.06 (A) 6.38 (A) 4.32 0.82 (C) 8.34 (A) 7.52 2.64 (C) 6.69 (A) 4.05
aThe letter in parentheses denotes the major contributing species to the limit, where C stands for cation and A stands for anion.
The cathodic and anodic limits from the PCM calculations show a significant stabilization effect from the dielectric response, resulting in electrochemical windows that are significantly wider than the vacuum calculations. This stabilization is likely to be overestimated; in real ILs, the dielectric effect is likely to be highly anisotropic and highly dependent on the coordination of thefirst solvent shell of counterions. Hence, the value of the PCM in modeling ILs appears to be limited, especially because the PCM requires the dielectric constant as an external input, which might not be available for less-studied IL systems.
We believe our approach of using a combination of MD and DFT calculations with a Hartree potential alignment procedure provides the most accurate model of an IL among the three level of approximations considered. The overall liquid structure of an IL is explicitly modeled, and all interion interactions are taken into account. This approach can potentially be applied to the study of other liquid systems. The main drawback is the need to
find or develop forcefields that capture the essential physics of the liquid structure to perform the MD simulations. While this drawback would not be a major issue for well-studied systems such as organic solvents, it could prove to be a significant hurdle when studying novel systems. However, we note that, specifically for ILs, transferable force fields have been developed that can describe bulk and interfacial properties of a wide set of ILs accurately.86
Given the difficulties in comparing experimental data on electrochemical windows and the inherent limitations of DFT in characterizing unoccupied states, it is difficult for us to make a definitive statement on the accuracy of our calculations. Never-theless, our calculations are in broad agreement with experi-mental data and have provided crucial insights into the species limiting the redox stability of the solvent. Our calculations predict the cathodic stability of the TFSI anion to be limiting in the case of P13 TFSI, in agreement with previous experiments and calculations.1In addition, our work predicts the anodic stability of the cation to be a limiting factor in BMIM PF6, BMIM BF4,
and P13 PF6. In particular, the relative instability of the BMIM
anion toward oxidation could account for the similar measured electrochemical windows for BMIM-based ILs; the BMIM cation could be limiting at both the anodic and the cathodic limits.
To our knowledge, no experimental or theoretical work thus far has shown any indication of possible cation anodic instability. This“gap”in the literature is particularly puzzling to us as there is experimental evidence in the literature that points to possible cation anodic instability. For example, Buzzeo et al.14measured 1-ethyl-3-methyl imidazolium (EMIM) TFSI and 1-hexyl-3-methyl imidazolium (HMIM) trifluorotris-(pentafl uoro-ethyl)-phosphate (FAP) to have very similar anodic limits, despite the fact that FAP is expected to be significantly more anodically stable than the TFSI anion. Although none of these specific ions with the exception of TFSI were investigated in our work, our results suggest that the imidazolium-based cations (EMIM and HMIM) could possibly be the limiting species at the anodic limits for these ILs. It is our wish that this prediction would be verified by future experimental work. For instance, Kroon et al.87 pre-viously conducted an extensive theoretical and experimental investigation into the decomposition pathways and products forN,N-butylmethylpyrrolidinium TFSI and BMIM BF4ILs at
the cathodic limit and showed that the predicted decomposition products are in excellent agreement with experiments. However, we have found no similar investigations on the anodic limit in the literature.
Our work therefore highlights that the prevalent assumption that the cation sets the cathodic limit and the anion sets the anodic limits may not be valid for a significant number of ILs. This could have implications in the design of ILs for electro-chemical applications in that it is no longer sufficient to optimize the respective ion for the respective limits; the cathodic and anodic limits of both cation and anion must be taken into account.
’CONCLUSION
In this work, we investigated the cathodic and anodic limits of six room-temperature ionic liquids (ILs) formed from the BMIM and P13 cations, and the PF6, BF4, and TFSI anions using a
combination of MD simulations and DFT calculations. In our approach, all interion interactions were taken into account by explicitly modeling the entire liquid structure. The relative cathodic and anodic limits of BMIM PF6, BMIM BF4, BMIM
TFSI, and P13 TFSI obtained from our approach are in fairly good agreement with existing experimental data. The cation- and anion-projected DOS obtained not only predict the TFSI anion to be less stable than the P13 cation against reduction (in agree-ment with previous findings1), but also provide surprising evidence of possible cation anodic instability. We predict the aromatic BMIM cation to be less stable against oxidation than the respective anions in BMIM PF6and BMIM BF4, and the P13
cation to be less stable against oxidation than the PF6anion in
P13 PF6. We also compared the predictions of our approach with
that of simpler approximations based on the calculated electron affinities and ionization energies of isolated ions using in vacuo or PCM calculations. We found that the PCM overstabilized the ions, resulting in predicted electrochemical windows that are much larger than those measured in experiments, while in vacuo calculations are insufficient to achieve quantitative accuracy due to the lack of local environment effects.
’ASSOCIATED CONTENT
b
S Supporting Information. Hartree potential plots for the room-temperature ionic liquids investigated (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.’AUTHOR INFORMATION
Corresponding Author *E-mail: [email protected].
’ACKNOWLEDGMENT
This work was supported by E. I. du Pont de Nemours & Co. through the DuPontMIT Alliance program. We would like to thank William L. Holstein and Steve R. Lustig from DuPont for their useful insights and assistance. This research was supported in part by the National Science Foundation through TeraGrid resources provided by the Pittsburgh Supercomputing Center.
’REFERENCES
(1) Howlett, P. C.; Izgorodina, E. I.; Forsyth, M.; Macfarlane, D. R. Z. Phys. Chem.2006,220, 1483–1498.
(2) Xu, K.Chem. Rev.2004,104, 4303–4418.
(3) McEwen, A. B.; McDevitt, S. F.; Koch, V. R.J. Electrochem. Soc.
(4) McEwen, A. B.; Ngo, H. L.; LeCompte, K.; Goldman, J. L. J. Electrochem. Soc.1999,146, 1687–1695.
(5) Lewandowski, A.; Galinski, M. J. Phys. Chem. Solids 2004, 65, 281–286.
(6) Garcia, B.; Lavallee, S.; Perron, G.; Michot, C.; Armand, M. Electrochim. Acta2004,49, 4583–4588.
(7) Matsumoto, H.; Matsuda, T.; Miyazaki, Y.Chem. Lett.2000, 1430–1431.
(8) Nakagawa, H.; Izuchi, S.; Kuwana, K.; Nukuda, T.; Aihara, Y. J. Electrochem. Soc.2003,150, A695–A700.
(9) Sakaebe, H.; Matsumoto, H.; Tatsumi, K.Electrochim. Acta2007, 53, 1048–1054.
(10) Galinski, M.; Lewandowski, A.; Stepniak, I.Electrochim. Acta
2006,51, 5567–5580.
(11) Hapiot, P.; Lagrost, C.Chem. Rev.2008,108, 2238–2264. (12) Zhou, F.; Cococcioni, M.; Kang, K.; Ceder, G.Electrochem. Commun.2004,6, 1144–1148.
(13) Ma, X.; Kang, B.; Ceder, G.J. Electrochem. Soc.2010,157, A925. (14) Buzzeo, M.; Hardacre, C.; Compton, R.ChemPhysChem2006, 7, 176–180.
(15) Rogers, E. I.; S~ac~aeljukic~ac~ae, B.; Hardacre, C.; Compton, R. G. J. Chem. Eng. Data2009,54, 2049–2053.
(16) Prendergast, D.; Grossman, J. C.; Galli, G.J. Chem. Phys.2005, 123, 014501.
(17) Koch, V.; Dominey, L.; Nanjundiah, C. J. Electrochem. Soc.
1996,143, 798–803.
(18) Ue, M.; Murakami, A.; Nakamura, S.J. Electrochem. Soc.2002, 149, 1572–1577.
(19) Vollmer, J. M.; Curtiss, L. A.; Vissers, D. R.; Amine, K.J. Electro-chem. Soc.2004,151, A178–A183.
(20) Zhang, S.; Sun, N.; He, X.; Lu, X.; Zhang, X.Green Chem.2006, 35, 1475.
(21) Tasaki, K.; Kanda, K.; Kobayashi, T.; Nakamura, S.; Ue, M. J. Electrochem. Soc.2006,153, 2192–2197.
(22) Johansson, P.Phys. Chem. Chem. Phys.2007,9, 1493–1498. (23) Ong, S. P.; Ceder, G.Electrochim. Acta2010,55, 3804–3811. (24) Maeshima, H.; Moriwake, H.; Kuwabara, A.; Fisher, C. a. J. J. Electrochem. Soc.2010,157, A696.
(25) Ngo, H.; LeCompte, K.; Hargens, L.; McEwen, A.Thermochim. Acta2000,357, 97–102.
(26) Martin, R. M.Electronic Structure: Basic Theory and Practical Methods; Cambridge University Press: New York, 2004; Vol. 1, p 648. (27) Tomasi, J.; Cammi, R.; Mennucci, B.; Cappelli, C.; Corni, S. Phys. Chem. Chem. Phys.2002,4, 5697–5712.
(28) Tomasi, J.; Mennucci, B.; Cammi, R.Chem. Rev.2005,105, 2999–3093.
(29) Cossi, M.; Rega, N.; Scalmani, G.; Barone, V.J. Chem. Phys.
2001,114, 5691.
(30) Cossi, M.; Scalmani, G.; Rega, N.; Barone, V.J. Chem. Phys.
2002,117, 43.
(31) Cossi, M.; Barone, V.J. Chem. Phys.2000,112, 2427. (32) Cossi, M.; Barone, V.J. Chem. Phys.2001,115, 4708. (33) Cossi, M.; Barone, V.; Robb, M. A.J. Chem. Phys.1999,111, 5295.
(34) Cossi, M.; Barone, V.J. Phys. Chem. A2000,104, 10614–10622. (35) Cammi, R.; Mennucci, B.; Tomasi, J.J. Phys. Chem. A1999, 103, 9100–9108.
(36) Cammi, R.; Mennucci, B.; Tomasi, J.J. Phys. Chem. A2000, 104, 5631–5637.
(37) Mennucci, B.; Tomasi, J.J. Chem. Phys.1997,106, 5151. (38) Mennucci, B.; Cances, E.; Tomasi, J.J. Phys. Chem. B1997, 101, 10506–10517.
(39) Barone, V.; Cossi, M.; Tomasi, J.J. Comput. Chem.1998,19, 404–417.
(40) Barone, V.; Cossi, M.J. Phys. Chem. A1998,102, 1995–2001. (41) Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J.Chem. Phys. Lett.1998,286, 253–260.
(42) Barone, V.; Cossi, M.; Tomasi, J.J. Chem. Phys.1997,107, 3210.
(43) Mennucci, B.; Tomasi, J.J. Chem. Phys.1997,107, 3032. (44) Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J.Chem. Phys. Lett.
1996,255, 327–335.
(45) Miertus, S.; Tomasi, J.Chem. Phys.1982,65, 239–245. (46) Miertus, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129.
(47) Smith, W.; Yong, C.; Rodger, P.Mol. Simul.2002,28, 385–471. (48) Smith, W.; Forester, T. R.J. Mol. Graphics1996,14, 136–141. (49) Smith, W.Mol. Simul.2006,32, 933–933.
(50) Jorgensen, W. L.; Maxwell, D. S.; Tirado-Rives, J.J. Am. Chem. Soc.1996,118, 11225–11236.
(51) Canongia Lopes, J. N.; Padua, A. A. H.J. Phys. Chem. B2004, 108, 16893–16898.
(52) Canongia Lopes, J. N.; Deschamps, J.; Padua, A. A. H.J. Phys. Chem. B2004,108, 11250–11250.
(53) Canongia Lopes, J. N.; Deschamps, J.; Padua, A. A. H.J. Phys. Chem. B2004,108, 2038–2047.
(54) Breneman, C. M.; Wiberg, K. B.J. Comput. Chem.1990,11, 361–373.
(55) Berendsen, H. J. C.; Postma, J. P. M.; van Gunsteren, W. F.; DiNola, A.; Haak, J. R.J. Chem. Phys.1984,81, 3684.
(56) Kresse, G.; Joubert, D.Phys. Rev. B1999,59, 1758–1775. (57) Kresse, G.; Furthmuller, J. Phys. Rev. B 1996, 54, 11169– 11186.
(58) Mrovec, M.; Albina, J.-M.; Meyer, B.; Els€asser, C.Phys. Rev. B
2009,79, 245121.
(59) Alkauskas, A.; Broqvist, P.; Devynck, F.; Pasquarello, A.Phys. Rev. Lett.2008,101, 106802.
(60) Shaltaf, R.; Rignanese, G.-M.; Gonze, X.; Giustino, F.; Pasquarello, A.Phys. Rev. Lett.2008,100, 186401.
(61) Frisch, M. J.; et al.Gaussian 03, revision C.02; Gaussian, Inc.: Pittsburgh, PA, 2003.
(62) Becke, A.J. Chem. Phys.1993,98, 5648–5652.
(63) Lee, C. T.; Yang, W. T.; Parr, R. G. Phys. Rev. B1988,37, 785–789.
(64) Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.; Pople, J. A. J. Chem. Phys.1998,109, 42.
(65) Scott, A. P.; Radom, L.J. Phys. Chem.1996,100, 16502–16513. (66) Weingartner, H.Z. Phys. Chem.2006,220, 1395–1405. (67) Lide, D. R., Ed.CRC Handbook of Chemistry and Physics, Internet Version 2011, 91st ed.; CRC Press: New York, 2010; pp 12114.
(68) Fitchett, B.; Rollins, J.; Conboy, J.J. Electrochem. Soc.2004, 151, E219–E225.
(69) Randstr€om, S.; Montanino, M.; Appetecchi, G.; Lagergrenb, C.; Morenoa, A.; Passerini, S.Electrochim. Acta2008,53, 6397–6401.
(70) O^aaz~ad~at’Mahony, A. M.; Silvester, D. S.; Aldous, L.; Hardacre, C.; Compton, R. G.J. Chem. Eng. Data2008,53, 2884–2891.
(71) Lewandowski, A.; Stepniak, I.Phys. Chem. Chem. Phys.2003, 5, 4215.
(72) Cohen, A.; Mori-Sanchez, P.; Yang, W.Phys. Rev. B2008, 77. (73) Sham, L.; Schl€uter, M.Phys. Rev. Lett.1983,51, 1888–1891. (74) Wang, C.; Pickett, W.Phys. Rev. Lett.1983,51, 597–600. (75) Chan, M.; Ceder, G.Phys. Rev. Lett.2010,105, 196403. (76) Heyd, J.; Scuseria, G. E.; Ernzerhof, M.J. Chem. Phys.2003, 118, 8207.
(77) Heyd, J.; Scuseria, G. E.; Ernzerhof, M.J. Chem. Phys.2006, 124, 219906.
(78) Heyd, J.; Scuseria, G. E.J. Chem. Phys.2004,121, 1187–92. (79) Lewandowski, A.; Swiderska-Mocek, A. J. Appl. Electrochem.
2010,40, 515–524.
(80) Barisci, J.; Wallace, G.; MacFarlane, D.; Baughman, R. Electro-chem. Commun.2004,6, 22–27.
(81) Fernicola, A.; Croce, F.; Scrosati, B.; Watanabe, T.; Ohno, H. J. Power Sources2007,174, 342–348.
(82) Reichardt, C.Green Chem.2005,7, 339.