Copyright1999 by the Genetics Society of America
Characterization of the Repeat-Tract Instability and Mutator Phenotypes
Conferred by a
Tn3
Insertion in
RFC1
, the Large Subunit of the
Yeast Clamp Loader
Yali Xie,* Chris Counter
†and Eric Alani*
*Section of Genetics and Development, Cornell University, Ithaca, New York 14853-2703 and†Department of Pharmacology and
Cancer Biology, Department of Radiation Oncology, Duke University Medical Center, Durham, North Carolina 27710
Manuscript received August 3, 1998 Accepted for publication October 22, 1998
ABSTRACT
The RFC1 gene encodes the large subunit of the yeast clamp loader (RFC) that is a component of eukaryotic DNA polymerase holoenzymes. We identified a mutant allele of RFC1 (rfc1::Tn3) from a large collection of Saccharomyces cerevisiae mutants that were inviable when present in a rad52 null mutation background. Analysis of rfc1::Tn3 strains indicated that they displayed both a mutator and repeat-tract instability phenotype. Strains bearing this allele were characterized in combination with mismatch repair (msh2D, pms1D), double-strand break repair (rad52), and DNA replication (pol3-01, pol30-52, rth1D/rad27D) mutations in both forward mutation and repeat-tract instability assays. This analysis indicated that the
rfc1::Tn3 allele displays synthetic lethality with pol30, pol3, and rad27 mutations. Measurement of forward
mutation frequencies in msh2Drfc1:Tn3 and pms1Drfc1:Tn3 strains indicated that the rfc1::Tn3 mutant
displayed a mutation frequency that appeared nearly multiplicative with the mutation frequency exhibited by mismatch-repair mutants. In repeat-tract instability assays, however, the rfc1::Tn3 mutant displayed a tract instability phenotype that appeared epistatic to the phenotype displayed by mismatch-repair mutants. From these data we propose that defects in clamp loader function result in DNA replication errors, a subset of which are acted upon by the mismatch-repair system.
M
UTATIONS in genes that are involved in DNA A major type of chromosomal instability that has been replication and repair often result in chromo- identified in yeast, in bacteria, and in cancer cells is somal instabilities such as base pair substitutions and repeat-tract instability (reviewed inCrouse1996; Kolod-frameshifts as well as insertion, deletion, and re- ner1996;ModrichandLahue1996;Siaet al. 1997b).arrangement events (i.e., Schaaper and Dunn 1987; This instability is thought to result mainly from the AguileraandKlein1988;Strandet al. 1993;McAlear failure to repair DNA slippage events that occur during
et al. 1996;Tranet al. 1996;GreenandJinks-Robert- the replication of repetitive DNA sequences such as son1997). Mutations that confer these phenotypes have those that contain mono-, di-, tri-, and tetranucleotide been directly identified in Escherichia coli and Saccharo- repeats. In both yeast and mammalian cells, mutations
myces cerevisiae using a variety of chromosome instability in mutS and mutL homolog mismatch-repair genes result assays (i.e., Feinstein and Low 1986; Aguilera and in a large increase (100- to 10,000-fold) in the rate of Klein1988; Michaels et al. 1990; Jeyaprakash et al. repeat-tract instability (Strandet al. 1993;Tranet al.
1994). Such basic research approaches have also led to 1997;Umaret al. 1998). This increase is thought to be a better understanding of the underlying chromosome due to the inability of these mutants to repair small stability defects that have been observed in patients who loop insertion/deletion mutations that occur during suffer from particular inherited diseases. For example, DNA slippage (reviewed in Crouse 1996; Kolodner the phenotypes exhibited by mismatch-repair-defective 1996; ModrichandLahue1996;Siaet al. 1997b). In
E. coli and S. cerevisiae strains provided evidence indi- yeast, mutations in DNA replication genes that encode
cating that the underlying cause for a large percentage the polymerase processivity factor PCNA (POL30) and of hereditary nonpolyposis colorectal cancers was a de- the flap endonuclease (RTH1/RAD27) have also been fect in mismatch repair (LevinsonandGutman1987; shown to confer an increase in repeat-tract instability at Strandet al. 1993;Crouse1996; reviewed inKolodner a rate that is similar to that observed in mismatch-repair-1996;ModrichandLahue1996).
defective mutants (Johnsonet al. 1995, 1996;Umaret al. 1996;Kokoskaet al. 1998). Mutations in other DNA
replication genes, such as those that encode the Pold (POL3), and Polε(POL2) DNA polymerases, however, Corresponding author: Eric Alani, Section of Genetics and
Develop-resulted in only modest increases in repeat-tract
instabil-ment, Cornell University, 459 Biotechnology Bldg., Ithaca, NY
14853-2703. E-mail: [email protected] ity (Johnsonet al. 1995; Tranet al. 1997; Kokoskaet
al. 1998). These data have led to the proposal that some man1989;Burgers1991;FienandStillman1992; Stillman 1994).
DNA replication factors function directly in the mis-match-repair pathway to repair loop insertion/deletions
In this study we tested strains bearing the rfc1::Tn3 that result from DNA slippage events while others act
allele alone or in combination with mismatch repair and at the level of DNA replication to prevent the formation
DNA replication mutations for defects in chromosome of these events (Kokoskaet al. 1998; reviewed inSiaet
stability. As described below, the rfc1::Tn3 mutation
con-al. 1997b).
ferred a mutator phenotype that appeared multipli-The above observations, in conjunction with the
obser-cative with mismatch-repair mutations. In repeat-tract vation that many cancer cells display repeat-tract
insta-stability assays the rfc1::Tn3 mutation conferred anz 10-bilities that are unlinked to previously identified repair
fold increase in the frequency of dinucleotide repeat-and replication genes, encouraged us to initiate screens
tract instability that appeared to be epistatic to the phe-in yeast to identify chromosomal phe-instability mutants
notype observed in mismatch-repair-defective mutants. (Liuet al. 1995). We hoped to identify additional factors
Taken together, our data are consistent with the idea that are involved in preventing mutagenic replication
that defects in clamp loader function result in DNA errors by preventing DNA slippage events or by
facilitat-replication errors, a subset of which are identified and ing the repair of these slippages through
mismatch-repaired by the mismatch-repair system. repair mechanisms. In one screen we searched for
mu-tants that displayed an increase in the frequency of both repeat-tract insertion/deletion and base pair
substitu-MATERIALS AND METHODS tion/frameshift events; unfortunately, this screen only
Media and chemicals: E. coli strains were grown in
Luria-identified mutants that displayed mutations in the
pre-Bertani (LB) broth or on LB agar, which was supplemented viously characterized mutS (MSH2) and mutL (PMS1,
with 100 mg/ml ampicillin when required (Miller 1972).
MLH1) homolog genes and the RAD27 gene (Y. Xie,
Yeast strains were grown in either YPD or minimal selective L. SchvaneveldtandE. Alani,unpublished data). In media (Roseet al. 1990). Selective media contained 0.7% yeast a second screen that is the focus of this article, we nitrogen base, 2% agar, 2% glucose, and 0.09% of a drop-out mix that lacks the amino acid used for selection. Sporulation searched for mutants that were inviable in a
double-media (SPM) were prepared as described previously (Detloff strand break-repair-deficient (rad52) background and
et al. 1991). When required, canavanine was included in mini-also displayed a mutator phenotype (Counter et al. mal selective media at 60 mg/liter (Roseet al. 1990).
5-fluoro-1997). This screen was conducted on the basis of two orotic acid (5-FOA) was purchased from U.S. Biologicals and observations made in E. coli: used as described previously (Boeke et al. 1984). When
re-quired, methyl-methane sulfonate (MMS; Aldrich Chemical, 1. Certain DNA replication mutants display chromo- Milwaukee) was included in YPD media at 0.017% (v/v).
E. colistrains:DH5a[F9phi80, dLacZD(lacZYA-argF), U169, some instability defects such as chromosomal
break-recA1, endA1, hsdr17 (r2K, m1K), lambda2, thi1, gyrA, relA1] ages that are lethal in recombination-deficient
back-and KC8 (gammax14862, MK121, leuB600, trpC9830, pyrF::Tn5, grounds (Michelet al. 1997). hisB463, Dlacx74, StrA, galU, K ) were used to amplify and
2. Strains that lack dam methylase, and are thus defec- manipulate all plasmids described in this article.
tive in strand discrimination during mismatch re- S. cerevisiaestrains: The genotypes of all strains used in these studies are shown in Table 1. With the exception of pair, display a mutator phenotype and are inviable
NKY 1068, EAY 561, and DNR53, all strains were derived from in recombination deficient (recA2) backgrounds
the isogenic FY strain background (Winston et al. 1995). (McGraw and Marinus 1980). This inviability, DNR53 is also an S288C-derived strain that contains the which can be rescued by mutations in the mutS, rfc1::Tn3 allele (Burnset al. 1994;Counteret al. 1997). This strain also contains a rad52::HIS3 mutation and is viable
be-mutL, and mutH mismatch-repair genes, is thought
cause it contains an ARS1, CEN4, URA3, plasmid bearing the to be caused by unrepairable double-strand breaks
wild-type RAD52 gene (Counteret al. 1997). NKY 1068 is an in dam2recA2strains that form as the result of MutH
SK1-derived strain (Kane and Roth 1974) that was kindly incising both template and newly replicated strands provided by Douglas Bishop and EAY 561 is a rfc1::Tn3 deriva-(McGraw andMarinus 1980;Au et al. 1992). Us- tive of NKY 1068.
The msh2D::TRP1, msh2D::hisG, pms1D::hisG, rad52D::URA3, ing this second screen we identified and
character-rad52D::LEU2, and rad27D::HIS3 alleles contain complete or ized a transposon Tn3 insertion allele of RFC1, a
nearly compete coding region deletions of their respective gene that encodes the large subunit of the highly
genes and were introduced into FY23 and FY86 by single-step conserved RFC complex that functions in eukaryo- transplacement. The primer sequences that were used to make tic DNA replication and repair. During DNA repli- polymerase chain reaction (PCR)-amplified DNA fragments containing the rad27D::HIS3 allele were described by Tish-cation, RFC interacts with the sliding clamp PCNA
koffet al. (1997) and the rad52D::URA3 and rad52D::LEU2 at the replication fork primer terminus in steps that
disruption plasmids were kindly provided by Todd Milne and require adenosine 59-triphosphate (ATP).
Forma-Dennis Livingston, respectively. All of the other disruption tion of the RFC-PCNA-primer terminus complex plasmids were made in the Alani laboratory and are available then promotes efficient DNA synthesis by both the upon request. Double mutant combinations of the alleles de-scribed in Table 1 were made by standard crosses (Roseet al.
Still-1990). The pol3-01 and pol30-52 alleles were introduced by Nucleic acid and protein techniques:All restriction endonu-cleases were purchased from New England Biolabs (Beverly, two-step transplacement (Roseet al. 1990). Vectors used to
introduce the pol3-01 (Y IPAM26) and pol30-52 (pBL241-52) MA) and used according to manufacturers’ specifications. Taq and Expand polymerases were purchased from Perkin-Elmer alleles into the FY strain background were kindly provided by
Akio Sugino and Peter Burgers, respectively. The rfc1::Tn3 Cetus (Norwalk, CT) and Boehringer Mannheim (Indianapo-lis), respectively. Plasmid DNA was isolated by alkaline lysis allele was introduced into the FY and NKY strain backgrounds
by single-step transplacement using DNA that had been PCR and all DNA manipulations were performed as described pre-viously (Maniatiset al. 1982). Yeast chromosomal DNA was
amplified from DNR53 chromosomal DNA using RFC1
spe-cific primers. The phenotype of rfc1::Tn3 in the different strain prepared as described byHolm et al. (1986). Preparations
backgrounds was identical with respect to mutator phe- were made from 5-ml yeast cultures grown to saturation in notype and cold, MMS, and UV sensitivity. All of the alleles YPD. The purified chromosomal DNA was dissolved in 50ml that were introduced by single-step transplacement were con- of double-distilled water and stored at2208prior to use. firmed by PCR analysis of chromosomal DNA isolated from Polymerase chain reaction (PCR) was performed as de-the transformed strains (primers available upon request). scribed previously (Saikiet al. 1985) and amplification
condi-The introduction of the pol30-52 mutation into the FY strain tions and primer sequences for the different reactions are was confirmed by sequencing PCR-amplified DNA frag- available upon request. The basic reaction was performed for ments containing the POL30 open reading frame. The pres- 30 cycles using a denaturation step of 30 sec at 948, an anneal-ence of the pol3-01 mutation was confirmed by digesting the ing step of 30 sec at 588, and a polymerization step of 2 min PCR-amplified POL3 gene from candidate strains with EcoRV at 728. Reactions were performed using Taq polymerase in 25 (recognition site is lost) or BstUI (recognition site is gained). ml with 5 pmol of each primer andz1mg of yeast DNA. The Detailed information about these restriction enzyme digestion DNA primer synthesis and DNA sequencing were performed protocols is available upon request. at the Cornell Biotechnology Analytical/Synthesis facility.
Genetic techniques:Yeast were transformed with DNA
us-ing the lithium acetate method as described byGeitz and
Schiestl(1991). Tetrads were dissected on YPD plates
imme-RESULTS diately after zymolyase treatment using previously established
methods (Roseet al. 1990). In the tetrad analysis described Identification of arfc1allele containing aTn3::LEU2
in Table 3, all tetrads that yielded four, three, and sometimes
insertion: We screened strains mutagenized with Tn3 two and one viable spores were examined for relevant genetic
transposon insertions for those that were inviable in a markers by PCR, by segregation of a linked marker (i.e.,
rad27D::HIS3), or by phenotype (i.e., mutator phenotype, double-strand break-repair-deficient rad52 null back-MMSs).
ground (Burnset al. 1994;Howellet al. 1994;Counter Mutation frequencies shown in Table 2 were determined by
et al. 1997). A collection of 836 yeast mutants that were
measuring the frequency of forward mutation to canavanine
identified from z300,000 colonies was examined for resistance (i.e.,Reenan and Kolodner 1992). Repeat-tract
instability frequencies (Tables 2 and 4) were determined by mutator phenotypes on canavanine plates and a single measuring frameshift events that resulted in resistance to candidate (DNR53, Table 1) was identified (Roseet al. 5-FOA in strains containing the plasmid pSH44[(GT)16T-URA3, 1990; Counter et al. 1997). Sequencing of the DNA ARSH4, CEN6, TRP1] (HendersonandPetes1992). In both
that flanked the Tn3 insertion in this candidate revealed the mutator and repeat-tract instability studies, tested strains
that the Tn3 element was located at bp 756 in the RFC1 were streaked to form single colonies on selective minimal
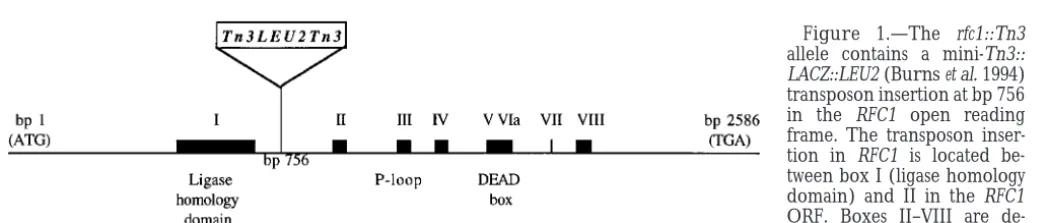
plates containing 2% glucose. Eleven independent colonies open reading frame (ORF) between homology boxes I were suspended in water and appropriate dilutions were then (ligase homology domain) and II (Figure 1;Cullmann plated onto minimal media with or without canavanine or
et al. 1995). The allele created by the insertion is referred
5-FOA. The median frequency of canavanine and 5-FOA
resis-to as rfc1::Tn3. Previously,Howellet al. (1994) showed
tance was determined for each experiment and the average
of three-to-six independent experiments is presented for each that the RFC1 gene product is required for cell viability; strain. however, plasmids containing a deletion variant of the The length of GT repeat tracts was determined by sequenc- RFC1 gene that lacks the entire aminoterminal ligase ing pSH44-derived plasmids recovered from independently
homology domain (D1-273) can weakly complement the isolated 5-FOAr colonies (Rose et al. 1990). Plasmids were
cold-sensitive phenotype of rfc1-1 mutants (Howell et
sequenced using the 240 sequencing primer described by
HendersonandPetes(1992). To examine large insertion/ al. 1994; Figure 1). Consistent with this observation was
deletion alterations in the CAN1 gene, the complete open the finding that the ligase homology domain of the reading frame of the CAN1 gene was amplified by PCR from
human homolog of RFC1 is not required for PCNA chromosomal DNA isolated from independently identified
interactions or replication functions in vitro (Uhlmann canr colonies. Amplified DNA was digested with Hph1 and
then electrophoresed on a 2% TAE-agarose gel. et al. 1997). On the basis of this information we
hypothe-Mitotic recombination frequencies were determined by mea- size that in rfc1::Tn3 strains a cryptic promoter within the suring the frequency of His1colonies in wild-type (NKY 1068) Tn3 element is driving expression of an aminoterminal-and rfc1:Tn3 (EAY 561) strains bearing a his4X-ADE2-his4B
cas-truncated version of the RFC1. sette (Bishopet al. 1992). From each strain 13 independent
Characterization of the rfc1::Tn3 phenotype: The colonies were plated using the appropriate dilutions onto
minimal media with or without histidine. The median fre- rfc1::Tn3 allele that was identified in DNR53 was
intro-quency of His1recombinants was determined. duced into the FY and SK-1 strain backgrounds (Table The genetic data presented in Table 2 and in the text were
1;materials and methods) and tested in DNA repair analyzed using the Mann-Whitney test statistic where P values
and mutator assays. This strain displayed a 19-fold in-,0.05 are considered significant (Pfaffenbergerand
TABLE 1
Strains used in this study
Strain Genotype Source
DNR53 MATa leu2-3, 112 lys2D201 trp1D1 ura3-52 his3D200 rad52::HIS3 C. Counter
rfc1::Tn3::LEU2 pRAD52 (ARS-CEN, URA3)
FY 23 MATa ura3-52 leu2D1 trp1D63 F. Winston
FY 86 MATaura3-52 leu2D1 his3D200 F. Winston EAY 255 MATaura3-52 leu2D1 his3D200 rad52D::LEU2 Lab collection EAY 265 MATaura3-52 leu2D1 his3D200 msh2D::TRP1 rad52D::LEU2 Lab collection EAY 281 MATa ura3-52 leu2D1 trp1D63 msh2D::hisG Lab collection EAY 310 MATa ura3-52 leu2D1 trp1D63 pms1D::hisG Lab collection EAY 432 MATa ura3-52 leu2D1 trp1D63 rad52D::URA3 Lab collection EAY 545 MATaura3-52 leu2D1 his3D200 rad27D::HIS3 This study EAY 546 MATa ura3-52 leu2D1 trp1D63 rfc1::TN3::LEU2 This study EAY 547 MATaura3-52 leu2D1 his3D200 rfc1::Tn3::LEU2 This study EAY 549 MATaura3-52 leu2D1 his3D200 pol30-52 This study EAY 550 MATaura3-52 leu2D1 trp1D63 pol30-52 This study EAY 552 MATa ura3-52 leu2D1 trp1D63 his3D200 pol30-52 msh2D::hisG This study EAY 554 MATaura3-52 leu2D1 trp1D63 his2D200 rfc1::Tn3::LEU2 msh2D::hisG This study EAY 556 MATa ura3-52 leu2D1 trp1D63 his3D200 rfc1::Tn3::LEU2 pms1D::hisG This study EAY 565 MATa ura3-52 leu2Dtrp1D63 pol3-01 Lab collection NKY 1068 MATaho::LYS2 lys2 ura3 leu2::hisG ade2::LK his4X-ADE2-his4B D. Bishop EAY 561 MATaho::LYS2 lys2 ura3 leu2::hisG ade2::LK his4X-ADE2-his4B This study
rfc1::Tn3::LEU2
(P50.034), was sensitive to UV and the alkylating agent repair mutants, suggesting that these two functions act in series in the repair of replication errors. The PCNA MMS, was cold sensitive for growth at 148, and displayed
a 9.5-fold higher median frequency of mitotic His1re- (POL30) and RAD27 replication factor genes have also been implicated in mismatch avoidance and/or correc-combinants compared to wild type (4.331024for wild
type vs. 4.131023for rfc1::Tn3) in an intrachromosomal tion, as rad27Dand certain pol30 (pol30-52 and pol30-104) mutants display a mutator/slippage phenotype that is gene conversion assay (Table 2;materials and
meth-ods; data not shown;Bishopet al. 1992). The replication similar to that observed in msh2D mutants (Strand et al. 1993;Johnsonet al. 1996;Umaret al. 1996;Kokoska and repair defects that were observed in rfc1::Tn3 strains
were similar to those described for rfc1 conditional mu- et al. 1998). Finally, genetic analyses of RFC1 and the
flap endonuclease RAD27 have revealed that alleles of tants (Howellet al. 1994;McAlearet al. 1996).
The rfc1::Tn3 mutation is synthetically lethal with both are lethal in a rad52 null background and are also synthetically lethal with each other (Table 3;Tishkoff rad52D,rad27D,pol30-52, andpol3-01mutations:Several
lines of genetic evidence have begun to reveal the com- et al. 1997;MerrillandHolm1998). This information, in conjunction with recent data from the Holm labora-plex interplay of replication and repair functions in
both the generation and repair of mutagenic replication tory, where they showed that rfc1 mutants accumulate small single-stranded DNA fragments (Merrill and errors (misincorporation or repeat-tract
insertion/dele-tion events). Studies ofMorrisonet al. (1993) revealed Holm1998), supports the idea that rad27D and rfc al-leles are both defective in the maturation of Okazaki that a polymerasedproofreading mutant (pol3-01)
dis-played multiplicative mutator defects with mismatch- fragments.
Figure 1.—The rfc1::Tn3 allele contains a mini-Tn3::
LACZ::LEU2 (Burnset al. 1994)
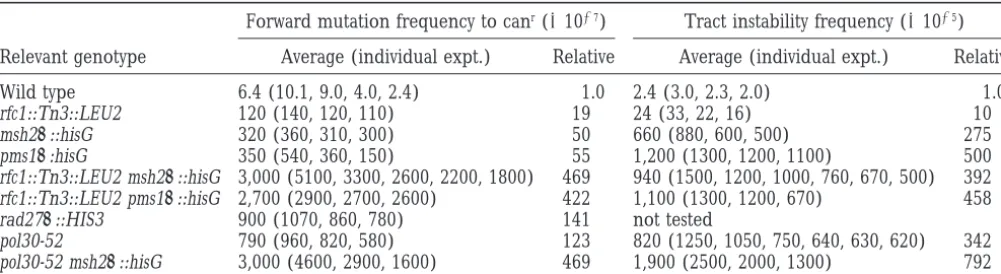
TABLE 2
Median frequency of forward mutations and dinucleotide repeat-tract instability in wild type,rfc1::Tn3, pms1D, msh2D, rad27D,andpol30-52strains
Forward mutation frequency to canr(31027) Tract instability frequency (31025) Relevant genotype Average (individual expt.) Relative Average (individual expt.) Relative
Wild type 6.4 (10.1, 9.0, 4.0, 2.4) 1.0 2.4 (3.0, 2.3, 2.0) 1.0
rfc1::Tn3::LEU2 120 (140, 120, 110) 19 24 (33, 22, 16) 10
msh2D::hisG 320 (360, 310, 300) 50 660 (880, 600, 500) 275
pms1D:hisG 350 (540, 360, 150) 55 1,200 (1300, 1200, 1100) 500
rfc1::Tn3::LEU2 msh2D::hisG 3,000 (5100, 3300, 2600, 2200, 1800) 469 940 (1500, 1200, 1000, 760, 670, 500) 392
rfc1::Tn3::LEU2 pms1D::hisG 2,700 (2900, 2700, 2600) 422 1,100 (1300, 1200, 670) 458
rad27D::HIS3 900 (1070, 860, 780) 141 not tested
pol30-52 790 (960, 820, 580) 123 820 (1250, 1050, 750, 640, 630, 620) 342
pol30-52 msh2D::hisG 3,000 (4600, 2900, 1600) 469 1,900 (2500, 2000, 1300) 792
Wild type (FY23), rfc1::Tn3:: LEU2 (EAY 546), msh2D::hisG (EAY 281), pms1D::hisG (EAY310), rfc1::Tn3::LEU2 msh2D::hisG
(EAY 554), rfc1::Tn3::LEU2 pms1D::hisG (EAY556), rad27D::HIS3 (EAY 545), pol30-52 (EAY 550), and pol30-52 msh2D::hisG (EAY 552)
strains were tested in the canrand 5-FOArassays as described inmaterials and methods. For each row the indicated strain was directly tested in the forward mutation assay and then transformed with pSH44 for testing in the repeat-tract instability assay. In each experiment, 11 independent colonies were tested. The average median frequency in each assay is also presented relative to the wild-type frequency.
The above information encouraged us to explore the mutant strains that were analyzed in the mutator and repeat-tract instability assays described below. Double interplay of rfc1::Tn3 with mutants in these unlinked
replication and repair functions. Haploid strains con- mutant combinations (i.e., crosses 1–5, Table 3) were classified as inviable on the the basis of the following: taining the rfc1::Tn3, rad27D, rad52D, msh2D, pms1D,
pol3-01, and pol30-52 mutations were mated to each (1) The segregation patterns of tetrads bearing four (PD), three (TT), and two (NPD) viable spores fit the other and tetrads from the resulting diploids were
exam-ined for spore viability, segregation of markers, and expected pattern for double mutant lethality in the case where two genes are segregating independently of each mutator and repeat-tract instability phenotypes (Tables
2–4). Double mutant combinations (i.e., crosses 6–9, other (1 PD: 4 TT: 1 NPD). This pattern is also mani-fested in reduced spore viability. (2) The inviability of Table 3) were classified as viable on the basis of the
following: (1) The majority of tetrads dissected con- double mutant combinations was confirmed by genotyp-ing all spore clones in tetrads containgenotyp-ing four and three tained four viable spores (91–99% spore viability). (2)
Genotyping analysis of several four-spore viable tetrads viable spores, and in some cases tetrads that contained two or one viable spores were genotyped. In cases of from each cross resulted in the identification of double
TABLE 3
Tetrad analysis of strains containingrfc1::Tn3::LEU2, pol30-52, pol3-01, rad27D, rad52D, msh2D,andpms1Dmutations
No. of tetrads Spore
viability
Cross Relevant genotype 4:0 3:1 2:2 1:3 0:4 Total (%)
1. EAY 5463EAY 549 rfc1::Tn33pol30-52 6 15 7 0 1 29 71.6 2. EAY 5463EAY 545 rfc1::Tn33rad27D::HIS3 12 23 6 0 0 41 78.2 3. EAY 5473EAY 565 rfc1::Tn33pol3-01 3 19 5 0 0 27 73.1 4. EAY 5493EAY 565 pol30-523pol3-01 3 5 5 3 1 17 58.8 5. EAY 5463EAY 265 rfc1::Tn33msh2D::TRP1 rad52D::URA3 6 36 20 1 3 66 66.0 6. FY 863EAY 432 Wild type3rad52D::URA3 23 1 5 0 0 29 90.5 7. EAY 5473EAY 281 rfc1::Tn33msh2D::hisG 37 1 0 0 0 38 99.3 8. EAY 5473EAY 310 rfc1::Tn33pms1D::hisG 36 2 0 0 0 38 98.7 9. EAY 5493EAY 281 pol30-523msh2D::hisG 16 1 1 0 0 18 95.0
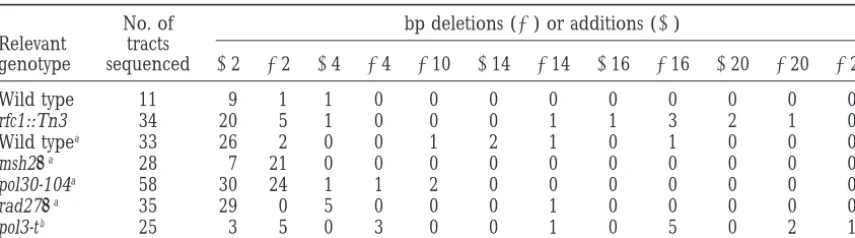
TABLE 4
Distribution of poly(GT) tract alterations in wild type,rfc1:Tn3::LEU2, msh2D, rad27D, pol3-t, andpol30strains
No. of bp deletions (2) or additions (1) Relevant tracts
genotype sequenced 12 22 14 24 210 114 214 116 216 120 220 222
Wild type 11 9 1 1 0 0 0 0 0 0 0 0 0
rfc1::Tn3 34 20 5 1 0 0 0 1 1 3 2 1 0
Wild typea 33 26 2 0 0 1 2 1 0 1 0 0 0
msh2Da 28 7 21 0 0 0 0 0 0 0 0 0 0
pol30-104a 58 30 24 1 1 2 0 0 0 0 0 0 0
rad27Da 35 29 0 5 0 0 0 1 0 0 0 0 0
pol3-tb 25 3 5 0 3 0 0 1 0 5 0 2 1
aData fromJohnsonet al. (1995, 1996).
bData fromKokoskaet al. (1998). In this data set five other alterations were observed in the URA3 coding region.
synthetic lethality, no spore clones were identified that mutations. This observation was also confirmed by show-ing that a msh2Dderivative of DNR53 (relevant genotype contained both mutations.
In control crosses, genotyping and spore viability anal- rfc1::Tn3, msh2D, rad52D, pRAD52 ARS-CEN URA3) was
not viable on 5-FOA media that selected for the loss of ysis demonstrated that rfc1::Tn3 rad52D, rad27Drad52D,
and pol30-52 rad52Ddouble mutants were inviable (data the pRAD52 plasmid (data not shown).
Therfc1::Tn3mutant displays a repeat-tract instability
not shown); this was expected because rad27Drad52D
strains were previously shown to be inviable (Tishkoff phenotype: The phenotype of the rfc1::Tn3 allele, as well as previous studies indicating that mismatch repair
et al. 1997), and different mutant alleles of the RFC1
(rfc1-1; Merrill and Holm 1998) and POL30 (pol30- and DNA replication mutants displayed an increased frequency of repeat-tract instability, encouraged us to
104;MerrillandHolm1998) genes were shown to be
synthetically lethal with rad52 null mutations. pol3-01 test whether the rfc1::Tn3 mutation confers a similar defect (Table 2;Strandet al. 1993;Johnsonet al. 1996; rad52Ddouble mutants were found to be viable (data
not shown); this result was also expected because previ- Kolodner 1996;Umar et al. 1996; Tranet al. 1997).
We measured the frequency of tract instability in an ous analysis indicated that the rad52 null mutation did
not exhibit synthetic lethality with mutations in the assay that detects frameshift events resulting in resis-tance to 5-FOA. These tests were performed in FY23-pola, pold, and polεpolymerase genes (Merrilland
Holm1998). rfc1::Tn3 strains were also mated to strains or FY86-derived strains containing the GT repeat-tract plasmid pSH44 [(GT)16T-URA3, ARSH4, CEN6, TRP1 ] bearing the pol30-52, rad27D, and pol3-01 mutations. As
shown in Table 3, rfc1::Tn3 pol30-52 (Cross 1), rfc1::Tn3 (Hendersonand Petes 1992). As shown in Table 2, the rfc1::Tn3 allele displayed a moderate repeat-tract
rad27D::hisG (Cross 2), and rfc1::Tn3 pol3-01 (Cross 3)
double mutants were inviable because poor spore viabil- instability phenotype (10-fold increased, P50.049) that was lower than that observed in mismatch-repair mu-ity (59–78%) was observed in tetrad analysis and no
spore clones were obtained that contained both muta- tants but was similar to that observed in strains bearing the DNA polymerase mutations pol3-01 and pol3-t (Tran tions.
As shown in Tables 2 and 3, rfc1::Tn3 msh2D::hisG (Cross et al. 1997;Kokoskaet al. 1998).
pSH44-derived plasmids obtained from independent 7) and rfc1::Tn3 pms1D::hisG (Cross 8) double mutants
were viable and displayed the MMSsand coldsphenotype 5-FOAr rfc1::Tn3 colonies were sequenced to examine the repeat-tract sequence changes that had occurred conferred by the rfc1::Tn3 allele and a mutator
pheno-type that was nearly equivalent to the product of the (Table 4). The majority (20/34) of tract alterations in
rfc1::Tn3 strains were 1-repeat insertion mutations. The
mutator frequencies of the individual mutants. The
via-bility of these double mutants encouraged us to test remaining alterations comprised one group (6/34) con-sisting of 1- or 2-repeat insertion/deletion mutations whether, analogous to the suppression of dam2 recA2
lethality by mutS2 mutations, a msh2 mutation could and another group (8/34) consisting of larger 7- to 10-repeat insertion/deletion mutations. This spectrum of suppress the lethality observed in rfc1::Tn3 rad52 double
mutants (McGrawandMarinus1980). In crosses be- tract alterations was somewhat similar to that observed in wild-type and rad27D strains in that the majority of tween a rfc1::Tn3 strain and rad52Dand msh2Drad52D
strains (Table 3, Cross 5; data not shown), no spore tract alterations in all three backgrounds were single repeat insertion mutations. However, compared to the clones were recovered that contained both (rfc1::Tn3
Figure 2.—The rfc1::Tn3 mutation does not result in large insertion/deletion muta-tions in the CAN1 gene. Inde-pendent canrcolonies were ob-tained from rad27D::HIS3
(EAY 545) and rfc1::Tn3 (EAY 547) strains, and a DNA frag-ment containing the CAN1 open reading frame was PCR amplified from each of these strains and incubated with HphI. Restriction enzyme-digested DNA fragments were electrophoresed in a 2% TAE-agarose gel. Lanes marked M contain a DNA marker and lanes 11 and 22 display DNA fragments from wild-type cansstrains. Digestion of the amplified CAN1 DNA with HphI resulted in 490-, 411-, 314-, 252-, 249-, and 207-bp fragments that could be detected by gel electrophoresis. Two smaller bands of 87 and 46 bp could not be detected. Lanes 1–10 and 12–21 display HphI-digested CAN1 DNA from canrrad27D::HIS3 and rfc1::Tn3 strains, respectively.
number of larger tract insertions/deletions (8/34 for Lanes 12–21) or 8 canrpol30-52 strains (data not shown). This finding is also consistent with results ofMcAlear
rfc1::Tn3 vs. 1/35 for rad27D).
The spectrum of repeat-tract insertion/deletion events et al. (1996), which showed that the rfc1-1 mutation
conferred a mutator phenotype that resulted principally in the rfc1::Tn3 strain differed from that observed in
mismatch repair (msh2D) and other DNA replication from an elevated frequency of base pair substitution mutations.
(pol30-104 and pol3-t) defective strains (Table 4 and
Johnson et al. 1995, 1996; Kokoska et al. 1998). In Double mutant analysis indicated a synergistic rela-tionship between rfc1 and mismatch-repair mutations:
msh2D strains only single repeat insertion/deletions
were observed, with the majority of these events con- To test whether RFC1 is required during mismatch re-pair, we examined the mutation frequency of rfc1::Tn3 sisting of deletions (Johnsonet al. 1996). In pol30-104
strains, which display a mutator and repeat-tract instabil- strains in combination with mismatch repair and other replication mutations in both forward mutation and ity phenotype similar to that observed in
mismatch-repair mutants, the vast majority (56/58) of tract al- tract instability assays. As shown in Table 2, in the for-ward mutation assay, the frequency of mutations in terations were one- or two-repeat insertion/deletion
mutations, with a similar number of insertions and dele- rfc1::Tn3 msh2D(469-fold increase) and rfc1::Tn3 pms1D (422-fold increase) double mutant strains appeared to tions (Johnsonet al. 1996). In pol3-t strains, which
con-tain a mutation in polymerasedthat is thought to reduce nearly equal the product of the mutation frequencies of the individual mutations (msh2D, 50-fold increase; the rate of DNA elongation, there was an approximately
equal distribution of small and large repeat insertion/ pms1D, 55-fold increase; rfc1::Tn3, 19-fold increase). While results in the forward mutation assay indicated a deletion mutations and a smaller number of alterations
that were presumably not due to repeat-tract instability nearly multiplicative relationship for defects in the clamp loader and mismatch-repair genes, the results (Kokoskaet al. 1998).
Recent analysis of rad27D strains revealed that, in from the tract instability assay were less clear. As shown in Table 2, frequency of tract instability in msh2D, pms1D, addition to repeat-tract length instability, these strains
also displayed a high frequency of insertion/deletion and pol30-52 strains (275- to 500-fold) was much higher than in rfc1::Tn3 strains (10-fold). The frequencies of events at the CAN1 and LYS2 loci (.14 bp, the majority
of which were duplications) (Johnsonet al. 1995;Tish- tract instability in rfc1::Tn3 msh2Dand rfc1::Tn3 pms1D double mutants, however, were similar to those observed koffet al. 1997;Kokoskaet al. 1998). The similarity in
the spectrum of tract instability events in rfc1::Tn3 and in msh2Dor pms1D strains (rfc1::Tn3 msh2Dvs. msh2D,
P 50.25; rfc1::Tn3 pms1Dvs. pms1D, P50.66).
rad27D mutants and the fact that rfc1::Tn3 rad27D,
rfc1::Tn3 rad52Dand rad27Drad52Dstrains are inviable Double mutant analysis also indicated that rfc1::Tn3
pms1D and rfc1::Tn3 msh2D mutants were viable but (Table 3 andTishkoff et al. 1997) encouraged us to
examine whether similar types of chromosomal re- rfc1::Tn3 pol30-52 double mutants were inviable (Table
3). We were interested in understanding this result be-arrangements could be detected in rfc1::Tn3 strains. We
examined rfc1::Tn3, rad27D::HIS3, and pol30-52 strains cause pol30-52 mutants display a strong mismatch-repair defect that appears to be epistatic to the defect observed for the presence of large insertion/deletion mutations
in the CAN1 locus (Figure 2). As predicted, a difference in msh2 and pms1 mutants (Umaret al. 1996). However,
given the known role of PCNA and RFC in DNA replica-in the size of at least one CAN1-derived fragment was
observed in CAN1 genes amplified from 9 out of 10 tion, this result suggests that pol30-52 strains display defects in cellular functions such as DNA replication canr rad27D strains (Figure 2, lanes 1–10). However,
no changes were observed in the size of CAN1 gene that are in addition to or are different from those involv-ing mismatch repair. Analysis of msh2Dpol30-52 double
mutants in the forward mutation and repeat-tract in- RFC and mismatch-repair functions act in series in a single repair pathway, as proposed for pol3-01 and pms1 stability assays also supported this conclusion. In both
assays, the msh2Dpol30-52 mutant displayed a mutator double mutants in Morrison et al. (1993), or act in
distinct DNA repair pathways that compete for the same and repeat-tract instability phenotype that appeared to
be roughly additive when compared to the mutator phe- substrates (Haynes and Kunz 1981). On the basis of the known function of RFC as a clamp loader in DNA notype observed for each of the single mutations (Table
2). This observation supports the idea that the mutator replication, we favor the idea that defects in the clamp loader result in replication errors that are acted upon phenotype exhibited by pol30-52 and msh2D mutants
reflects the action of gene products functioning in paral- by mismatch repair (acting in series; Morrisonet al.
1993). The types of mutations found in a rfc1-1 strain lel, noncompeting pathways (HaynesandKunz1981;
Morrison et al. 1993). In such a scenario, the pol30- that confers repair and replication defects similar to
52 mutation confers replication errors as the result of those observed in rfc1::Tn3 strains provides further sup-defects in both mismatch repair and DNA replication. port for this idea (McAlear et al. 1996). The rfc1-1
The finding that the pol30-52 mutation, unlike mis- mutation was shown to confer an increase in base pair match-repair mutations, confers sensitivity to DNA-dam- substitutions that are likely to have formed from base aging agents and, like rad27D::HIS3 and rfc1::Tn3, is pair mismatches that are substrates for mismatch repair synthetically lethal with rad52 null mutations, supports (reviewed inKolodner1996).
this idea; moreover, pol30-52 is defective in homotri- A similar relationship between mismatch repair and meric interactions and is also defective in in vitro DNA RFC functions was not observed in the repeat-tract insta-replication reactions (Table 3; Ayyagari et al. 1995). bility assay because the rfc1::Tn3 msh2 or rfc1::Tn3 pms1 Further support is provided from the work ofKokoska double mutants displayed a mutator phenotype that was
et al. (1999, accompanying article) in which they ana- indistinguishable from that observed in msh2 or pms1 lyzed the effect of the pol30-52 mutation on micro- and single mutants. The different phenotypes in these two minisatellite instability and found that, in addition to assays were not unexpected considering that RFC1, conferring a defect in mismatch repair, the pol30-52 MSH2, and PMS1 are likely to be functioning in multi-mutation conferred a minisatellite instability pheno- subunit DNA replication and repair machines and it is type. They hypothesized that this phenotype was the difficult to determine which effects are direct and which result of the pol30-52 mutation affecting DNA replica- are indirect. Three possible explanations for these dif-tion by increasing the rate of DNA polymerase slippage. ferences are as follows:
1. Repeat-tract instability in msh2 and pms1 mutants, as
DISCUSSION measured by the (GT)
14-T-URA3 detection assay, is already occurring at a saturating level and so an in-We identified the rfc1::Tn3 allele in a screen for
muta-crease in these events could not be detected in dou-tions that are lethal in a rad52 null background. This
ble mutant combinations. We believe that this is not analysis indicated the rfc1::Tn3 allele conferred a
muta-the case because higher frequencies of repeat-tract tor phenotype, an elevated recombination frequency,
instabilities have been observed in pol30-52 msh2D sensitivity to UV and MMS, cold sensitivity, and synthetic
strains and even higher frequencies have been re-lethality with rad52, rad27D, and pol30 mutations. The
ported in msh2Dstrains containing mononucleotide phenotypes conferred by the rfc1::Tn3 allele were similar
repeat tracts. (Table 2; Siaet al. 1997a).
to those reported for previously characterized rfc1 alleles
2. The increase in repeat-tract instability in rfc1::Tn3 (Moiret al. 1982;Howell et al. 1994; McAlearet al.
strains resulted not from DNA polymerase slippage 1994). In addition, we showed that rfc1::Tn3 displays
events but from an increase in unequal sister chroma-a repechroma-at-trchroma-act instchroma-ability phenotype. This observchroma-ation
tid exchanges (Strand et al. 1993). The fact that
encouraged us to test genetic interactions between the
the rfc1::Tn3 allele confers a hyper-recombination
rfc1::Tn3 mutation and mutations in other replication
phenotype provides support for this idea. In such a (RAD27, POL3) and repair (MSH2, PMS1) genes.
scenario one would expect the repeat-tract instability In the canavanine mutator assay, a nearly
multiplica-observed in rfc1::Tn3 strains to be dependent on tive effect on mutation frequency was observed when
RAD52 function. Unfortunately, this hypothesis
can-the rfc1::Tn3 mutation was analyzed in combination with
not be tested as rfc1::Tn3 mutants are lethal in rad52
msh2Dand pms1Dmutations. A multiplicative
relation-null backgrounds. ship was previously observed in pol3-01 pms1 double
3. The repeat-tract instability phenotype in rfc1::Tn3 mutants; this observation led Morrison et al. (1993)
strains could result from an increase in DNA slippage to propose that DNA replication errors resulting from
events that are not recognized by, or occur indepen-defects in polymerasedproofreading functions are
re-dently of, the mismatch-repair system. The fact that paired by the mismatch-repair pathway. Because our
z25% of the tract alterations in rfc1::Tn3 strains were data displayed a relationship that was almost, but not
be repaired by the mismatch-repair pathway provides ent from that observed in rad27D and pol3-t mutants where the effect of these mutations on repeat-tract insta-some support for this idea (Siaet al. 1997a), as do
recent observations suggesting that the pol30-52 mu- bility was dependent on the size of the repeat unit (Kokoskaet al. 1998). (2) Larger DNA rearrangements,
tation increases the rate of repeat-tract instability
through mechanisms that appear independent of as analyzed by DNA sequencing or by restriction digest analysis of the CAN1 gene, occurred at high frequency mismatch repair [Table 2;Kokoskaet al. (1999)].
in rad27D strains but were not observed in rfc1::Tn3
Synthetic lethality analysis and chromosome instabil- or pol30-52 strains (Figure 2; Tishkoffet al. 1997). In ity assays indicate that therfc1,pol30, andrad27muta- summary, while each of these mutations appears to de-tions interact genetically but display unique chromo- crease the efficiency of DNA replication in ways that some instability phenotypes: Genetic analysis of rfc1, are likely to stall the DNA replication fork, their overall
pol30, and rad27 mutations indicated that they were all effect on chromosomal instability appears to be unique
synthetically lethal with mutations in the RAD52 recom- for each mutation and is not well understood at this binational repair pathway and that rfc1 pol30 and rfc1 time.
rad27 double mutants were also inviable (Merrilland In this study we observed that the pol3-01 mutation, Holm1998). Additional analysis also showed that mu- which causes a defect in the polymerase-dexonuclease tations in all three of these genes resulted in a muta- proofreading function, was synthetically lethal with the tor phenotype as well as sensitivity to DNA-damaging rfc1::Tn3 mutation (Table 3). Previous analysis by Mor-agents. One hypothesis that is consistent with the above rison et al. (1993) showed that haploid pol3-01 pms1 data is that mutations in each of these genes result in double mutants are inviable because of the accumula-the stalling of accumula-the replication fork, thus leading to accumula-the tion of a catastrophic number of mutations. While the formation of double-strand breaks that are repaired by inviability of pol3-01 rfc1::Tn3 mutants might be ex-the RAD52 double-strand break repair system. This hy- plained in this manner, we believe that this is not the pothesis is partly based on the findings in E. coli that case because msh2, pms1, and pol3-01 single mutants all mutations that disrupt replication fork movement result display similar mutation frequencies and rfc1::Tn3 msh2 in the formation of double-strand breaks that are lethal and rfc1::Tn3 pms1 double mutants are viable (Mor-in recomb(Mor-ination-deficient backgrounds and the obser- rison et al. 1993; Kolodner1996;Umar et al. 1996). vation that rfc1 mutants grow slowly and are delayed A similar conclusion was reached by Kokoska et al. in progressing through S phase (Howell et al. 1994; (1998) to explain why pol3-01 rad27D double mutants
McAlearet al. 1996;Michelet al. 1997). In addition, are inviable. They hypothesized that the inviability of
recent studies by the Holm laboratory (Merrill and pol3-01 rad27Dmutants was not due to mutational load Holm1998) indicated that rad27, pol30, and rfc1 mu- because diploids homozygous for both mutations were tants accumulate small single-stranded DNA fragments also inviable. In cases where mutational load was sus-during DNA replication in vivo, suggesting that they are pected as the cause of haploid inviability, diploids homo-defective in Okazaki fragment maturation. zygous for the mutational load mutations were found to On the basis of the phenotypes of the mutants de- be viable, presumably because recessive lethal mutations scribed above and the observation that Pol30p physically occur less frequently in diploid cells (Morrisonet al. interacts with the RFC and Rad27p, one might have 1993). On the basis of the observation that pol3-01 expected the spectrum of chromosomal instability de- rad27Ddouble mutants are inviable and the fact that the fects in these mutants to be similar (Burgers1991;Li RAD27 gene product is required for Okazaki fragment
et al. 1995;Uhlmannet al. 1997). However, as outlined processing, Kokoska et al. (1998) proposed that the
below, the chromosome instability profiles of the pol30, proofreading exonuclease function of POL3 was
some-rad27, and rfc1 mutants are different. (1) The frequency how required for Okazaki fragment processing. The fact of repeat-tract instability in rfc1::Tn3 strains was much that rfc1, pol30, and rad27Dmutants all display Okazaki lower than was observed in rad27Dor pol30-52 mutants fragment processing defects and rfc1 and rad27D muta-(Table 2;Johnsonet al. 1995, 1996; Umaret al. 1996; tions are inviable in a pol3-01 background is consistent Tishkoffet al. 1997;Kokoskaet al. 1998;Merrilland with this idea (MerrillandHolm1998).
Holm1998) and the spectrum of repeat-tract changes RFC is unlikely to play a direct role in mismatch
differ-strand discrimination candidates. However, double mu- viability but are specifically defective in mismatch repair. It is unlikely, for example, that we would have detected tant analysis of the mismatch-repair mutations msh2 and
pms1 and the rfc1::Tn3 mutation indicated that the RFC mutations in the POL30 gene, the product of which is a strand discrimination candidate, using Tn3 or UV was unlikely to play a role in mismatch repair because
the mutator phenotype of rfc1 was not epistatic to, but mutagenesis screens (i.e., pol30-52) because POL30 is essential for viability and only rare mutations in POL30 was in series with, the mutator phenotypes conferred
by msh2 and pms1 mutations. The repeat-tract instability that were found by targeted mutagenesis display both a strong tract instability and mutator phenotype (Ayya-phenotype conferred by the rfc1::Tn3 mutation was weak
relative to the mutator phenotype as judged by canavan- gariet al. 1995). This information suggests that
fraction-ation of cell extracts that catalyze mismatch repair in ine papillation assays and in comparison with the
repeat-tract instability phenotype observed in msh2Dand pol30- vitro might be a more effective way to identify new
mis-match-repair components.
52 strains (Strand et al. 1993; Umar et al. 1996). A
mutant defective in strand discrimination would be ex- We thank Elizabeth Evans, Todd Milne, Lori Schvaneveldt, Tanya pected to function downstream of mismatch recogni- Sokolsky, Daniel Smith, and Barbara Studamire for providing advice, reagents, and/or technical assistance, Elizabeth Evans, Tom Petes,
tion steps and would therefore be expected to display an
Daniel Smith, Tanya Sokolsky, and Barbara Studamire for helpful
equally strong mutator and tract instability phenotype
discussions and comments on the manuscript, and Tom Petes and
(reviewed in Kolodner1996). In addition, the
spec-Michael Liskay for sharing unpublished data. E.A. and Y.X. were
trum of repeat-tract insertion and deletion events ob- supported by National Institutes of Health grant GM53085 and U.S. served in rfc1::Tn3 strains was different from that ob- Department of Agriculture Hatch Grant NYC-186424.
served in msh2 strains because a large number of tract alterations in rfc1::Tn3 strains involved .14 bp
inser-tion/deletions: such alterations were not observed in LITERATURE CITED
mismatch-repair mutants (Table 4;Strandet al. 1993;
Aguilera, A.,andH. L. Klein,1988 Genetic control of
intrachro-Johnson et al. 1995). Finally, unlike in E. coli where mosomal recombination in Saccharomyces cerevisiae. I. Isolation dam2recA2 double mutant lethality can be suppressed and genetic characterization of hyper-recombination mutations.
Genetics 119: 779–790.
by a mutation in mutS, in the analogous situation in
Au, K. G., K. WelshandP. Modrich,1992 Initiation of
methyl-yeast the msh2D mutation was unable to suppress the directed mismatch repair. J. Biol. Chem. 267: 12142–12148. lethality of rfc1::Tn3 rad52 double mutants (seeresults; Ayyagari, R., K. J. Impellizzeri, B. L. Yoder, S. L. GaryandP. M.
Burgers,1995 A mutational analysis of the yeast proliferating
McGraw and Marinus 1980). This observation
pro-cell nuclear antigen indicates distinct roles in DNA replication
vides further support that the RFC is not playing a strand
and DNA repair. Mol. Cell. Biol. 15: 4420–4429.
discrimination role analogous to that of dam methylase. Bishop, D. K., D. Park, L. XuandN. Kleckner,1992 DMC1: a
meiosis-specific yeast homolog of E. coli recA required for
recom-As described in the Introduction, we performed
an-bination, synaptonemal complex formation, and cell cycle
pro-other screen aimed at identifying new mutations in
mis-gression. Cell 69: 439–456.
match-repair genes. We screened 70,000 cells mutagen- Boeke, J. D., F. LacrouteandG. R. Fink,1984 A positive selection ized with ultraviolet light for those that displayed both for mutants lacking orotidine-59-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197:
a repeat-tract instability and a mutator phenotype. The
345–346.
51 mutants that were identified all contained mutations Burgers, P. M.,1991 Saccharomyces cerevisiae replication factor C. II. in either the MLH1, PMS1, or MSH2 genes. In a subse- Formation and activity of complexes with the proliferating cell nuclear antigen and with DNA polymerases delta and epsilon. J.
quent screen involving 220,000 UV-mutagenized cells
Biol. Chem. 266: 22698–22706.
that was designed to avoid detection of these three Burns, N., B. Grimwade, P. B. Ross-Macdonald, E. Y. Choi, K. genes, only mutations in RAD27 were identified (Y. Xie, Finberg et al., 1994 Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces
cerevis-L. Schvaneveldt and E. Alani, unpublished data).
iae. Genes Dev. 8: 1087–1105.
Why were mutations in genes that are likely to play Counter, C. M., M. Meyerson, E. N. EatonandR. A. Weinberg, a role in mismatch repair (i.e., helicases, single-strand 1997 The catalytic subunit of yeast telomerase. Proc. Natl. Acad.
Sci. USA 94: 9202–9207.
binding proteins, and exonucleases) not identified?
Crouse, G. F.,1996 Mismatch repair systems in Saccharomyces
cerevis-One possibility is that mismatch-repair functions are iae, pp. 411–448 in DNA Damage and Repair: Biochemistry, Genetics overlapping or are redundant because studies in E. coli and Cell Biology, edited byJ. NickoloffandM. Hoekstra.
Hu-mana Press, Clifton, NJ.
have suggested that at least three exonucleases
partici-Cullmann, G., K. Fien, R. B. Kobayashi andB. Stillman,1995
pate in mismatch repair (Harriset al. 1998). Another
Characterization of the five replication factor C genes of
Saccharo-possibility is that any remaining uncharacterized mis- myces cerevisiae. Mol. Cell. Biol. 15: 4661–4671.
Detloff, P., J. SieberandT. D. Petes,1991 Repair of specific base
match-repair factors also play essential roles during
pair mismatches formed during meiotic recombination in the
DNA replication: in such a scenario, a transposon
muta-yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 11: 737–745.
genesis scheme might not allow for the identification Feinstein, S. I.,andK. B. Low,1986 Hyper-recombining recipient
strains in bacterial conjugation. Genetics 113: 13–33.
of an essential mismatch-repair protein such as a
single-Fien, K.,andB. Stillman,1992 Identification of replication factor
strand binding protein or a DNA helicase, while a
ge-C from Saccharomyces cerevisiae : a component of the leading-strand
nome-wide UV mutagenesis might not have been effi- DNA replication complex. Mol. Cell. Biol. 12: 155–163. Geitz, R. D.,andR. H. Schiestl,1991 Applications of high
ciency lithium acetate transformation of intact yeast cells using late small single-stranded DNA fragments during DNA synthesis. Genetics 148: 611–624.
single-stranded nucleic acids as carrier. Yeast 7: 253–263.
Green, C. N.,andS. Jinks-Robertson,1997 Frameshift intermedi- Michaels, M. L., C. CruzandJ. H. Miller,1990 mutA and mutC:
two mutator loci in Escherichia coli that stimulate transversions. ates in homopolymer runs are removed efficiently by yeast
mis-match repair proteins. Mol. Cell. Biol. 17: 2844–2850. Proc. Natl. Acad. Sci. USA 87: 9211–9215.
Michel, B., S. D. ErlichandM. Uzest,1997 DNA double-strand
Harris, R. S., K. J. Ross, M. J. LombardoandS. M. Rosenberg,
1998 Mismatch repair in Escherichia coli cells lacking single- breaks caused by replication arrest. EMBO J. 16: 430–438.
Miller, J.,1972 Experiments in Molecular Genetics. Cold Spring Harbor
strand exonucleases ExoI, ExoVII, and RecJ. J. Bacteriol. 180:
989–993. Laboratory Press, Cold Spring Harbor, NY.
Modrich, P.,andR. S. Lahue,1996 Mismatch repair in replication
Haynes, R. H.,andB. A. Kunz,1981 DNA repair and mutagenesis,
pp. 371–414 in The Molecular and Cellular Biology of the Yeast Sacchar- fidelity, genetic recombination and cancer biology. Annu. Rev. Biochem. 65: 101–133.
omyces: Life Cycle and Inheritance, edited byJ. N. Strathern, E. W.
JonesandJ. R. Broach.Cold Spring Harbor Laboratories, Cold Moir, D., S. E. Stewart, B. C. OsmondandD. Botstein,1982 Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties Spring Harbor, NY.
Henderson, S. T.,andT. D. Petes,1992 Instability of simple se- and pseudo-revertant studies. Genetics 100: 547–563.
Morrison, A., A. L. Johnson, L. H. JohnstonandA. Sugino,1993 quence DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 12: 2749–
2757. Pathway correcting DNA replication errors in Saccharomyces
cerevis-iae. EMBO J. 12: 1467–1473. Holm, C., D. W. Meeks-Wagner, W. L. FangmanandD. Botstein,
1986 A rapid, efficient method for isolating DNA from yeast. Pfaffenberger, R. C.,andJ. H. Patterson,1977 Statistical Methods for Business and Economics. Richard D. Irwin Inc., Homewood, IL.
Gene 42: 169–173.
Howell, E. A., M. A. McAlear, D. RoseandC. Holm,1994 CDC44: Reenan, R. A. G.,andR. D. Kolodner,1992 Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and a putative nucleotide-binding protein required for cell cycle
pro-gression that has homology to subunits of replication factor C. MSH2 genes: evidence for separate mitochondrial and nuclear
functions. Genetics 132: 975–985. Mol. Cell. Biol. 14: 255–267.
Jeyaprakash, A., J. W. WelchandS. Fogel,1994 Mutagenesis of Rose, M. D., F. WinstonandP. Hieter,1990 Methods in Yeast Genet-ics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
yeast MW104-1B strain has identified the uncharacterized PMS6
DNA mismatch repair gene locus and additional alleles of existing NY.
Saiki, R. K., S. Scharf, F. Faloona, K. B. Mullis, G. T. Hornet al., PMS1, PMS2 and MSH2 genes. Mutat. Res. 325: 21–29.
Johnson, R. E., G. K. Kovvali, L. PrakashandS. Prakash,1995 1985 Enzymatic amplification of beta-globin genomic se-quences and restriction site analysis for diagnosis of sickle cell Requirement of the yeast RTH1 59to 39 exonuclease for the
stability of simple repetitive DNA. Science 269: 238–240. anemia. Science 230: 1350–1354.
Schaaper, R. M., andR. L. Dunn,1987 Spectra of spontaneous
Johnson, R. E., G. K. Kovvali, S. N. Guzder, N. S. Amin, C. Holm
et al., 1996 Evidence for involvement of yeast proliferating cell mutations in Escherichia coli strains defective in mismatch correc-tion: the nature of in vivo DNA replication errors. Proc. Natl. nuclear antigen in DNA mismatch repair. J. Biol. Chem. 271:
27987–27990. Acad. Sci. USA 84: 6220–6224.
Sia, E. A., R. J. Kokoska, M. Dominska, P. GreenwellandT. D. Kane, S. M.,andR. Roth,1974 Carbohydrate metabolism during
ascospore development in yeast. J. Bacteriol. 118: 8–14. Petes,1997a Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol.
Kokoska, R. J., L. Stefanovic, H. T. Tran, M. A. Resnick, D. A.
Gordeninet al., 1998 Destabilization of yeast micro- and minisa- 17:2851–2858.
Sia, E. A., S. Jinks-Robertson andT. D. Petes, 1997b Genetic tellite DNA sequences by mutations affecting a nuclease involved
in Okazaki fragment processing (rad27) and DNA polymerased control of microsatellite stability. Mutat. Res. 383: 61–70.
Stillman, B., 1994 Smart machines at the DNA replication fork. (pol3-t). Mol. Cell. Biol. 18: 2779–2788.
Kokoska, R. J., L. Stefanovic, A. B. Buermeyer, R. M. Liskayand Cell 78: 725–728.
Strand, M., T. A. Prolla, R. M. LiskayandT. D. Petes,1993
Desta-T. D. Petes,1999 A mutation of the yeast gene encoding PCNA
(pol30-52) destabilizes both microsatellite and minisatellite DNA bilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365: 274–276. sequences. Genetics 151: 511–519
Kolodner, R.,1996 Biochemistry and genetics of eukaryotic mis- Tishkoff, D. X., N. Filosi, G. M. GaidaandR. D. Kolodner,1997 A novel mutation avoidance mechanism dependent on S. cerevisiae match repair. Genes Dev. 10: 1433–1442.
Levinson, G.,andG. A. Gutman,1987 High frequencies of short RAD27 is distinct from DNA mismatch repair. Cell 88: 253–263. Tran, H. T., D. A. GordeninandM. A. Resnick,1996 The preven-frameshifts in poly-CA/TG tandem repeats borne by
bacterio-phage M13 in Escherichia coli K-12. Nucleic Acids Res. 15: 5313– tion of repeat-associated deletions in Saccharomyces cerevisiae by mismatch repair depends on size and origin of deletions. Genetics 5338.
Li, X., J. Li, J. Harrington, M. R. LieberandP. M. Burgers,1995 143:1579–1587.
Tran, H. T., J. D. Keen, M. Kricker, M. A. ResnickandD. A.
Gorde-Lagging strand DNA synthesis at the eukaryotic replication fork
involves binding and stimulation of FEN-1 by proliferating cell nin, 1997 Hypermutability of homonucleotide runs in mis-nuclear antigen. J. Biol. Chem. 270: 22109–22112. match repair and DNA polymerase proofreading yeast mutants.
Liu, B., N. C. Nicolaides, S. Markowitz, J. K. Willson, R. E. Par- Mol. Cell. Biol. 17: 2859–2865.
sonset al., 1995 Mismatch repair gene defects in sporadic colo- Tsurimoto, T., andB. Stillman, 1989 Purification of a cellular rectal cancers with microsatellite instability. Nat. Genet. 9: 48–55. replication factor, RF-C, that is required for coordinated synthesis
Maniatis, T., E. F. FritschandJ. Sambrook,1982 Molecular Clon- of leading and lagging strands during simian virus 40 DNA
replica-ing: A Laboratory Manual. Cold Spring Harbor Laboratory Press, tion in vitro. Mol. Cell. Biol. 9: 609–619.
Cold Spring Harbor, NY. Uhlmann, F., J. Cai, E. Gibbs, M. O’DonnellandJ. Hurwitz,1997
McAlear, M. A., E. A. Howell, K. K. EspenshadeandC. Holm,1994 Deletion analysis of the large subunit p140 in human replication Proliferating cell nuclear antigen (pol30) mutations suppress rfc1 factor C reveals regions required for complex formation and mutations and identify potential regions of interaction between replication activities. J. Biol. Chem. 272: 10058–10064. the two encoded proteins. Mol. Cell. Biol. 14: 4390–4397. Umar, A., A. B. Buermeyer, J. A. Simon, D. C. Thomas, A. B. Clark McAlear, M. A., K. M. TuffoandC. Holm,1996 The large subunit et al., 1996 Requirement for PCNA in DNA mismatch repair at
of replication factor C (Rfc1p/Cdc44p) is required for DNA a step preceding DNA resynthesis. Cell 87: 65–73.
replication and DNA repair in Saccharomyces cerevisiae. Genetics Umar, A., J. I. Risinger, W. E. Glaab, K. R. Tindall, J. C. Barrett
142:65–78. et al., 1998 Functional overlap in mismatch repair by human
McGraw, B. R.,andM. G. Marinus,1980 Isolation and characteriza- MSH3 and MSH6. Genetics 148: 1637–1646.
tion of Dam1revertants and suppressor mutations that modify Winston, F., C. DollardandS. L. Ricupero-Hovasse,1995 Con-secondary phenotypes of dam-3 strains of Escherichia coli K-12. struction of set of convenient Saccharomyces cerevisiae strains that Mol. Gen. Genet. 178: 309–315. are isogenic to S288C. Yeast 11: 53–55.
Merrill, B. J.,and C. Holm,1998 The RAD52 recombinational