Abstract
BEHOF, WILLIAM JAMES. Progress Towards the Synthesis of Novel Graphite Derivatives from a Solution Processable Poly-Cyano Precursor Polymer. (Under the direction of Dr. Christopher B. Gorman.)
In this thesis, the synthetic progress towards the development of a novel potentially conducting ladder polymer, through the direct writing of a soluble precursor polymer, is presented. The proposed ladder polymer was synthesized through three steps; the synthesis of a poly-cyano monomer for polymerization, the synthesis of a poly-cyano precursor polymer, and a one step cascading cyclization of a poly-cyano polymer.
Progress Towards the Synthesis of Novel Graphite Derivatives from a Solution Processable Poly-Cyano Precursor Polymer
by
William James Behof
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Doctor of Philosophy
Chemistry
Raleigh, North Carolina 2010
APPROVED BY:
_________________________________ Dr. David A. Schultz
_________________________________ Dr. Lin He
_________________________________ Dr. Alexander Deiters
Biography
The author was born in the city of Park Ridge, Illinois to James and Judy Behof on March 19, 1982. After graduating from the public primary schools, William attended Grayslake
Community College; he then transferred to the University of Illinois at Chicago (UIC) and graduated with a bachelors of science degree in biochemistry. While attending UIC he
participated in undergraduate research with the advisement of Dr. Duncan Wardrop. In the spring of 2005 he was accepted into the department of chemistry graduate program at North Carolina State University. Before leaving for North Carolina, William married his wife Judy
on July 30th 2005. At NC State he pursued his Doctor of Philosophy degree under the advisement of Dr. Christopher B. Gorman. In his time at NC State William and Judy
welcomed the birth of their first son, Dakota James, on June 07, 2006 and the adoption of their second son, Talon Jackson. At the time of graduation they were expecting the birth of their third son, Austin Reese. William received his Doctor of Philosophy degree in
Acknowledgements
I must first acknowledge God, without him I would not be here and none of this would be possible.
I would like to think that I could come up with the right words to thank my wife, parents and family, but I cannot find words strong enough to show my love, gratitude and thanks to
them. I could not have done this without my family as they had my back and supported me in whatever I had to do. I have a great gratitude towards my parents for raising me the way they did. Thank you dad for surviving the fall, I couldn’t of have gone through grad school
without you. Judy, I want to thank you for dealing with me on the other end and for helping me survive. A special thanks to my Uncle Bill for giving me my work ethic, teaching me to
measure twice, cut once and to watch out for the brake cleaner. To my Aunt Mary Ellen and Uncle Bill Wade, thank you for encouraging me to go as far as I can go.
I would like to acknowledge Dr. Christopher B. Gorman, who took me into, and supported
me for the past three years on a research assistant grant. You have taught me how to write and overcome my lack of experience in the field of synthetic chemistry. A special thanks to
A great thanks goes out to my friends and group members at NC State. I greatly appreciate the great deal of time Dr. Kusum Chandra spent tutoring me. This help was invaluable and I would not be the chemist I am today without it. A special thanks to Dr.
Jennifer Ayres for putting up with me in lab. Dr. Joseph Fritz, Dr. Ned Bowen and Dr. David Dickson, from the University of Illinois at Chicago, helped me decide to go to
graduate school. My friends and alliances at NC State, Wesleigh Edwards, Justin Kennemur, Joe DeSousa, Rachel Kudgus, Matt Thompson, Rob Schmidt, Deborah Bromfield, Michelle Clark and Alan Harvell, who all made my time a little bit easier. I would also like to
acknowledge Gorman group members; Dr. Anil Sharma, Eric Tucker, Lebo Xu, Jenelle Willett, Molly Brannock and Dr. Feng Lu for teaching me how to fashion my arguments and
discussions related to our work.
I need to thank Aaron Wallace, Steve Yamnitz and Bill Sanderson (Go VOLS) for helping live as a christian. Steve, thank you for being a best man at my wedding and for all the less
than intelligent adventures we had as children. Aaron, besides being a consistent fishing buddy, there is no way to show my thanks and gratitude to what you and Mel have done for Judy and I.
“How well I have learned that there is no fence to sit on between heaven and
hell. There is a deep, wide gulf, a chasm, and it that chasm is no place for any
Table of Contents
List of Tables ... xii
List of Figures ... xiv
List of Schemes ... xviii
List of Abbreviations and Terms ... xxi
Chapter 1 ... 1
1.1 Size Reduction and Moore’s Law ...2
1.2 Photolithography ...3
1.3 Electromigration ...4
1.4 Organic Conducting Materials...5
1.5 Direct Writing ...9
1.6 Proposed Ladder Polymer ...10
1.7 Perspectives and Goals of Dissertation Research ...14
1.8 References ...17
Chapter 2 ... 21
2.1 Introduction ...22
2.2 Results and Discussion ...24
2.2.1 Assessment of Various Nucleophile and Electrophile Combinations ...24
2.2.2 Optimization of Couplings Employing Benzyl Cyanide Nucleophiles ...29
2.4 Experimental Section ...47
2.5 References ...58
Chapter 3 ... 60
3.1 Introduction ...61
3.2 Results and Discussion ...63
3.2.1 A-A monomer Formation Through TMS-Acetonitrile Nucleophiles ...63
3.2.2 Precursor Polymer Formation Through TMS-Acetonitrile Nucleophiles ...65
3.2.3 A-B Monomer Synthesis Through Ethyl Cyanoacetate Anions ...68
3.2.4 Polymerization of A-B Monomer ...71
3.3 Conclusion ...76
3.4 Experimental ...77
3.5 References ...87
Chapter 4 ... 90
4.1 Introduction ...91
4.2 Results and Discussion ...95
4.2.1 Synthesis of Cyano Precursor Oligomers ...95
4.2.2 Cyclization Under Acidic Conditions ...97
4.2.3 Cyclization With Excess n-BuLi ...103
4.2.4 The Effect of Ketene Imine Intermediates on Cyclization ...105
4.2.5 Photo-physical Properties ...113
4.2.7 Thermal Properties ...117
4.2.8 Crystal Structures ...118
4.3 Conclusion ...124
4.4 Experimental ...125
4.5 References ...139
Appendix ...143
Appendix A ...144
A.1 Introduction ...145
A.2 Results and Discussion ...146
A.2.1 Temperature Profile of Si and SiO2 in Toluene ...146
A.2.2 Microwave Effects on the Growth of Polymer Brushes ...148
A.2.3 Thickness Optimization of Polymer Brushes ...152
A.3 Conclusion ...154
A.4 Experimental ...155
A.5 References ...157
Appendix B ...160
B.1 Experimental for (64) C26H15Br3N4 (x08082) ...161
B.2 Experimental for (65) C27.25H18.25Br2Cl3.75N5 (x08100) ...165
B.3 Experimental for (68) C20H19N3 (x09086) ...170
B.5 Experimental for (76) C33H33N3 (x09060) ...180
B.6 Acknowledgement ...184
Appendix C ...185
C.1 1H NMR of Compound 11 ...186
C.2 13C NMR of Compound 11 ...187
C.3 1H NMR of Compound 12 ...188
C.4 13C NMR of Compound 12 ...189
C.5 1H NMR of Compound 14 ...190
C.6 13C NMR of Compound 14 ...191
C.7 1H NMR of Compound 16 ...192
C.8 13C NMR of Compound 16 ...193
C.9 1H NMR of Compound 18 ...194
C.10 13C NMR of Compound 18 ...195
C.11 1H NMR of Compound 19 ...196
C.12 13C NMR of Compound 19 ...197
C.13 1H NMR of Compound 20 ...198
C.14 13C NMR of Compound 20 ...199
C.15 1H NMR of Compound 21 ...200
C.16 13C NMR of Compound 21 ...201
C.17 1H NMR of Compound 22 ...202
C.19 1H NMR of Compound 23 ...204
C.20 13C NMR of Compound 23 ...205
C.21 1H NMR of Compound 24 ...206
C.22 13C NMR of Compound 24 ...207
C.23 1H NMR of Compound 25 ...208
C.24 13C NMR of Compound 25 ...209
C.25 1H NMR of Compound 26 ...210
C.26 13C NMR of Compound 26 ...211
C.27 1H NMR of Compound 27 ...212
C.28 13C NMR of Compound 27 ...213
C.29 1H NMR of Compound 30 ...214
C.30 13C NMR of Compound 30 ...215
C.31 1H NMR of Compound 34 ...216
C.32 13C NMR of Compound 34 ...217
C.33 1H NMR of Compound 35 ...218
C.34 13C NMR of Compound 35 ...219
C.35 1H NMR of Compound 36 ...220
C.36 13C NMR of Compound 36 ...221
C.37 1H NMR of Compound 37 ...222
C.38 13C NMR of Compound 37 ...223
C.40 13C NMR of Compound 38 ...225
C.41 1H NMR of Compound 39 ...226
C.42 13C NMR of Compound 39 ...227
C.43 1H NMR of Compound 40 ...228
C.44 13C NMR of Compound 40 ...229
C.45 1H NMR of Compound 41 ...230
C.46 13C NMR of Compound 41 ...231
C.47 1H NMR of Compound 43 ...232
C.48 13C NMR of Compound 43 ...233
C.49 1H NMR of Compound 51 ...234
C.50 13C NMR of Compound 51 ...235
C.51 1H NMR of Compound 52 ...236
C.52 13C NMR of Compound 52 ...237
C.53 1H NMR of Compound 53 ...238
C.54 13C NMR of Compound 53 ...239
C.55 1H NMR of Compound 54 ...240
C.56 13C NMR of Compound 54 ...241
C.57 1H NMR of Compound 55 ...242
C.58 13C NMR of Compound 55 ...243
C.59 1H NMR of Compound 56 ...244
C.61 1H NMR of Compound 57 ...246
C.62 13C NMR of Compound 57 ...247
C.63 1H NMR of Compound 58 ...248
C.64 13C NMR of Compound 58 ...249
C.65 1H NMR of Compound 67 ...250
C.66 13C NMR of Compound 67 ...251
C.67 1H NMR of Compound 68 ...252
C.68 13C NMR of Compound 68 ...253
C.69 1H NMR of Compound 69 ...254
C.70 13C NMR of Compound 69 ...255
C.71 1H NMR of Compound 70 ...256
C.72 13C NMR of Compound 70 ...257
C.73 1H NMR of Compound 74 ...258
C.74 13C NMR of Compound 74 ...259
C.75 1H NMR of Compound 75 ...260
C.76 13C NMR of Compound 75 ...261
C.77 1H NMR of Compound 76 ...262
C.78 13C NMR of Compound 76 ...263
C.79 1H NMR of Compound 77 ...264
List of Tables
Chapter 2
Table 2-1. Efficacy of aryl organo-boranes as the nucleophile ... 25
Table 2-2. Efficacy of acetonitrile anions as the nucleophile ... 27
Table 2-3. Results of coupling of 22 with 10 under Pd-mediated coupling conditions. ... 38
Table 2-4. Variation of Base ... 41
Table 2-5. Variation of solvent and then further variation of base. ... 44
Table 2-6. Variation of ligand ... 46
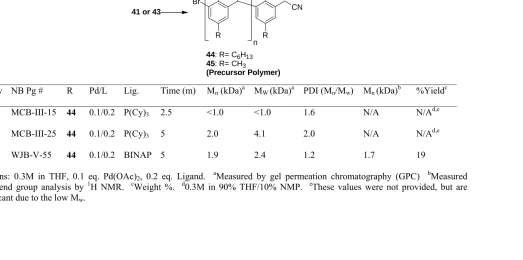
Chapter 3 Table 3-1. Synthesis of the A-A monomer with TMSCH2CN ... 64
Table 3-2. Polymerization of 41 and 43 ... 72
Table 3-3. Polymerization of 41 and 43 (continued) ... 73
Chapter 4 Table 4-1. Failed attempts to effect the cascading cyclization under single nucleophile conditions in acid.a ... 102
Table 4-2. Efficacy of ketene-imine intermediates with various nucleophiles ... 109
Table 4-3. Photo-physical data for molecules 67, 68, 69, 70 and 72 ... 115
Appendix B Table B-1. Summary of Crystal Data for (64) x08082 ... 163
Table B-3. Summary of Crystal Data for (68) x09086 ... 173
Table B-4. Summary of Crystal Data for (69) x09034 ... 178
List of Figures
Chapter 1
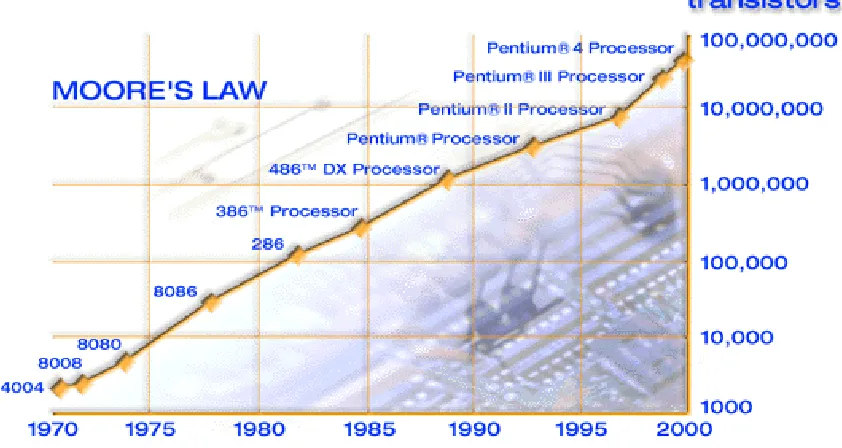
Figure 1-1. Moore’s Law (A prediction that transistor density would double every other
year) ... 3
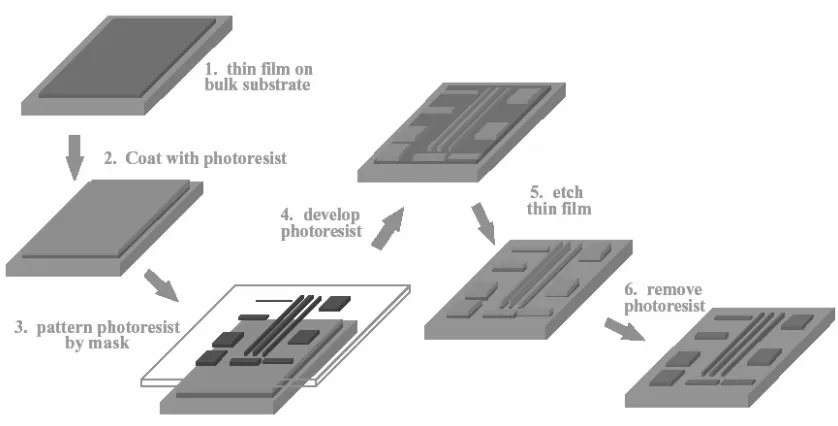
Figure 1-2. The current photo-lithographic patterning process in which, chips are made. .... 4
Figure 1-3. Electromigration of the gold atoms, which leads to the failure of the metal
connection. ... 5
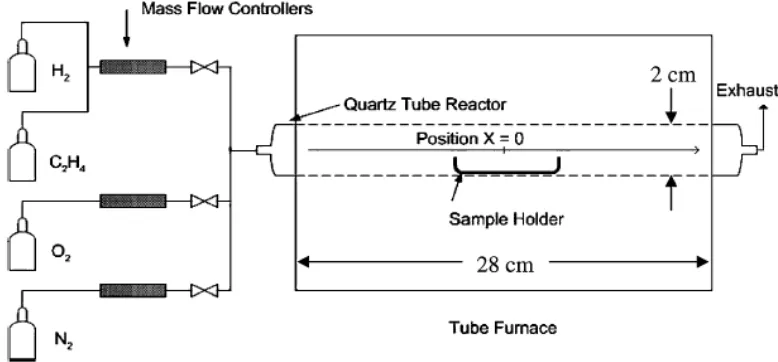
Figure 1-4. Specialized reactor needed for the synthesis of graphite ... 6
Figure 1-5. Structure of pentance (A); Structure of substituted heptacene (B) ... 7
Figure 1-6. Examples of known precursor ladder polymers and reaction conditions to form a
fully aromatic ladder polymer. ... 8
Figure 1-7. Schematic depiction of direct writing. Conversion of an insulating material
directly into a conducting material upon application of light would require no metallization, strip or lift-off steps. ... 9
Figure 1-8. Fully cyclized ladder polymer end result ... 10
Figure 1-9. Advantages of poly amino-pyridine systems ... 11
Chapter 2
Figure 2-1. Reaction of molecules 9 and 10. (top) After 43 m (bottom) after 2 h. ... 33
Figure 2-2. In-situ formation and reaction of the organo-zinc nucleophile ... 34
Chapter 3
Figure 3-1. 1H-NMR spectrum of 44 (Table 3-3,Entry 6) ... 74
Figure 3-2. 1H NMR (d6-DMSO) of insoluble material. The large peak at 5.0 ppm is due to
Chapter 4
Figure 4-1. ESI mass spectra illustration the partial and full cyclization of molecule 57 as
well as problematic amide formation ... 99
Figure 4-2. Crystal structure of the products isolated from the reaction of molecule 24 with
36 wt% HBr/HOAc, the alternatively cyclized molecule 64 (Left) and partially
cyclized trimer 65 (Right) ... 101
Figure 4-3. UV-Vis absorption spectrum of molecules 67-70 and 72 in CH3CN (left). .... 113 Figure 4-4. Emission spectra of molecules 67-70 in CH3CN (left) and UV-Vis, emission and
excitation spectra of molecule 69 in CH3CN (right). ... 114
Figure 4-5. Cyclic voltammetry of molecules 67, 68 and 69 thin films on the platinum
electrode in propylene carbonate containing 0.1M TBAPF6. Scanned from +1.5
V to -2.0 V vs. Ag+/AgNO3 reference for 2 cycles, scan rate = 100 mV s-1. .... 116
Figure 4-6. TGA scan of 70 from 0oC to 600oC ... 118
Figure 4-7. Structure of molecule 68 (top left), ORTEP drawing of molecule 68 showing
naming and numbering scheme (top right). Ellipsoids are at the 50% probability level, (bottom left) ace to edge stacking of 68, dimerization through hydrogen
bonding (bottom right). ... 121
Figure 4-8. Structure of molecule 69 (top left), ORTEP drawing of molecule 69 showing
naming and numbering scheme (top right). Ellipsoids are at the 50% probability level, (bottom left) Stacking of molecule 69, Dimerization through hydrogen
bonding (bottom right). ... 122
Figure 4-9. Structure of molecule 76 (top left), ORTEP drawing of molecule 76 showing
naming and numbering scheme (top right). Ellipsoids are at the 50% probability level, (bottom left) Stacking of molecule 76, (bottom right) Dimerization through
hydrogen bonding. ... 123
Appendix A
Figure A-2. Temperature profiles of neat toluene, toluene with SiO2 and with Si during
irradiation at 100 watts for 2 minutes. ... 147
Figure A-3. IR temperature photographs and orientations in microwave cavity: A) vertical
photograph of position B (side picture shown for clarity) under 100W in toluene; C) vertical photograph of position D under irradiation at 100W in toluene. ... 148
Figure A-4. Si and SiO2 wafer orientation in the microwave vial sample holder. ... 150 Figure A-5. Thickness of PMMA brushes on both Si (Black) and SiO2 (Red) after
microwave irradiation for 2 m in toluene/methyl methacrylate 1:1 v/v. ... 151
Figure A-6. AFM micrographs, 10µm x 10µm, of SiO2 (A) and Si (B) wafers after
irradiation at 100 watts for 30 seconds in toluene/methyl methacrylate 1:1 v/v 152
Figure A-7. Graph indicating the effect of wattage on brush thickness after 2 minutes of
growth for PMMA. ... 153
Figure A-8. Graph indicating the effect of wattage on brush thickness after 2 minutes of
growth for PMMA. ... 154
Appendix B
Figure B-1. ORTEP drawing of (64) x08082 showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 162
Figure B-2. ORTEP drawing of (65) x08100 molecule A showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 166
Figure B-3. ORTEP drawing of (65) x08100 molecule B showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 167
Figure B-5. ORTEP drawing of (68) x09086 molecule B showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 172
Figure B-6. ORTEP drawing of (69) x09034 molecule A showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 176
Figure B-7. ORTEP drawing of (69) x09034 molecule B showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 177
Figure B-8. ORTEP drawing of (76) x09060 showing naming and numbering scheme. Ellipsoids are at the 50% probability level and hydrogen atoms were drawn with arbitrary radii for clarity. ... 181
List of Schemes
Chapter 1
Scheme 1-1. ... 13
Scheme 1-2. ... 14
Scheme 1-3. ... 15
Scheme 1-4. ... 16
Chapter 2 Scheme 2-1. ... 23
Scheme 2-2. ... 26
Scheme 2-3. ... 28
Scheme 2-4. ... 30
Scheme 2-5. ... 31
Scheme 2-6. ... 32
Scheme 2-7. ... 36
Scheme 2-8. ... 37
Scheme 2-9. ... 43
Chapter 3 Scheme 3-1. ... 62
Scheme 3-2. ... 63
Scheme 3-4. ... 66
Scheme 3-5. ... 67
Scheme 3-6. ... 69
Scheme 3-7. ... 70
Scheme 3-8. ... 71
Scheme 3-9. ... 76
Chapter 4 Scheme 4-1. ... 92
Scheme 4-2. ... 93
Scheme 4-3. ... 94
Scheme 4-4. ... 95
Scheme 4-5. ... 96
Scheme 4-6. ... 97
Scheme 4-7. ... 98
Scheme 4-8. ... 104
Scheme 4-9. ... 106
Scheme 4-10. ... 107
Scheme 4-11. ... 107
Scheme 4-12. ... 108
Scheme 4-14. ... 117
Appendix A
List of Abbreviations and Terms
Abbreviations, Symbol or Term ExplanationΦF Quantum yield of fluorescence
Å Angstrom
AIBN 2,2’-azobisisobutyronitrile
Ag+/AgNO3 Silver/Silver Nitrate reference electrode
aq Aqueous
bp Boiling Point
bs Broad singlet
Br Bromine
Brine Saturated solution of sodium chloride
Bu Butyl
n-BuLi n-butyllithium
oC Degree(s) Celsius
13C NMR Carbon-13 nuclear magnetic resonance
spectroscopy
cm-1 Reciprocal centimeters
COSY Correlation Spectroscopy
CV Cyclic Voltammetry
δ Chemical shift in ppm from tetramethylsilane
DCM Dichloromethane
dd Doublet of doublets
DMAP 4-(N,N-dimethylamino) pyridine
DMF Dimethylformamide
DMSO Dimethylsulfoxide
dba Dibenzylideneacetone
dt Doublet of triplets
EAS Electronic absorption spectra
EBG Estimated Band Gap
Equiv Equivalents
Et Ethyl
Et2O Diethyl ether
EtOAc Ethyl Acetate
EtOH Ethanol
eV Electron Volts
g Grams
GC Gas Chromatography
GPC Gel permeation chromatography
h Hours
H+ Proton or protic acid
HOAc Acetic Acid
Hex Hexanes
1H NMR Proton nuclear magnetic resonance spectroscopy
HPLC High performance liquid chromatography
HMDS Hexamethyldisilazide
HOMO Highest Occupied Molecular Orbital
HRMS High resolution mass spectrometry
H2SO4 Sulfuric Acid
Hz Hertz
i-Pr Isopropyl
IR Infrared Spectroscopy
J Coupling constant
λMAX The lowest energy transition
λONSET Emission The emission peak onset
λONSET Absorption The λMAX peak onset
L Liter
LDA Lithium diisopropylamide
LiTMP Lithium tetramethylpiperidide
LUMO Lowest Unoccupied Molecular Orbital
µ Micro
M Molar
m Multiplet
M+ Parent molecular ion
MeCN or CH3CN Acetonitrile
MeOH Methanol
mg Milligrams
MHz Megahertz
m Minutes
mL Milliliters
mmol Millimole
mol Mole
Mn Number average molecular weight
Mp Melting point
Mw Weight Average molecular weight
N Normal
n Degree of polymerization
NBS N-bromo succinimide
NH4OAc ammonium acetate
NMP N-methylpyrrolidinone
nm Nanometer
PDI Poly-dispersity index
Ph Phenyl
ppm Parts per million
Pr Propyl
q Quartet
R Alkyl group
rt Room temperature
s Singlet
t Triplet
TBAPF6 Tetrabutylammoniumhexafluorophosphate
TBDMS Tertbutyldimethylsilyl
TGA Thermal gravitametric analysis
THF Tetrahydrofuran
TIPS Tri-iso-propylsilyl
TMS Trimethylsilyl
V Volts
Chapter 1
1.1
Size Reduction and Moore’s Law
Figure 1-1. Moore’s Law (A prediction that transistor density would double every other year)
1.2
Photolithography
method. Due to the thickness of both the thin film and the photo-resist, patterning thin lines creates low aspect ratios which results in mechanically and electrically unstable structures. The aspect ratio also decreases with the number of times the chip gets washed and etched.
Figure 1-2. The current photo-lithographic patterning process in which, chips are made.
1.3
Electromigration
through a wire. While electromigration does not affect the most recent chips, multiple fans are required to keep these devices cool and decrease the likelihood of device failure. However, as feature sizes are reduced, failure due to electromigration will become more and more likely.
Figure 1-3. Electromigration of the gold atoms, which leads to the failure of the metal connection.
1.4
Organic Conducting Materials
conductivity of copper (5.8x105 (S/cm)).15 Graphite, however, has a conductivity of 2.2x103 (S/cm)15,16 and doped poly(acetylene) has shown conductivities as high as 3.6x102 (S/cm).17,18 These are figures of merit that may be difficult to match or exceed, however. Fused ring graphitic or aromatic ladder materials also have the potential to maximize the density of intermolecular packing compared to typical, conjugated organics because they are fused and thus required to be planar. In addition, these systems are computationally shown to have properties that are similar to highly conducting materials, e.g. zero band gap.19
Graphite, however, is not a material that is amenable to patterning even though it is used in many other applications.20-25 Graphite is not melt or solution processable, and only a few techniques are reported for its synthesis.26,27 The best known method to produce graphite uses a nickel catalyst at 750-900 K in a ethylene/oxygen rich atmosphere (Figure 1-4).24 Although the reported procedure is the mildest, it is not compatible with nanometer-scale patterning.
The synthesis of small oligomer acenes (fused linear carbon chains) was explored to avoid the processing problems associated with graphite. Small molecule linear acenes, e.g. pentacene (Figure 1-5, A), have been synthesized as an alternative to graphite and show very impressive properties (hole mobilites upward of 5 cm2/V*s).28-31 However, only substituted hexacene and heptacene (Figure 1-5, B) have been synthesized, as un-substituted linear acenes have not been isolated due poor solubility and stability.28 The synthesis of longer acene systems is increasingly difficult because of the needed substitution.
TIPS
TIPS
A B
Figure 1-5. Structure of pentance (A); Structure of substituted heptacene (B)
We hypothesize that this solid-state conversion might be performed locally to create patterns, as discussed in more detail in the next section. A candidate precursor conversion requires mild chemistry that does not involve local heating or harsh reagents, which might facilitate unwanted side reactions. Ideally, a photo-chemically initiated process could most likely be adapted to established photolithography procedures.
R1 R1 R2 R2 n n R1 R2 R2 R1 R1 R2 R2 R1 N N N N BocHN R(O)C C(O)R n N N N N N N R R n NHBoc
CF3CO2H
ZnCl2
115oC
Figure 1-6. Examples of known precursor ladder polymers and reaction conditions to form a fully aromatic ladder polymer.
1.5
Direct Writing
Although the ladder polymers described above require conditions that are too harsh for nanometer-scale patterning, they suggest a very useful idea for patterning. The precursor polymer is typically soluble in organic solvents while the ladder polymer is not. It is not only of convenience that the precursor polymer and the ladder polymer have two different solubilities in organic solvents, but also allows the user to write patterns onto materials in a much more efficient way. The soluble precursor polymer can be coated onto a substrate and, upon local laser irradiation38,39 or other type of conversion,40-44 it would be converted to the insoluble, desired, conductive material. The starting precursor could then be washed away.
Figure 1-7. Schematic depiction of direct writing. Conversion of an insulating material directly into a conducting material upon application of light would require no metallization, strip or lift-off steps.
waste generated.45 This method has been tested with metallo-polymers and resulted in 12 nm structures with high aspect ratios.45
1.6
Proposed Ladder Polymer
As fused, sp2 hybridized materials have several advantages over non-fused polymers, we have chosen to pursue the synthesis of a novel ladder material from a soluble precursor polymer. The proposed poly-amino-iso-quinoline ladder polymer is illustrated in Figure 1-8. As expressed before, the proposed ladder polymer is anticipated to have high conductivity due to the extended conjugation throughout the material. This hypothesis is supported by computations illustrating that fused amino-iso-quinolines are capable of being either n and p-type materials46 with electron mobilities close to that of other carbon acenes.47
N HN N HN N HN NH
X
R R R R
CN
n
Figure 1-8. Fully cyclized ladder polymer end result
(N-NH) units (Figure 1-9, C).55 Third, the electronic properties can be tuned through protonation,56 and deprotonation (Figure 1-9, D).10,57
Ligands Metal Atoms Ligands N N N N N N N N N N N N 4 N N
C6H13
C6H13
N N
C6H13
C6H13 CSA H H A B C D N N N N H H H H
Figure 1-9. Advantages of poly amino-pyridine systems
Scheme 1-1.
CN A
CN
B
N HN N HN N N NH2
X
R R R R
CN
n
CN CN CN CN CN
R R R
X CN
R CN
CN
n
Tautomerization Polymerization
Precursor Polymer
Ladder Polymer A-B Monomer
N HN N HN N N NH
X
R R R R
CN
1.7
Perspectives and Goals of Dissertation Research
The long term goal of this project is the development of a novel, potentially conducting, material through the direct writing of a soluble precursor polymer. In this thesis, the synthesis of a soluble precursor polymer is presented. In addition, optimal ring closing conditions for discrete oligomers of the aforementioned precursor polymer are also reported.
Since the proposed reactions are relatively unexplored, several challenges exist with the synthetic route presented in Scheme 1-1. The three challenges that are addressed here are:
1. Optimization of a model coupling in application to a polymerization
2. Synthesis of an highly acidic poly-cyano precursor through an anionic polymerization 3. Development of a route for the efficient ring closure of poly-cyano oligomers
Chapter 2 discusses possible and optimal coupling reactions for the precursor polymer and illustrates that highly electron deficient mono-aryl, di-aryl and bis-diaryl acetonitriles can be synthesized effectively using either a nucleophilic aromatic substitution (NAS) or a palladium-mediated coupling pathway (Scheme 1-2). It is shown that the synthesis of di-aryl acetonitriles most conveniently proceeded via NAS while palladium-mediated coupling is required to prepare the bis-diaryl acetonitrile efficiently. These reactions were extremely sensitive to steric and ligand electronic effects.
Scheme 1-2.
X
CN CN CN
In Chapter 3, the synthesis of a novel poly-(cyano)-precursor polymer through a palladium catalyzed coupling is reported (Scheme 1-3). The synthesis of the polymer is possible through a short and efficient synthesis of the monomerwhere the critical step is the selective NAS of an ethyl cyanoacetate anion. The precursor polymer was synthesized in modest yield and molecular weight. In addition, it is also illustrated that optimal conditions from the previous study (Chapter 2) may not be directly applicable to the polymerization.
Scheme 1-3.
CN CN CN
C6H13 C6H13 Br
CN Br
CN CN
C6H13
n
Scheme 1-4.
N HN N
C4H9 NH2
CN CN CN CN
N N
Br NH2 H2N Br
36 wt% HBr/HOAc
n-BuLi
1.8
References
(1) Intel. http://www.intel.com/research/silicon/mooreslaw.htm
(2) Intel. http://www.intel.com/pressroom/kits/quickreffam.htm#Celeron (3) Intel. http://www.intel.com/pressroom/archive/releases/20040830net.htm (4) Intel.
http://www.intel.com/technology/architecture-silicon/32nm/index.htm?iid=tech_sil+32nm
(5) Monceaux, C. Masters Thesis, North Carolina State University, 2004. (6) Pang, X.; Kriman, A. M.; Bernstein, G. H. J. Electrochem. Soc.2002, 149, G103-G108.
(7) Schneider, G.; Hambach, D.; Niemann, B.; Kaulich, B.; Susini, J.; Hoffmann, N.; Hasse, W. Appl. Phys. Lett.2001, 78, 1936-1938.
(8) Durkan, C.; Schneider, M. A.; Welland, M. E. J. Appl. Phys.1999, 86, 1280-1286.
(9) Shiraishi, K.; Rajca, A.; Pink, M.; Rajca, S. J. Am. Chem. Soc.2005, 127, 9312-9313.
(10) Tal, S.; Blumer-Ganon, B.; Kapon, M.; Eichen, Y. J. Am. Chem. Soc.2005, 127, 9848-9854.
(11) Payne, M. M.; Parkin, S. R.; Anthony, J. E. J. Am. Chem. Soc.2005, 127, 8028-8029.
(12) Zhang, C. Y.; Tour, J. M. J. Am. Chem. Soc.1999, 121, 8783-8790.
(13) Jadhav, A. V.; Gulgas, C. G.; Gudmundsdottir, A. D. Eur. Polym. J. 2007, 43, 2594-2603.
(16) Jaszczak, D. J. A. http://www.phy.mtu.edu/~jaszczak/graphprop.html (17) Chiang, C. K.; Fincher, C. R.; Park, Y. W.; Heeger, A. J.; Shirakawa, H.; Louis, E. J.; Gau, S. C.; Macdiarmid, A. G. Phys. Rev. Lett.1977, 39, 1098-1101.
(18) Chiang, C. K.; Druy, M. A.; Gau, S. C.; Heeger, A. J.; Louis, E. J.;
Macdiarmid, A. G.; Park, Y. W.; Shirakawa, H. J. Am. Chem. Soc.1978, 100, 1013-1015. (19) Bendikov, M.; Duong, H. M.; Starkey, K.; Houk, K. N.; Carter, E. A.; Wudl, F. J. Am. Chem. Soc.2004, 126, 7416-7417.
(20) Baughman, R. H. Science2006, 312, 1009-1010.
(21) Lee, J.; Kim, J.; Hyeon, T. Adv. Mater.2006, 18, 2073-2094.
(22) Makarova, O. V.; Mancini, D. C.; Moldovan, N.; Divan, R.; Tang, C. M.; Ryding, D. G.; Lee, R. H.; Science2003, 182-186.
(23) Miller, T. M.; Kwock, E. W.; Baird, T.; Hale, A. Chem. Mat.1994, 6, 1569-1574.
(24) Phillips, J.; Shiina, T.; Nemer, M.; Lester, K. Langmuir2006, 22, 9694-9703. (25) Schueller, O. J. A.; Brittain, S. T.; Marzolin, C.; Whitesides, G. M. Chem. Mat.1997, 9, 1399-1406.
(26) Brooks, J. D.; Taylor, G. H. Nature1965, 206, 697-699.
(27) Zakhidov, A. A.; Baughman, R. H.; Iqbal, Z.; Cui, C. X.; Khayrullin, I.; Dantas, S. O.; Marti, I.; Ralchenko, V. G. Science1998, 282, 897-901.
(28) Anthony, J. E. Chem. Rev.2006, 106, 5028-5048.
(29) Duong, H. M.; Bendikov, M.; Steiger, D.; Zhang, Q. C.; Sonmez, G.; Yamada, J.; Wudl, F. Org. Lett.2003, 5, 4433-4436.
(30) Tang, M. L.; Mannsfeld, S. C. B.; Sun, Y. S.; Becerril, H. A.; Bao, Z. N. J. Am. Chem. Soc.2009, 131, 882-883.
(32) Forrester, A. R.; Irikawa, H.; Thomson, R. H.; Woo, S. O.; King, T. J. J. Chem. Soc., Perkin Trans. 11981, 1712-1720.
(33) Kawaguchi, K.; Nakano, K.; Nozaki, K. J. Org. Chem.2007, 72, 5119-5128. (34) Rumynskaya, I. G.; Romanova, E. P.; Agranova, S. A.; Frenkel, S. Y. Acta Polym.1991, 42, 250-253.
(35) Goldfinger, M. B.; Crawford, K. B.; Swager, T. M. J. Am. Chem. Soc.1997, 119, 4578-4593.
(36) Goldfinger, M. B.; Swager, T. M. J. Am. Chem. Soc.1994, 116, 7895-7896. (37) Yamaguchi, S.; Swager, T. M. J. Am. Chem. Soc.2001, 123, 12087-12088. (38) Kordas, K.; Bali, K.; Leppavuori, S.; Uusimaki, A.; Nanai, L.; Science2000, 399-404.
(39) Lee, C. W.; Kim, Y. B.; Lee, S. H. Chem. Mat.2005, 17, 366-372. (40) Asakura, T.; Yamato, H.; Matsumoto, A.; Murer, P.; Ohwa, M. J. Photopolym. Sci. Technol.2003, 16, 335-345.
(41) Ogura, T.; Higashihara, T.; Ueda, M. J. Photopolym. Sci. Technol.2009, 22, 429-435.
(42) Salavagione, H. J.; Miras, M. C.; Barbero, C. Macromol. Rapid Commun.
2006, 27, 26-30.
(43) Shirai, M.; Suyama, K.; Okamura, H.; Tsunooka, M. J. Photopolym. Sci. Technol.2002, 15, 715-730.
(44) Wang, M. X.; Jarnagin, N. D.; Lee, C. T.; Henderson, C. L.; Yueh, W.; Roberts, J. M.; Gonsalves, K. E. J. Mater. Chem.2006, 16, 3701-3707.
(45) Clendenning, S. B.; Aouba, S.; Rayat, M. S.; Grozea, D.; Sorge, J. B.; Brodersen, P. M.; Sodhi, R. N. S.; Lu, Z. H.; Yip, C. M.; Freeman, M. R.; Ruda, H. E.; Manners, I. Adv. Mater.2004, 16, 215-219.
(47) Eng, M. P.; Albinsson, B. Angew. Chem.-Int. Edit.2006, 45, 5626-5629. (48) Berry, J. F.; Cotton, F. A.; Lei, P.; Lu, T. B.; Murillo, C. A. Inorg. Chem.
2003, 42, 3534-3539.
(49) Gaillard, S.; Elmkaddem, M. K.; Fischmeister, C.; Thomas, C. M.; Renaud, J. L. Tetrahedron Lett.2008, 49, 3471-3474.
(50) Gong, H. Y.; Zhang, X. H.; Wang, D. X.; Ma, H. W.; Zheng, Q. Y.; Wang, M. X. Chem.-Eur. J.2006, 12, 9262-9275.
(51) Hasan, H.; Tan, U. K.; Lin, Y. S.; Lee, C. C.; Lee, G. H.; Lin, T. W.; Peng, S. M. Inorg. Chim. Acta2003, 351, 369-378.
(52) Lee, M. H.; Cho, B. K.; Yoon, J.; Kim, J. S. Org. Lett.2007, 9, 4515-4518. (53) Vellis, P. D.; Mikroyannidis, J. A.; Lo, C. N.; Hsu, C. S. J. Polym. Sci., Part A: Polym. Chem.2008, 46, 7702-7712.
(54) Wan, Y. J.; Niu, W. J.; Behof, W. J.; Wang, Y. F.; Boyle, P.; Gorman, C. B. Tetrahedron2009, 65, 4293-4297.
(55) Keinan, S.; Ratner, M. A.; Marks, T. J. Chem. Phys. Lett.2004, 392, 291-296. (56) Hancock, J. M.; Jenekhe, S. A. Macromolecules2008, 41, 6864-6867.
Chapter 2
Optimization of Applicable Coupling Conditions to
the Synthesis of the Precursor Polymer
Behof, W. J.; Brannock, M.C.; Morrison, G.; Gorman, C.B. “Synthesis of a Poly-Cyano
2.1
Introduction
As part of a research program in the synthesis of cyano-containing polymers, we became interested in synthesizing oligomers and polymers with the general repeat unit shown in
Scheme 2-1 (Middle). The diaryl methane subunit can be formed through a benzyl/phenyl
linkage as shown in Scheme 2-1. Scheme 2-1 also shows the two logical bond
deconstructions that could potentially give rise to this linkage. The substituent on the aryl
group could be nucleophilic (M) while the substituent at the benzylic position would then be the leaving group (LG) (Scheme 2-1, top). Alternatively, the substituent positions could be
Scheme 2-1. M Nu CN CN CN + CN E R R LG
CN CN CN
R R LG E CN CN CN + CN Nu R R M
CN CN CN
R R
CN
CN CN CN
R R
CN CN CN
R R n n m m n m
Precursor Polymer
Previous attempts by Gorman group members to synthesize the precursor polymer
(Scheme 2-1), were unsuccessful. These results lead us to believe the coupling reaction was
ineffective.1 Therefore, a two-step strategy was developed to find an effective coupling approach. First, a quick assessment of commercially available or easily synthesized leaving
Herein, we illustrate the efficacy of organo-boranes, organo-lithiums, acetonitrile anions and benzyl anions as nucleophilic partners in the coupling shown in Scheme 2-1. Of all the
methods attempted, use of benzyl anion nucleophiles appeared to be most effective. Thus,
both a nucleophilic aromatic substitution and a palladium mediated pathway using the benzyl anion nucleophiles were optimized. Choice of base, solvent, and catalyst/ligand will be
shown to be key parameters to optimize for a successful coupling. Finally, an optimal, high yielding route will be illustrated.
2.2
Results and Discussion
2.2.1 Assessment of Various Nucleophile and Electrophile Combinations
The first task is to explore potential couplings, which are applicable to the synthesis of the
precursor polymer, with commercially available starting materials. This is needed because there is a vast amount of reported reactions concerning C-C bond formation. The nucleophilic partners that will be explored herein are: organo-boranes, organo-lithiums,
acetonitrile anions and aryl acetonitrile anions.
2.2.1.1 Organo-borane nucleophiles
Organo-borane nucleophiles are of interest because of their functional group tolerance and mild conditions for coupling reactions that utilize them. Nobre et al. illustrated the cross
acids, at both elevated and room temperature.2 The conditions reported in that work were used as a starting point to test the coupling between benzyl bromide 2 and the aryl boronic
acid 1 (Table 2-1). Variations in temperature, time and heating method on the efficacy of the
coupling were also investigated.
Table 2-1. Efficacy of aryl organo-boranes as the nucleophile
CN
Br B(OH)2
CN CN CN CN
+
CN
1 2 3
Entry NB Pg # Pd:Lig Heating Temp (oC) Time (h) Yield [%] of 3a
1 WJB-IV-11 0.01/0.02 Ther. 80 19 16
2 WJB-IV-96 0.02/0.04b Ther. 25 72 0 b,c 3 WJB-IV-97 0.02/0.04 Mic. 130 3 min 0b,c,3
Conditions and Reagents: toluene, K3PO4, Pd(OAc)2, PPh3. aIsolated yield, bMolecule 2
decomposed, yields of 3 were estimated by TLC and NMR to be very low.
A modest 16% yield of molecule 3 was isolated under the reported conditions (entry 1).
However, debromination of molecule 2 was observed as a major side product by NMR and
thin layer chromatography (TLC). The temperature of the reaction was reduced in order to minimize the de-bromination of molecule 2 (entry 2). Although, there was no decomposition
of molecule 2, there was also no formation of molecule 3. The large loss of molecule 2 at
low yields.4,5 This coupling was not explored further. In addition, it is suspected that molecule 2 should only be used at room temperature or lower to minimize debromination.
2.2.1.2 Organo-lithium nucleophiles
Aryl lithium nucleophilic reagents were tested because of their high nucleophilicity, potential to react via a non-palladium mediated pathway, and presumed reactivity at low
temperatures. Pirrung et al. reported the efficient synthesis of di-aryl methanes through,
in-situ generated and electron deficient, aryl lithium reagents with benzyl halides.6 However, when these conditions were applied to the coupling of molecule 1 with the in-situ generated aryl lithium of 4, molecule 3 was isolated only in modest yield (Scheme 2-2).
Scheme 2-2.
CN
Br Br
CN CN CN CN CN
+
2 3
4
n-BuLi, THF
Temp= -78oC, 17% Temp= -100oC, 28%
Possible reasons for the low yields include intermolecular lithium halogen exchange, steric
2.2.1.3 Acetonitrile anion nucleophiles
Carbon-carbon bond formation could proceed through carbanion coupling with a halide
electrophile. Culkin et al. employed acetonitrile anions to synthesize diphenyl acetonitriles from bromo-benzene while showing they have dual functions. First, a single equivalent of acetonitrile anion can act as a nucleophile and form the desired benzyl cyanide. Upon
formation of the benzyl cyanide, a second equivalent of acetonitrile anion can deprotonate the more acidic benzyl proton, creating a benzyl nucleophile which then can undergo another
coupling with molecule 4.7
Table 2-2. Efficacy of acetonitrile anions as the nucleophile
CN CN CN CN
+ Br
H H
CN
4 5 3
Entry NB Pg # Cat Lig Base Solvent Temp(oC) Producta
1 WJB-I-2 Pd2dba3 BINAP NaHMDS DMF 90 0
2 WJB-I-93 Pd(OAc)2 BINAP LiHMDS Toluene 25 0
3 WJB-I-88 Pd2dba3 P(tBu)3 LiHMDS Toluene 25 0
4 WJB-I-89 Pd2dba3 P(tBu)3 LiHMDS Toluene 0 0
Conditions and Reagents: 2 eq. of base, 0.1/0.2 Pd:Ligand. aEstimated by both TLC and
NMR.
Couplings were performed under the conditions reported by Culkin et al. and with slight variations (Table 2-2, Entries 1-3). These yielded no product. Addition of the anion of 5 at
0oC (Entry 4) resulted in the absence of molecules 3 and 4, raising questions about the
observation is supported by literature reports of nucleophilic attack by acetonitrile anions at the electrophilic aryl cyanide carbon.8-10 Therefore, utilization of acetonitrile anions as a nucleophilic coupling partner was not a practicable route.
2.2.1.4 Benzyl cyanide nucleophiles
Deprotonation of PhCH2CN or o-CNPhCHCN and subsequent use as a nucleophilic
equivalent has had several successful precedents. You and Verkade11,12 illustrated coupling of phenyl acetonitrile anions to aryl halides in the presence of a proazaphosphatrane ligand.
Culkin and Hartwig7,13 showed that the anion of benzyl nitriles could undergo a palladium-mediated coupling to aryl halides and studied the mechanism of this transformation in some detail (Scheme 2-3). Wan et al. has reported couplings between aryl chlorides and phenyl
acetonitrile anions through a nucleophilic aromatic substitution pathway (NAS).14 Therefore we pursued this method further because of these successful precedents and the significant
limitations described with the aforementioned (Section 2.2.1-3) methods.
Scheme 2-3.
CN
R= H, 93% R=t-Bu, 98%
R + K-OtBu, Pd(OAc)2
BINAP or Verkade Cat. Br
2.2.2 Optimization of Couplings Employing Benzyl Cyanide Nucleophiles
Given the precedents described in Section 2.2.1.4, a cyanobenzyl nucleophile was selected
as the nucleophile for this coupling. Moreover, o-cyanophenyl benzyl nitrile (o -CNC6H5CH2CN) is relatively acidic, (pKa = 19.2 in dimethyl sulfoxide (DMSO)).15 The
resulting carbanion would then be a suitable nucleophile in a carbon-carbon coupling
reaction. The pKa determination of similar molecules containing such groups will be reported below. Furthermore, the arene is relatively electron deficient which renders it a
good electrophile. As reported, Hartwig et al.,6 Satoh et al.14 and Verkade et al.10, 11 employed, with high efficiency, phenyl acetonitrile for monoarylations and acetonitrile for di-arylations.
The examples given above suggest several challenges and potential opportunities in the carbon-carbon bond forming reaction under study here. First, since our proposed arene is so
electron deficient, can simple nucleophilic aromatic substitution compete with palladium-mediated coupling? This question may also be relevant in some of the examples shown above. Second, in the work above, limitations were observed when ortho substituents were
present on the arene electrophile.16 The presence of o-cyano groups in our target might thus be an issue. Third, since the methine proton in the product (e.g. Ph2(CN)CH) should be more
acidic than the proton that must be removed in the starting material (e.g. Ph(CN)CH2), can
Fourth, are reactions where Ph2(CN)C- (Scheme 2-4, right) acts as a nucleophile
problematic?
Scheme 2-4.
X
CN CN
R R
CN
R CN CN
X
CN CN
R R
CN
R CN CN
2.2.2.1 Nucleophilic aromatic substitution
In the work of Hartwig et al.7,13,17 and Verkade et al.11,12 discussed above, both electron rich and electron deficient aryl halide substrates were explored. In the coupling of interest here, the aryl halide is electron deficient, begging the question as to whether, in this case, a
nucleophilic aromatic substitution (NAS) reaction would be applicable. We tested this in three ways. First, a reaction from the literature was repeated under NAS conditions. Then from this, two new coupling reactions were explored.
First, the NAS pathway was investigated in the case of an electron deficient arene. Culkin and Hartwig reported formation of molecule 8 from p-bromo benzonitrile (6) in 99% yield in
the presence of palladium acetate (Pd(OAc)2) and
2,2’-bis(diphenylphosphino)-1,1’-binaphthyl (BINAP).7 You and Verkade reported a similar reaction using p-chloro benzonitrile (7) to achieve molecule 8 in 92% yield in the presence of Pd(OAc)2 and a
palladium/ligand under the same conditions and obtained 42% (X = Br) and 72% (X = Cl) yield, respectively. Thus, palladium-mediated coupling does result in a higher yield. However, in this case, the results provide evidence for competition between NAS and the
palladium pathway.
Scheme 2-5.
CN
X
CN
NC
6: X= Br
7: X= Cl
8 i or ii
Conditions and Reagents: (CH3)2CHCN, sodium hexamethyldisilazide (NaHMDS),
toluene (i) 6, 100⁰C, 1 h, 42% (ii) 7, 90⁰C 2 h, 72%.
We then turned to the NAS of 2,6-dichloro benzonitrile (9)with o-cyanophenyl acetonitrile
(10).When two equivalents of molecule 10 were reacted withmolecule 9 in the presence of
sodium tert-butoxide (Na-OtBu) (Scheme 2-6), only the mono-coupled product 11 was
obtained in 91% yield. When molecule 11 was isolated and reacted with molecule 10 for a
longer period of time (72 h) and in excess (3.2 equivalents) base, a small amount (13 %) of bis-coupled product 12 was isolated. This reaction was clearly inefficient for the formation
Scheme 2-6.
CN Cl Cl
CN
CN CN
Cl
11 9
CN CN
CN CN CN
12
CN CN
+
10
i ii
Conditions and Reagents: (i) Na-OtBu, THF, µW, 155oC, 5 m, 91%; (ii) Na-OtBu, 10,
THF, reflux, 72 h, 13%.
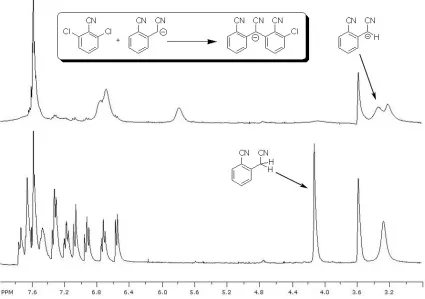
In an attempt to understand why this coupling was inefficient, the reaction was followed by nuclear magnetic resonance (NMR). Molecule 10 (1.4 equiv.) and sodium tert-butoxide (1.2 equiv.) were mixed in d8-tetrahydrofuran (THF). The benzylic peak at 4.15 ppm shifted to
3.45 ppm and broadened considerably as would be expected for deprotonation of molecule
10. Then, 2,6-dichloro-benzonitrile (9) electrophile (1.0 equiv) was introduced. After 43 m,
the NMR spectrum (Figure 2-1, top) showed both nucleophile and electrophile co-existing in
solution with the emergence of new peaks. After 2 h, Figure 2-1, bottom showed no anion
as evidenced by the disappearance of the peak at 5.7 ppm. Moreover, a re-emergence of the
benzyl protons peak at 4.1 ppm was observed. Thus, it was concluded that the anion was gone. Moreover, no methine peak indicative of the products 11 or 12 was observed around
6.2 ppm. Instead, a series of well-separated peaks relatively upfield for aromatic protons were observed. These were assigned to the anion of 11 as the same spectrum could be
Figure 2-1. Reaction of molecules 9 and 10. (top) After 43 m (bottom) after 2 h.
2.2.2.2 Reduction of basicity through metal complexation
The effects of benzyl anion metal complexation were explored because organo-zinc
reagents are milder carbanions but are still quite nucleophilic. Benefits of milder reagents include a potential shift in the anion equilibrium. Second, the organo-zinc could be less likely to attack the aryl cyanide. After deprotonation of molecule 10 in d8-THF ZnBr2 was
added. The organo-zinc reagent subsequently formed (Figure 2-2), as suggested by the
density around the aryl ring. This is expected as the C-ZnBr bond should be more covalent than that of a Na+ PhCHCN- salt. However, after heating at 60oC for 2 h there was no formation of molecule 11. Thus, the organo-zinc reagent appeared to be insufficiently
reactive in this case. Additionally, complete deprotonation of the methine proton was observed when molecule 11 was added. Similar experiments performed with CeCl3 in place
of ZnBr2 also failed to yield the desired product.
2.2.2.3 Palladium-mediated coupling
Given the poor results when attempting to prepare molecule 12 via NAS, palladium
mediated-coupling was explored. Hartwig et al. showed that Pd(OAc)2/BINAP could couple
phenyl acetonitrile anions with p-tBu-bromobenzene.7 These conditions were used as the starting point to optimize the reaction for the formation of molecule 12. Because it is the
second coupling that is challenging, the reaction of molecule 11 with molecule 10 was
explored. The mono iodo and bromo derivatives of molecule 9 are proposed as electrophiles
since palladium-mediated couplings typically are more efficient with aryl bromides and iodides than chlorides. The synthesis of molecule 18 was challenging because even though
molecule 16 could be synthesized from molecule 15 the subsequent conversion to the aryl
cyanide failed (Scheme 2-7).18 This method is currently being explored by other Gorman
group students. Electrophilic aromatic substitution from molecule 14, which was synthesized
from the di-ortho lithium metalation of molecule 13,19 with iodine mono-chloride (ICl) was
ineffective.20 However, molecule 18 could be synthesized from a di-lithium halogen
Scheme 2-7.
CN TMS TMS
C6H13 CN
C6H13
13 14
I I
NH2
C6H13 NH2
C6H13
16 17 CN I I CH3 18 CN Br Br CH3 15
Li-TMP, I2 88%
I2, AgNO3 Quant.
ICl, CH2Cl2
0%
CuSO4, K3CuCN4,
H2SO4, NaNO2, 0%
n-BuLi, I2 70%
CN I I
C6H13
CN I I
C6H13
.
The di-phenyl methane nucleophilic partners 21-23 were synthesized through NAS of
molecules 17, 18 and 20 with molecule 10 (Scheme 2-8). Molecule 20 was synthesized
through bromination of 4-hexyl aniline to form molecule 19. This is then followed by
Scheme 2-8. CN X X R CN CN CN R X CN CN + NH2 R Br Br
19: R= C6H13 20: R= C6H13, X= Br, 10% 17: R= CH3, X= Br 18: R= CH3, X= I
21: R= C6H13, X= Br, 60% 22: R= CH3, X= Br, 92% 23: R= CH3, X= I, 90%
i ii
Conditions and Reagents: (i) CuSO4, K3CuCN4, H2SO4, NaNO2; (ii). K-OtBu, DMF, µW,
100oC, 100W, 25 m.
Reaction of molecules 22 and 10 under palladium-mediated coupling was then explored.
The results are shown in Table 2-3. A maximum yield of 62% was obtained. Comparison of
entries 1 and 2 in Table 2-3 indicate that use of the aryl bromide indeed does result in a
higher yield. Comparison of entries 2 and 3 indicate that NAS is indeed also less efficient on the aryl bromide compared to palladium mediated coupling. Comparison of entries 3 and 4
indicate that microwave heating was more efficient than thermal heating and gave a much higher yield of product. Moreover, extending the reaction time under thermal heating showed no further increase in yield when the reaction time was increased from 4 h to 6 h.
Table 2-3. Results of coupling of 22 with 10 under Pd-mediated coupling conditions.
CN CN CN CN CN
Br
CN CN CN CN CN
10 22
24
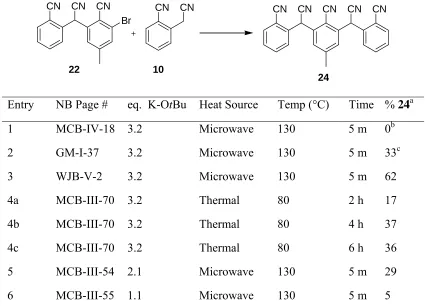
Entry NB Page # eq. K-OtBu Heat Source Temp (°C) Time % 24a
1 MCB-IV-18 3.2 Microwave 130 5 m 0b
2 GM-I-37 3.2 Microwave 130 5 m 33c
3 WJB-V-2 3.2 Microwave 130 5 m 62
4a MCB-III-70 3.2 Thermal 80 2 h 17
4b MCB-III-70 3.2 Thermal 80 4 h 37
4c MCB-III-70 3.2 Thermal 80 6 h 36
5 MCB-III-54 2.1 Microwave 130 5 m 29
6 MCB-III-55 1.1 Microwave 130 5 m 5
Conditions: 0.3M THF, 1.2 eq. 10, 0.1 eq. Pd(OAc)2, 0.2 eq. BINAP. aValues obtained
from HPLC with an estimated error of ± 5%. bMolecule 9 was used instead of molecule 22. cNo Pd catalyst or BINAP were added.
2.2.2.4 Variation of base and effect of cation counterion complexation
The yield of a palladium-catalyzed coupling reaction can be greatly influenced by the base
employed. Furthermore, the data above indicate that excess base is required. This requirement likely results because (1) both molecules 22 and 24 are deprotonated
preferentially to molecule 10 and (2) any equilibrium between molecules 22- or 24- and
true. You and Verkade showed that the reaction between bromobenzene and benzyl cyanide to form diphenyl acetonitrile could be accomplished in 93 % yield using 1.4 equivalents of sodium hexamethyl disilazide.11,12 The tabulated pKa values of benzyl cyanide and diphenyl acetonitrile are 21.9 and 17.5, respectively.21 Thus, we speculated that there likely was some equilibrium between the anion of the product and that of the starting material in this case.
However, in our case, the o-cyano groups likely had an important influence on the pKa (and more importantly, the relative pKa) values of our starting materials and product.
To determine the relative acidity of the protons on molecules 10, 22 and 24, pKa
measurements in DMSO were performed, by a co-worker Molly Brannock, using the procedure developed by Bordwell et al.22 The pKa of molecule 10 was measured to be19.2.
Molecule 22 had a pKa of 13.6 and molecule 24 had pKa1 and pKa2 values of 13.2. The
similarity of the two pKa values for molecule 24 is consistent with the findings of
Streitwieser where unfused, diprotic structures were determined to have very similar, almost
indistinguishable, pKa values.23 Thus, if the anion of molecule 10 is produced, it is likely in
equilibrium with the anion of molecule 22 and the (di)anion of molecule 24 (Scheme 2-4).
Given the values, this equilibrium is likely to be unfavorable, resulting in the need to use
more than one equivalent of base. This need is in contrast to the results reported by You and Verkade above.
These pKa values may be only partially relevant, however. There are reports that suggest weak bases such as potassium carbonate (K2CO3)24 and dimethylamino pyridine (DMAP)25
molecules 10, 22 and 24 might be quite different. Indeed, based on computations, Ding et al.
suggest that neutral acids are typically eleven orders less acidic in THF than in DMSO.26 Furthermore, even if the anions of molecules 22 and 24 are produced, they may be innocent
or reactive, particularly depending on the solvent and counter-cation present. The reactivity of carbanions is particularly variable depending on these parameters.27 Thus, some variation of base was explored.
To explore variation of base in an efficient manner, reactions were conducted under otherwise identical conditions and analyzed by HPLC. Both the percentage of unreacted
molecule 22 and the percentage of molecule 24 are given in Table 2-4. Weak bases (entries
1-5) clearly were ineffective. Potassium tert-butoxide yielded the greatest conversion.
Addition of 18-crown-6, however, resulted in little recovered starting material or product. Thus, use of the larger potassium counterion favors product formation but complexation of the potassium tends to produce side products (e.g. loss of starting material without formation
Table 2-4. Variation of Base
CN CN CN CN CN
Br
CN CN CN CN CN
10 22
pKa (DMSO) = 13.6
24
pKa (DMSO) = 13.2
Yield % ofa
Entry NB Page # Base Recovered 22 24
1 GM-I-61 CsF 93 4
2 GM-I-29 NEt3 100 ndb
3 GM-I-31 Pyridine 97 ndb
4 GM-I-61 Ph2NH 98 1
5 WJB-V-4 Cs2CO3 87 8
6 GM-I-35 NaOMe 47 22
7 GM-I-50 Na-OiPr 53 1
8 GM-I-74 Li-OtBu 100 ndb
9 WJB-V-6 Na-OtBu 74 26
10 WJB-V-2 K-OtBu 15 62
11 MCB-III-53 K-O18-crown-6 tBu/0.1 eq. 3 37
Conditions: 0.3M in THF, 1.2 eq. 10, 3.2 eq. Base, 0.1 eq. Pd(OAc)2, 0.2 eq. BINAP,
130°C, µW, 100W, 5 m. aValues obtained from HPLC with an estimated error of ± 5%.
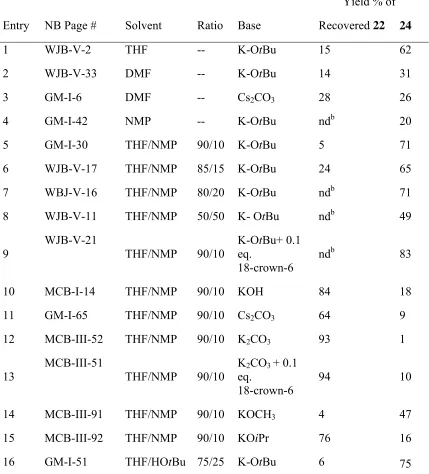
2.2.2.5 Variation of solvent / base combinations
As this reaction generates anions that must transmetallate to palladium, and as metalated
nitriles form both complex aggregates with counterions28 and also complex to palladium,7 the choice of counterion and solvent is likely to have a large influence on the efficiency of this reaction. Thus, several solvents were explored for further optimization of the coupling. The
results are presented in Table 2-5. The best conditions found in Section 2.2.2.4 are
reproduced as Entry 1. Inoh et al. used Cs2CO3/DMF to deprotonate p-nitro toluenes (pKa of
20.4 in DMSO),29 yet entries 2 and 3 in Table 2-5 indicate poor conversion and loss of
starting material when DMF was used. When molecule 24 was heated in DMF briefly,
decomposition was observed indicating that DMF is not a suitable solvent for this reaction.
N-Methylpyrrolidone (NMP) was tried as an alternative polar, aprotic solvent (Table 2-5,
entry 4). The desired product was obtained in 20% yield with no starting material recovered.
However, when mixed NMP/THF was used (Table 2-5, entries 5-9), excellent results were
obtained. In 90/10 THF/NMP in the presence of 0.1 eq. 18-crown-6, an 83% yield of product was obtained. Note that, in the presence of NMP, 18-crown-6 increased the reaction yield.
This behavior was not the case in pure THF. In addition, use of aryl iodide 23 as the
electrophile yielded less than optimal results. This is in sharp contrast to the notion that aryl
iodides are better electrophilic partners under palladium mediated catalysis. Use of molecule
21 as the electrophile illustrated that solubility of (di)anion 25 has little effect on the yield
Scheme 2-9.
CN CN CN CN CN
Br
CN CN CN CN CN
C6H13
C6H13
21 10 25
K-OtBu, Pd(OAc)2, BINAP
THF/NMP (90/10), µW 100W, 5 m, 62%
To test the efficiency of a milder base in this solvent system (Table 2-5, entries 10-13),
several other, weaker bases than K-OtBu were explored. The moderate conversion to molecule 24 using potassium hydroxide (KOH) (Table 2-5, entry 10), indicates that KOH
was strong enough for deprotonation, but this base was not efficient. It was suspected that
the formation of water upon the protonation of hydroxide anion might be hindering the further formation of product by inactivating the palladium. To test the effect of water on the yield, reactions containing 0.22, 0.44, and 1.1 equivalents of water were run. Yields were
found to change minimally from 71 % to 77% when 0.22 eq. were added and 77% to 71% when 0.44 equivalents were added, but decreased dramatically from 71% to 39% when 1.1
eq. were added. Carbonate bases were also explored but gave poor yields. The slightly less basic potassium methoxide and potassium iso-propoxide gave no advantage in yield. Furthermore, addition of tert-butyl alcohol (as a buffer) systematically decreased the yield of
Table 2-5. Variation of solvent and then further variation of base.
Yield % ofa Entry NB Page # Solvent Ratio Base Recovered 22 24
1 WJB-V-2 THF -- K-OtBu 15 62
2 WJB-V-33 DMF -- K-OtBu 14 31
3 GM-I-6 DMF -- Cs2CO3 28 26
4 GM-I-42 NMP -- K-OtBu ndb 20
5 GM-I-30 THF/NMP 90/10 K-OtBu 5 71
6 WJB-V-17 THF/NMP 85/15 K-OtBu 24 65
7 WBJ-V-16 THF/NMP 80/20 K-OtBu ndb 71
8 WJB-V-11 THF/NMP 50/50 K- OtBu ndb 49
9 WJB-V-21 THF/NMP 90/10 K-Oeq. tBu+ 0.1 18-crown-6
ndb 83
10 MCB-I-14 THF/NMP 90/10 KOH 84 18
11 GM-I-65 THF/NMP 90/10 Cs2CO3 64 9
12 MCB-III-52 THF/NMP 90/10 K2CO3 93 1
13
MCB-III-51
THF/NMP 90/10
K2CO3 + 0.1
eq.
18-crown-6 94 10
14 MCB-III-91 THF/NMP 90/10 KOCH3 4 47
15 MCB-III-92 THF/NMP 90/10 KOiPr 76 16
16 GM-I-51 THF/HOtBu 75/25 K-OtBu 6 75
Conditions: 1.2 eq. 10, 3.2 eq. Base, 0.1 eq. Pd(OAc)2, 0.2 eq. BINAP, 130°C, µW, 100W,
2.2.2.6 Effect of supporting ligand
Additional monodentate and bidentate phosphine ligands were tested for coupling
efficiency as shown in Table 2-6. The bidentate bis-diphenylphosphinoferrocene (dppf) was
used both in the presence and absence of 18-crown-6 (entries 2 and 3 respectively). A slight increase in yield was observed in the absence of 18-crown-6, so subsequent reactions were
run without this additive. Use of the bidentate diphenylphosphinobutane (dppb) and bis-diphenylphosphinoethane (dppe) resulted in similar yields (entries 4 and 5). Three
monodentate ligands were then explored (entries 6, 7, and 8). Tri-cyclohexylphosphine (P(Cy)3) provided the best results. The comparably poor result obtained with P(tBu)3