ABSTRACT
RYAN, JOSEPH ANTHONY. Quantification via Inductively-Coupled Plasma Optical Emission Spectroscopy (ICP-OES) of the Cellular Internalization and Nuclear Localization of Gold Nanoparticles Passivated with BSA-SV40 Large T NLS Conjugates after Incubation with Human Cervical Cancer (HeLa) Cells. (Under the direction of Dr. Stefan Franzen).
Quantification via Inductively-Coupled Plasma Optical Emission Spectroscopy (ICP-OES) of the Cellular Internalization and Nuclear Localization of Gold Nanoparticles
Passivated with BSA-SV40 Large T NLS Conjugates after Incubation with Human Cervical Cancer (HeLa) Cells
by
Joseph Anthony Ryan
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
Chemistry
Raleigh, North Carolina 2009
APPROVED BY:
________________________________ ______________________________ Dr. Stefan Franzen Dr. Edmond Bowden
BIOGRAPHY
TABLE OF CONTENTS
List of Tables ... ix
List of Figures ... xi
Chapter 1: Introduction ... 1
1.1. Introduction and Background. ... 2
1.2. Cancer. ... 2
1.2.1. Current Cancer Diagnostics. ... 3
1.2.2. Current Cancer Treatments. ... 4
1.3. Brief Review of Relevant Cellular Biology. ... 6
1.3.1. Eukaryotic Cell Structure. ... 6
1.3.2. Cell Membrane. ... 8
1.3.3. Cellular Internalization Mechanisms. ... 9
1.3.4. Nuclear Localization. ... 11
1.3.5. General Cellular Internalization Considerations. ... 11
1.4. Nanotechnology. ... 13
1.4.1. Colloidal Gold... 15
1.4.1.1. Synthesis of Gold Nanoparticles... 16
1.4.1.2. Stability of Gold Nanoparticles. ... 17
1.5. Existing Intracellular Delivery Vehicles. ... 18
1.5.2. Non-Viral Vectors. ... 20
1.5.3. Viral/Non-Viral Combination Vectors. ... 20
1.5.3.1. SV40 Large T NLS. ... 21
1.6. Summary. ... 22
1.7. References. ... 24
Chapter 2: Synthesis and Characterization of Large T-SMCC-BSA/Nanoparticle Complexes. ... 32
2.1. Introduction. ... 33
2.2. Results and Discussion. ... 36
2.2.1. Large T-SMCC-BSA Synthesis. ... 36
2.2.1.1. SMCC-BSA Conjugation. ... 36
2.2.1.2. Quantification of Large T Peptides per BSA-SMCC. ... 40
2.2.2. Passivation of Gold Nanoparticles with Large T/BSA Conjugates. ... 43
2.2.2.1. Quantification of Large T/BSA Conjugates per Nanoparticle. ... 43
2.2.2.2. Critical Coagulation Concentration (CCC) Tests. ... 46
2.2.2.2.1. Varying BSA:Nanoparticle Ratio. ... 48
2.2.2.2.2. Varying Large T/BSA:Nanoparticle Ratio. ... 50
2.2.2.2.3. Varying Large T:BSA Ratio. ... 51
2.2.2.2.4. Varying Temperature. ... 52
2.3. Conclusions. ... 53
2.4.1. Materials. ... 54
2.4.2. Methods. ... 55
2.4.2.1. SMCC-BSA Conjugation. ... 55
2.4.2.2. Quantification of Large T Peptides per BSA/SMCC. ... 58
2.4.2.3. Quantification of Large T/BSA Conjugates per Nanoparticle. ... 60
2.4.2.3.1. Preparation of BSA-MBS-[Ru(bipy)2bipy-C6H12-SH]2+ Conjugates. ... 60
2.4.2.3.2. Time-Resolved Emission Methods. ... 62
2.4.2.4. Critical Coagulation Concentration (CCC) Tests. ... 63
2.4.2.4.1. Varying BSA:Nanoparticle Ratio. ... 63
2.4.2.4.2. Varying Large T/BSA:Nanoparticle Ratio. ... 64
2.4.2.4.3. Varying Large T:BSA Ratio. ... 65
2.4.2.4.4. The Effect of Temperature Variation. ... 66
2.5. References. ... 68
Chapter 3. Incubations of Large T-SMCC-BSA/Nanoparticle Complexes with Human Cervical Cancer Cells. ... 70
3.1. Introduction. ... 71
3.1.1. The HeLa Cell Line. ... 71
3.1.2. Discussion of ICP-OES Method Development. ... 72
3.1.3. General Cellular Incubation Considerations. ... 73
3.2.1. ICP-OES Calibration Experiments. ... 73
3.2.2. Probing Optimal Time of Incubation. ... 78
3.2.3. Probing Optimal Number of Large T Peptides per Nanoparticle Complex and the Effect of Temperature on Uptake Efficiency. ... 80
3.2.4. Probing Effect of Particle Size and Excess Passivating Agent on Uptake Efficiency. ... 82
3.2.5. Pulse-Chase Experiments. ... 86
3.3. Conclusions. ... 87
3.4. Experimental. ... 88
3.4.1. Materials. ... 88
3.4.2. Methods. ... 89
3.4.2.1. Without Cells (ICP-OES Calibration Experiments). ... 89
3.4.2.2. Probing Optimal Time of Incubation. ... 92
3.4.2.3. Probing Optimal Number of Large T Peptides per Nanoparticle Complex and the Effect of Temperature on Uptake Efficiency. ... 93
3.4.2.4. Probing Effect of Particle Size and Excess Passivating Agent on Uptake Efficiency. ... 94
3.4.2.5. Pulse-Chase Experiments. ... 96
3.5. References. ... 97
4.1. Introduction. ... 99
4.1.1. Development of a Method for Nuclear Extraction from HeLa. ... 99
4.2. Results and Discussion. ... 101
4.2.1. Probing the Efficacy of Nuclear Association of Large T. ... 101
4.2.2. Examination of Centrifugation Effects on Resulting ICP-OES Signal. .. 102
4.2.3. Examination of Toxicity of Constructs. ... 104
4.3. Conclusions. ... 106
4.4. Experimental. ... 107
4.4.1. Materials. ... 107
4.4.2. Methods. ... 109
4.5. References. ... 111
Chapter 5: Conclusions. ... 112
5.1. Summary. ... 113
5.2. Questions for Further Study. ... 114
5.2.1. Regarding Construction and Quantification of Large T-SMCC- BSA/Gold Nanoparticle Complexes. ... 114
5.2.2. Regarding Delivery of Large T-SMCC-BSA/Gold Nanoparticle Complexes to Cells. ... 116
5.2.3. Regarding Sub-Cellular Fractionation of Cells Incubated with Large T- SMCC-BSA/Gold Nanoparticle Complexes. ... 118
LIST OF TABLES
Table 2.1. Chart showing solubility of BSA in solutions of varying percentages of three different organic solvents at 25 oC. ... 38 Table 2.2. General evaluation of SMCC binding to BSA. ... 39 Table 2.3. Number of SMCC molecules conjugated with BSA for samples to be used
in HeLa delivery experiments. ... 40 Table 2.4. Concentration of active cysteine residues determined via Ellman’s reagent
assay prior to conjugation to BSA:SMCC. ... 42 Table 2.5. Lifetimes and calculated populations. ... 45 Table 2.6. Large T peptides per BSA and per nanoparticle complex. ... 46 Table 2.7. CCC values of 15 nm diameter citrate- passivated gold nanoparticles after
mixing with varying amounts of BSA. ... 49 Table 2.8. CCC values of 15 nm diameter citrate-passivated gold nanoparticles after
mixing with varying amounts of large T-BSA conjugates. ... 51 Table 2.9. CCC values of 15 nm diameter citrate- passivated gold nanoparticles after
mixing with large T-BSA conjugates of varying large T concentration. .... 52 Table 2.10. CCC values of 15 nm diameter large T-BSA-passivated gold nanoparticles
determined after conjugation at room temperature (approximately 25 oC), then storage at varying temperatures for 12 hours. ... 53 Table 3.1. ICP-OES intensities and calculated number of citrate-stabilized 20 nm
diameter nanoparticles for standards prepared with and without acid
digestion. ... 73 Table 3.2. ICP-OES intensities of 15 nm diameter nanoparticle complexes constructed
with varying passivating layers and incubated on glass coverslips for
Table 3.3. ICP-OES intensities of HeLa cell samples incubated for 6 h at 37 °C and in 5 % CO2 with and without 15 nm-diameter gold nanoparticle complexes constructed using a 15:1 Large T/BSA (experimentally-determined ratio) conjugate. ... 75 Table 4.1. The number of 5 nm gold nanoparticle complexes (of varying large T
concentration) per HeLa cell detected via ICP-OES, including data from fractionation experiments. ... 101 Table 4.2. The percentages of 5 nm gold nanoparticle complexes detected via
LIST OF FIGURES
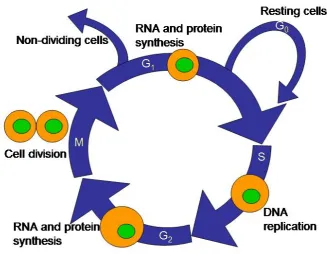
Figure 1.1. Drawing of eukaryotic cell cycle. ... 8 Figure 1.2. General intracellular delivery vehicle considerations. ... 12 Figure 2.1. Cartoon representing the general construction scheme of large
T/BSA-nanoparticle complexes. ... 33 Figure 2.2. Structures of m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS) and succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate
(SMCC). ... 35 Figure 2.3. Molar ratio of large T peptides bound per BSA versus molar ratio of large T
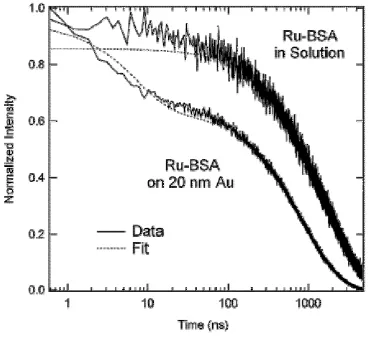
peptides initially added to SMCC-BSA samples. ... 42 Figure 2.4. Normalized intensity versus time for solutions of [Ru(bipy)2bipy-C6H12-S]2+
-BSA in solution and [Ru(bipy)2bipy-C6H12-S]2+-BSA + 20 nm diameter
gold colloids. ... 44 Figure 2.5. Potential energy versus nuclear separation; general description of DLVO
theory. ... 47 Figure 2.6. Fluorescamine structure before and after conjugation with primary
amines. ... 55 Figure 2.7. Molecular structures of cysteine and lysine. ... 56
Figure 2.8. Structure of DTNB (Ellman’s Reagent) and general mechanism. ... 59 Figure 3.1. Average number of 15 nm diameter gold nanoparticle complexes
(determined via ICP-OES analysis) per HeLa cell (determined via
cell-counting) vs incubation time. ... 78 Figure 3.2. Plot of the average number of 15 nm diameter nanoparticle complexes
per well as determined via ICP-OES analysis vs number of large T
peptides per gold nanoparticle complex. ... 80 Figure 3.3. Results of ICP-OES analyses of 6 h HeLa incubations with large
Figure 3.4. Plot of the average number of 15 nm diameter nanoparticle complexes per well as determined via ICP-OES analysis vs number of BSA (diamonds) or large T-BSA (squares) per 15 nm diameter gold
nanoparticle complex. ... 83 Figure 3.5. Plot of the average number of 15 nm diameter gold nanoparticle complexes
per well as determined via ICP-OES analysis vs chase time. ... 86 Figure 4.1. Percent viability (as determined via FACS; compared to cell samples
with no nanoparticle complexes delivered) versus large T per 5 nm
Chapter 1
1.1. Introduction and Background.
The main focus of this dissertation is the development of a gold nanoparticle-based
cellular delivery vehicle which targets the nuclei of cancerous cells, although it should be
noted that the technology described herein can also be used for other applications in drug
delivery and contrast agents. The description starts in this Chapter with a brief overview of
current cancer research followed by a discussion of existing delivery vectors. This will
include a brief review of the inherent cellular processes, of which these vectors are designed
to take advantage. The full details of the experiments, used to validate this specific delivery
vector and to test the efficacy of nuclear targeting, will be delineated in Chapters 2 – 4.
Finally, the last chapter of this dissertation is a summary of all the results from this research
and a discussion of future experiments which could be performed in the continuing of this
line of scientific inquiry.
1.2. Cancer.
Cancer research is one of the most prolific, intriguing, and vitally important areas of
contemporary scientific investigation. Research topics include a study of potential causes of
cancer,1,2,3,4,5 elucidation of new mechanisms by which cancer develops and is sustained,6,7,8,9,10 and, ultimately, the search for innovative techniques to detect and
selectively destroy it.11,12,13,14,15,16 Despite a massive effort in funding and focus of research regarding cancer in the United States for nearly forty years – since then-President Nixon
However, while scientific inquiries may not have yielded a proper cure yet, recent
endeavors have not only revealed some of the mechanisms involved in how certain cells
become cancerous, but also illuminated how much more complex and comprehensive the
disease can be compared to what was initially thought.18,19 For example, recent research20,21 has begun to define the dynamic chemical interplay between certain malignant tumors and
their host’s immune system and has also highlighted the limits of our understanding of
immune reactions in general.
1.2.1. Current Cancer Diagnostics.
Whether one is examining an X-ray slide for abnormally dense regions,22 analyzing
blood test results for biochemical byproducts of potential cancerous cells,23 interpreting anomalous computed tomographic (CT) scan data,24 physically examining suspect tissue during an endoscopic procedure,25 or even sifting through data from post-biopsy cytogenetic26 or immunohistochemistry analyses27 of potentially cancerous cells – the sooner
it is determined that some cells have become cancerous, the greater the chances of a patient’s
survival. The utility of novel biological probes capable of detecting small
changes/accumulations in cancerous (or pre-cancerous) conditions in general can not be
understated, and innovative probes useful in harmless, non-toxic cellular analysis would be
1.2.2. Current Cancer Treatments.
The most common methods of treating cancer patients typically entail surgery – when
practical – to remove as much of a cancerous mass from a patient as possible,28 combined with subsequent administration of one or more of the following: chemotherapy,29 radiation therapy,30 hormonal therapy,31 and immunotherapy.32 Additionally, photodynamic therapy has been shown to be quite effective at successfully treating certain types of skin cancers
without involving surgery.33
Some chemotherapeutic agents, such as cisplatin (cis
-diamminedichlorido-platinum(II), a square planar molecule), help kill cancer cells by associating with the
hereditary material – deoxyribonucleic acid (DNA) – in a cell and cross-linking DNA
strands, thereby interfering with DNA replication during cell division and ultimately
inducing apoptosis (programmed cell death).34 Other chemotherapeutic agents interfere with different facets of the mitotic process in a cell: for example, paclitaxel (a tetracyclic molecule
with a heptadecane skeleton) binds to tubulin, the “building block” molecule of a cell’s
cytoskeleton, thus preventing the cytoskeleton from disassembling as/when necessary to aid
in cell division.35 (Moreover, paclitaxel also aids in inducing apoptosis in cancer cells by binding to an apoptosis-stopping protein named Bcl2 (B-cell leukemia 2) present in many
cancer cells, subsequently allowing a cell’s intrinsic “kill switch” to function.)36 The
administration of both of these particular chemotherapeutic molecules (and many more like
them) would benefit greatly from the development of a more efficacious method of delivery
to their site of intended activity, which could lessen the negative impact upon otherwise
Radiotherapy is the use of ionizing radiation – potentially a beam of photons,30 electrons,37 protons,38 neutrons,39 ions40, or some combination41 – to damage the DNA of
cancerous cells. The DNA of the cells might be damaged directly by the beam, or indirectly
attacked by resulting free radicals of other nearby molecules (e.g.: water, O2) ionized by the
beam. However, certain larger tumors can develop a general resistance to radiotherapy when
these tumors outgrow their blood supply, thus developing a hypoxic environment (less O2 in
the cell translates to less DNA damage via beam). In these instances, one option is to use a
dose of a “radiosensitizing” agent42 – such as cisplatin43 – prior to beginning radiotherapy. Once again, any novel method of specifically targeting/transporting these agents to their sites
of cellular activity could limit their exposure to uptake by healthy cells surrounding
cancerous tissue, potentially lower the necessary doses of both radiotherapy and the
transported chemotherapeutic, and directly aid in the destruction of the cancerous cells.
Hormone therapy as a cancer treatment is an attempt to restrict the growth of certain
cancerous cells by either administering hormones which cause cells to actively undergo
apoptosis44 or removing a source of hormones which particular cancer cells (e.g. some breast45 or prostate46 tumors) need to continue their otherwise unregulated growth. Two drugs of this latter type are tamoxifen,47 ((Z)-2-[4-(1,2-diphenylbut-1-enyl)phenoxy]-N,N
-dimethyl-ethanamine) which competitively inhibits the binding of the hormone estrogen to
breast cancer cells, and letrozole,48 (4-[(4-cyanophenyl)-(1,2,4-triazol-1-yl)methyl]benzonitrile) which blocks the production of estrogen by binding to the heme of
the cytochrome P450 subunit of the aromatase enzyme responsible for producing estrogen.
attack cancerous cells, either through immunization against certain antigens commonly
expressed (or overexpressed) by cancerous cells,49 or by administering a therapeutic antibody
to actively promote an immune response in a given area of the body.50 Treating cancer through hormonal therapy or immunotherapy could be made more beneficial through the use
of any novel delivery vector which could aid in transporting the respective therapeutic
moieties to their cellular sites of consequence.
1.3. Brief Review of Relevant Cellular Biology. 1.3.1. Eukaryotic Cell Structure.51
All living organisms are comprised of cells, the simplest unit capable of independent
existence. Organisms can be classified into two fundamentally different types according to
the structure and complexity of their cells. Prokaryotes – kingdom Monera (bacteria and
cyanobacteria) – are single-celled organisms which have a plasma membrane confining the
contents of that cell to a discrete compartment, but lack distinct internal membrane systems
(organelles). The DNA in prokaryotic cells is generally confined to one or more nuclear
regions (but these regions are not bounded by a separate membrane), and some prokaryotic
cells have a cell wall or outer membrane enveloping the entire cell, including their plasma
membrane. Eukaryotic cells contain a membrane-bounded structure called a nucleus, which
is where the cell’s DNA is localized. Additionally, these cells have many other organelles
bounded by distinct membranes, and the structure and function of these organelles can vary
When growing cells reach a certain size, they either stop growing or divide. The life
cycle of cells – also referred to as the cell cycle – is an attempt to describe the general
activities of cells which are actively growing and dividing. This cycle is defined as the
period from the start of one division to the onset of the next, and a typical cell cycle diagram
is shown in Figure 1.1. The major defining parts of the cell cycle are the S phase (“S” for
“synthesis”) when DNA is replicated in preparation for division, and the actual cell division
itself – termed “mitosis,” the M phase. Separating these two events are “gap” phases – G1
and G2 – when the cell is manufacturing the mRNAs and proteins necessary to enter the next
phase. Cells which do not divide – i.e. mature red blood cells or neurons – are considered
perpetually in the G1 phase, and are therefore not engaged in the cell cycle. During the G1, S,
and G2 phases (collectively termed “interphase”), a cell will roughly double its mass, and the
end result of the M phase is two cells, which are typically (but not always) identical to each
other. Lastly, under certain conditions (i.e. starvation), a cell may exit the cell cycle and
Figure 1.1. Drawing ofeukaryotic cell cycle.
1.3.2. Cell Membrane.51
Cell membranes are phospholipid bilayers which form an external boundary around a
cell. The aqueous environment both inside and outside a cell helps form and maintain the
bilayer structure. Diverse proteins are embedded through the cell membrane (Figure 1.4),
and these proteins can function as mediators by which a cell can actively and selectively
internalize various moieties. It should be noted that while it is energetically unfavorable for
the phospholipids comprising the membrane to escape the bilayer formation, nothing
prevents individual molecules from moving laterally within the plane of the bilayer. The
membrane (including any unanchored trans-membrane proteins), in essence, behaves like a
1.3.3. Cellular Internalization Mechanisms.51
Since the topic of cellular internalization (that is, the ability/process of a cell to take
in some external species) is a quite extensive subject area, the main methods by which a cell
actively internalizes external moieties will be mentioned here, and only one will be examined
in more detail. It should be noted passive diffusion across a eukaryotic cell membrane is
possible for very small, uncharged molecules (e.g. water, O2), but ions and other larger
molecules must be actively transported across the membrane via a protein channel of some
form. Thus, cellular membranes are semi-permeable.
Phagocytosis is the process by which a cell engulfs a relatively large particle (e.g. cell
debris or some microorganism) and ingests these particles in a vesicle called a phagosome (>
250 nm in diameter). Phagocytosis is a specialized function of macrophages and certain
white blood cells. Most eukaryotic cells ingest fluid and small molecules by means of
relatively small vesicles (< 150 nm in diameter) in a process called pinocytosis. Primarily
using a process which involves the proteins clathrin, adaptin, and dynamin, a small portion of
the cell membrane is invaginated and pinched off into the cytoplasm, becoming a discrete
vesicle. These vesicles are then fused with other vesicles in the cell – endosomes and
lysosomes – in order to digest the internalized substance, while the vesicle proteins are then
recycled.
While pinocytosis can be a non-specific process, merely capturing any molecules
which happen to be in the extracellular environment near a burgeoning clathrin-coated
macromolecules. A complementary receptor on the surface of the cell may bind to a given
macromolecule and then be internalized by the cell via a clathrin-coated vesicle. This process
is called receptor-mediated endocytosis (RME), and is the method of cellular internalization
of macromolecules important to the homeostasis of cells, such as cholesterol, vitamin B12,
and iron, to name a few. However, RME can also be used by viruses attempting to infect
cells.52
Another way to look at the RME process is to consider the various forces at work at
the interface between a particle potentially able to be internalized by a cell via RME
(meaning the surface of the particle contains some concentration of a ligand complimentary
to a receptor on a cell membrane) and the outer side of a cell membrane.53 Forces which aid
in the internalization process could be: 1) forming new bonds between ligands on the particle
and receptors on the cell surface (driven by a reduction in the overall free energy of the
system); and 2) potential non-specific attractions between the cell surface and the particle.
Forces which hinder the internalization process could be: 1) the cell membrane bending to
surround the particle (including thermal fluctuations in the cell membrane potentially
opposed to such deformation); 2) the ability of receptor molecules to diffuse laterally along
the membrane to get to the site of particle-membrane interaction; 3) the degree to which a
ligand-receptor bond is able to stretch as a cell membrane is drawn around a particle; and 4)
potential non-specific repulsive forces between the cell surface and the particle. Though
there are many factors which would affect the relative contribution of one force over another
particle surface – as long as the attractive forces are greater than the repulsive forces, then the
particle will become enveloped by the cell membrane and internalized.
1.3.4. Nuclear Localization.51
The DNA in a eukaryotic cell is bounded by two concentric membranes referred to as
the nuclear membrane. This membrane is contiguous with the endoplasmic reticulum (ER),
and is perforated by nuclear pores. Each pore contains an intricate protein mechanism – a
nuclear pore complex – which restricts and regulates the flow of large molecules (i.e. RNAs,
proteins), while allowing for the free, non-specific passing of small water-soluble molecules
(i.e. ions, small metabolites) to and from the nucleus.
Any protein which is to be taken into a nucleus will necessarily contain a particular
amino acid sequence – a nuclear localization sequence – which will initiate a cascade of
events involving the binding of this protein with one or more transport molecules, which then
facilitate transport through the nuclear pore complex.
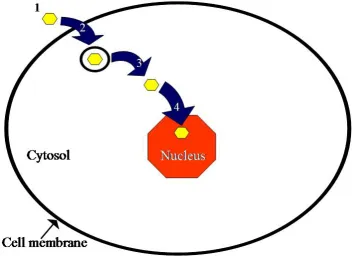
1.3.5. General Cellular Internalization Considerations.
In summary, any potential cellular internalization vector or drug delivery vehicle
intending to transport a molecular cargo of some sort to a cell’s nucleus (for example) will
Figure 1.2. General intracellular delivery vehicle considerations. The four major
considerations enumerated here are detailed in the text.
First, a potential vector must be both stable with regard to the in vitro or in vivo salt
concentrations surrounding the cells of interest, and with equal regard to the ability to protect
that vector’s “payload” from premature digestion or from some otherwise inactivating
process. Moreover, consideration should be given to the degree of toxicity the presence of
this potential vector may cause. Additionally, when working in vivo, the vector must be
capable of extended circulation and not subject to phagocytosis by other cells. Second, a
potential intracellular vector must be capable of getting into the cell of interest, meaning the
entire vector should be small enough to gain entry and, ideally, be targeted to increase both
uptake has occurred, this potential vector needs some method of escaping any endosomal
pathway to which it may be subjected, in order to avoid being digested or excreted. Finally,
this potential vector should ideally be targeted for localization at the site of intended activity
for the “payload.” While achieving localization in the nucleus of a cell is not the goal of
every cellular delivery vector,54,55 the active site of many drugs is in the nucleus and any attempts at novel genetic manipulation experiments are likely to need access to cell nuclei.
The vector described in this dissertation is intended for use with experiments which require
nuclear localization after internalization, and subsequently employs a method for gaining
efficient entry to the nucleus via the nuclear pore complex – a nuclear localization sequence
(NLS) described in detail in a later section.
1.4. Nanotechnology.
Establishing a greater understanding of nanoscopic materials has permitted their
subsequent utilization across many disciplines. In this dissertation, I am interested in
potential medical applications. Exciting advances in “gene chip” apparati56 are spurring the development of innovative diagnostic tools. Nanowires composed of various metals and
metal oxides are being used as “nanosensors” for the detection and monitoring of varying
biomaterials; most notably antibodies,57 DNA,58 viruses,59 and bacteria.60 Gold nanowires
Nanoparticles comprised of varying polymers65 have been developed and used to aid in the cellular internalization of a range of materials both in vitro66 and in vivo.67 However,
common issues with some such polymeric nanoparticles involve inefficiency of delivery68,69 or cytotoxicity,70,71 though very promising research using biodegradable polymers is currently being performed.72 Many metallic colloids have been put to common use in biological experiments. Quantum dots (nanoparticles commonly comprised of a
cadmium-selenide core and a zinc sulfide shell)73 are used frequently as “tracers” during intracellular experiments.74,75 Aluminum,76 iron,77 platinum,78 silver,79 and other metallic colloidal sols80,81 are also commonly used in many different bioapplications.
Unfortunately, using these materials as cellular delivery vectors can be subject to
many issues. They can exhibit low delivery efficiency, expensive and prohibitively
time-consuming to make, difficult to modify or fine-tune, can be quite toxic, or perhaps have other
inherent properties which make for problematic assays (i.e.: quantum dot “blinking”).
Moreover, to get around some of these limitations, many cellular uptake experiments are run
under complex/harsh conditions specifically in order to effect internalization of whatever
moiety is intended to be delivered; that is, many experiments are dependant upon the use of
chemical methods (i.e. digitonin, a chemical used to open pores in cellular membranes in
vitro),82 physical manipulation (i.e. microinjection: literally using a syringe to inject materials
into cells, thus bypassing the cellular membrane)83,84 or even electrical shocks (e.g. electroporation, which is the administering an electric shock to cells, causing pores in cellular
1.4.1. Colloidal Gold.
Colloidal gold has perhaps the longest history of medicinal use of all metallic
sols.86,87 In the interest of constructing a novel drug delivery vector capable of cellular internalization, gold nanoparticles certainly are a strong candidate for such use: the
biocompatibility of gold has been well established,88 gold nanoparticles are simple to make and easily size-tunable,89 the surface chemistry of gold nanoparticles permit facile and
straight-forward modification with different passivating ligands,90 many biologically significant moieties have been shown to complex readily and in a stable fashion with gold
nanoparticles,91 and their optical properties – specifically the presence of a strong localized surface plasmon resonance band92 – make for simple detection schema.
One potential problem93 involved with using inorganic nanoparticles (or any prospective non-biodegradable delivery vector, for that matter) in vivo: the inability of the
body to clear/excrete these particles. Since there is no way to easily dissolve metallic
nanoparticles in cells, these particles can accumulate in a cell (which may impede cell
growth), and the ability of the body to excrete these nanoparticles decreases dramatically
when the diameter of the particles is larger than ~ 5 nm.93 However, while this is a serious consideration in the overall development of any inorganic nanoparticle intracellular delivery
vehicle, the research described in this dissertation should be thought of as a preliminary in
vitro “proof of concept” that the nanoparticle vehicles synthesized will localize in the nuclei
of certain cells, and it is recognized that further refinement will be necessary to achieve
1.4.1.1. Synthesis of Gold Nanoparticles.
There have been many published methods of gold nanoparticle preparations in both
aqueous and organic solutions since Faraday’s groundbreaking work.86 Generally, tetrachloroauric (HAuCl4) is reduced to form spheres of reasonably uniform diameter in all
of these methods, but under widely-varying conditions: choice of reducing agent, differing
reagent concentrations or order of addition of these reagents, differing temperatures, and
mixing rate. All of these variations have been used to routinely synthesize spherical gold
nanoparticles from 1 to 200 nm,94 and to construct nanoparticles modified with a panoply of functional and/or protective ligands, including carboxylic acids,95,96 phosphenes,95 oligonucleotides,97 amines,98 proteins,99 enzymes,99 and small drug molecules.100
The most common method of synthesizing gold nanoparticles in aqueous solution is
using sodium citrate as the reducing agent of HAuCl4, first demonstrated by Turkevich et
al.101 Briefly, an aqueous solution of HAuCl4 is heated to 100 oC and kept at refluxing
conditions. While stirring vigorously, an amount of sodium citrate is added quickly,
resulting in the solution changing color from the initial yellow to clear, then to grey, and
finally to burgundy, which marks the end of the reaction. The mixture is kept at 100 oC for a further 15 minutes and subsequently cooled to room temperature, maintaining vigorous
stirring throughout. Varying the initial ratio of sodium citrate to HAuCl4 will determine the
ultimate size of the nanoparticles; larger ratios yield smaller particles. The resulting
nanoparticles will be approximately spherical, and have an overall negative surface charge,
counter ions in solution, form an electric double layer which causes nanoparticles to be
repelled from one another in solution, thus preventing the aggregation of the colloids.
1.4.1.2. Stability of Gold Nanoparticles. 102
A colloidal system is defined as a suspension of solid particles dispersed in a
continuous liquid phase. The nature and magnitude of interactions between particles in a
suspension greatly affect the stability of the system. These interactions are typically
characterized by the extent to which they either behave as a stabilizing or destabilizing force
towards the system. For example, van der Waals forces – the collection of universal
attractive forces between atoms and molecules – draw colloidal particles together while
electrostatic forces – arising from a charged double layer present at each particle’s surface –
cause particles to be repelled from one another. If the attractive forces between particles in
suspension become greater than the repulsive forces (e.g. if the distance between particles is
smaller than the sum of their electric double layer distances), the particles will fuse
irreversibly (flocculate) and precipitate.
Moreover, the addition of other species to a suspension – for example, a polymer –
can induce either stabilization or flocculation of the particles in suspension. If an added
polymer is non-adsorbing with respect to the particle surfaces, an attractive force may be
created between two particles if a polymer molecule is ejected from between two
approaching particle surfaces. Due to an osmotic pressure difference between the relatively
high concentration of polymer outside the area between the two surfaces, the surfaces are
However, at higher polymer concentrations, it is possible that polymer molecules would be
unable to be ejected from the area between two approaching surfaces. This could give rise to
a repulsive force with respect to the two surfaces as the polymer molecule(s) resist
compression; this is known as depletion stabilization. If an added polymer is capable of
simultaneously adsorbing to more than one particle in suspension, this polymer will hold the
particles together, giving rise to a phenomenon known as bridging flocculation. The degree
to which a polymer capable of adsorbing to a particle in suspension can create a thick, dense
adsorbed layer completely covering each individual particle will determine the amount of
steric stabilization afforded to the system. Using a polyionic polymer in a suspension where
the particles are oppositely-charged compared to the polymer will likely result in bridging,
and subsequent flocculation. However, adsorption of a polyionic polymer to neutral particles
in suspension then effectively adds an electrostatic stabilization mechanism to any steric
stabilization effects. This is known as electrosteric stabilization, and is very common in
biological systems.
1.5. Existing Intracellular Delivery Vehicles.
Scientists are continually developing and refining the ability to both deliver desired
therapeutic molecules to specific areas in the body and to selectively manipulate genetic
material in a living cell. Cancer research is just one of the many possible fields of
biomedical research which would benefit greatly from advances in targeted drug delivery and
tailored gene manipulation. In general, the development of an efficient, non-cytotoxic vector
nuclei – would be of great value with regard to typical experiments. A prospective reliable
delivery vector could prove invaluable in defining intracellular mechanisms (i.e. transport
mechanisms, feedback mechanisms in protein synthesis, etc.), examining known oncogenes
and tumor repressor mechanisms, or perhaps aid in the discovery of new dynamics of this
nature. To that end, there have been many different methods developed to deliver genetic
material or molecules of biological significance to locations within cells, and a brief review
of these vectors is in order.
1.5.1. Viral Vectors.
Many different methods of implementing targeted drug delivery and genetic
modification are currently being used and refined. For example, mammalian viruses –
retroviruses,103 adenoviruses,104,105 adeno-associated viruses,106,107 and even plant viruses108,109 – are themselves commonly being “re-outfitted” to function as a delivery vehicle for a payload of genetic material. This strategy is an attempt to modify the DNA of a
given cell; take an existing vehicle (a virus) which is known both to invade cells and deliver
genetic material with high efficiency, and merely replace the intrinsic, otherwise
cell-damaging/disease-causing bits with some desired genetic material. A vector of this
composition would seem to be ideal with respect to actually delivering some moiety of
consequence. Disappointingly, the major obstacles in using these modified viruses in vivo
are the propensity for the vectors themselves to elicit a strong immune response, and they can
also be quite toxic. Furthermore, the modification of these viruses is a non-trivial procedure,
can fit inside a native virus with a typical diameter of 30 – 100 nm. Also, uncontrolled
recombination can be a concern in using a viral vector, since each specific function of a
given virus’ inherent DNA/RNA may not always be clear.110 It should be mentioned that plant viruses are least limited by these considerations.
1.5.2. Non-Viral Vectors.
Other approaches to targeted genetic manipulation involve delivery methods of a
non-viral nature. Liposomes,111 hydrogels,112 and dendrimers113 have been used successfully as carriers for genetic material in cellular uptake experiments. Gene guns114 have been used to achieve the same effects, although this method is typically more for use with plant cell
transfection than with mammalian cells due to the more robust character of the exterior
membrane of plant cells. Even “naked” DNA (and cationic-packed DNA)115,116 has been shown to be taken up by certain cells under specific circumstances. However, all of these
methods – especially the latter three – can be inefficient, are often cytotoxic, are prone to
inserting DNA non-specifically in cells (increasing likelihood of tumor formation), and will
often only yield a transient transfection of the desired DNA instead of a permanent
change.117,118
1.5.3. Viral/Non-Viral Combination Vectors.
While there is much current research attempting to manipulate entire viruses in such a
fashion as to make them plausible cellular delivery vectors, various active regions of the
using relevant pieces of viruses as biological probes;119 one such piece being the NLS sequence of the simian virus 40 (SV40) large tumor (“large T”) antigen.
1.5.3.1. SV40 Large T NLS.
SV40, a polyomavirus, was discovered in 1959 as a contaminant in poliovirus
vaccines prepared using rhesus monkey renal cell cultures.120 It was found that this virus
could induce large tumors in rodents (hence the “large T” moniker) and immortalize many
types of cells in culture (including human cells). The entire SV40 large T antigen is an
oncoprotein known to both stimulate cell division and attenuate intrinsic cellular tumor
suppression mechanisms, such as the p53 regulatory mechanism.121 The large T NLS peptide
sequence – PKKKRKV – is a very small piece of the entire antigen (708 amino acids),122 and is itself the most thoroughly-characterized NLS.123 It has been shown to effectively penetrate cell nuclei using a mediated process via nuclear pore complexes.83
Some of the seminal studies examining nuclear penetration involving this peptide
sequence were performed using colloidal gold stabilized with a conjugate of large T bound to
larger proteins, notably bovine serum albumin (BSA).84,124 In this early research, these complexes were microinjected into both amoeba and BALB/3T3 cells, and differing
parameters (i.e. using different peptides, using point-mutated peptide sequences, varying
carrier protein) were manipulated to examine the extent to which nuclear localization would
or would not take place. The gold nanoparticle merely functioned as a marker for the
complexes’ location as determined via transmission electron microscopy (TEM) analyses
1.6. Summary.
There is a distinct need for innovative approaches to intracellular drug delivery and
genetic manipulation. Whether one desires to activate/suppress/change a given gene in a
particular cell, deliver a therapeutic molecule to a specific organelle within a cell, or even if
one merely wants to probe a cellular process of interest, then getting moieties of interest into
cells reliably, efficiently, and without generating significant toxicity is of paramount
importance.
This dissertation demonstrates that an intracellular delivery vehicle can be developed
using large T/BSA-gold nanoparticle complexes. In order to examine the potential
interaction of these complexes with cells cultured in vitro, human cervical cancer cells
(HeLa) were selected for analysis since they are a very widely-used cell line, easy to
maintain, and allow for facile transport of external moieties across their cell membrane.
Additionally, the degree to which complexes could efficiently localize in cell nuclei even
when administered merely by incubating cells in media containing these complexes was
demonstrated and quantified. Moreover, various parameters of particle synthesis (i.e.
number of large T peptides per BSA, overall stability of large T/BSA-colloidal gold
complexes, nanoparticle diameter, temperature, etc.) were tested to optimize the
internalization and subsequent nuclear localization of these complexes.
Using colloidal gold as a “scaffold” upon which one can place a stabilizing, non-toxic
substance (such as a serum albumin, for example) which itself can be quickly and relatively
easily modified with certain peptide sequences that target either specific cells or particular
inquiry detailed in this dissertation. There is a precedent for this idea – Feldherr et al used
such colloidal constructs in their ground-breaking examinations of nuclear uptake
mechanisms.83,84 However, the quantification of cellular internalization of exogenous nanoparticles has not been previously presented.
One other issue involved in intracellular experimentation: imaging methods used
when examining cells after incubation with colloidal gold delivery vectors (i.e.: video
enhanced color differential interference contrast (VECDIC) microscopic techniques, TEM,
confocal microscopy) have considerable limitations concerning potential delivery methods
themselves. For example, VECDIC and confocal microscopy are both limited in the size of
the discrete particles which can be detected, and TEM employs a random sectioning of cells
for examination, potentially leading to inaccurate reporting of internalization. To address
this issue, the use of inductively-coupled plasma – optical emission spectroscopy (ICP-OES)
as a routine detection method of nanoparticle complex internalization was defined and
developed. This technique provided quantitative information otherwise unavailable via other
common detection methods.
While the research presented on the following pages does not attempt to address all
the issues in designing the ideal delivery method or probe described previously, this research
can be seen as a “first step” with regard to both learning about issues associated with cellular
uptake of colloidal constructs and eventually addressing potential needs of cancer research in
1.7. References.
1 Hussain, S. P.; Hofseth, L. J.; Harris, C. C.
Nature Reviews, 2003, 3, 276 – 285.
2 Smith, J. S.; Green, J.; Berrington de Gonzalez, A.; Appleby, P.; Peto, J.; Plummer, M.;
Franceschi, S.; Beral, V. Lancet, 2003, 361, 1159 - 67.
3 Jones, P. A.; Baylin, S. B.
Nature Reviews: Genetics, 2002, 3, 415 – 428.
4 Kuper, H.; Adami, H.-O.; Trichopoulos, D.
Journal of Internal Medicine, 2000, 248, 171 –
183.
5 Clemons, M.; Goss, P.
New England Journal of Medicine, 2001, 344(4), 276 – 285.
6 Marks, P. A.; Rifkind, R. A.; Richon, V. M.; Breslow, R.; Miller, T.; Kelly, W. K.
Nature Reviews: Cancer, 2001, 1, 194 – 202.
7 Calle, E. E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M. J.
New England Journal of Medicine, 2003, 348(17), 1625 – 1638.
8 Ueda, H.; Ullrich, S. J.; Gangemi, J. D.; Kappel, C. A.; Ngo, L.; Feitelson, M. A.; Jay, G.
Nature Genetics, 1995, 9, 41 – 47.
9 Nigg, E. A.
Nature Reviews: Cancer, 2002, 2, 1 – 11.
10 Carmeliet, P.; Jain, R. K.
Nature, 2000, 407, 249 – 257.
11 Bach, P. B.; Kelley, M. J.; Tate, R. C.; McCrory, D. C.
Chest, 2003, 123(1), 72 – 82.
12 Ilic, D.; O’Connor, D.; Green, S.; Wilt, T.
Cancer Causes Control, 2007, 18, 279 – 285.
13 Otto, S. J.; Fracheboud, J.; Looman, C. W. N.; Broeders, M. J. M.; Boer, R.; Hendriks, J.
H. C. L.; Verbeek, A. L. M.; de Koning, H. J. Lancet, 2003, 361, 1411 – 1417.
14 Walsh, J. M. E.; Terdiman, J. P.
Journal of the American Medical Association, 2003, 289(10), 1288 – 1296.
15 Noteborn, M. H. M.; Zhang, Y.-H.; van der Eb, A. J.
Mutation Research, 1998, 447 – 455.
16 Lian, F.; Hu, K.-Q.; Russell, R. M.; Wang, X.-D.
Int. J. Cancer, 2006, 119, 2084 – 2089.
18 Tagscherer, K. E.; Fassil, A.; Campos, B.; Farhardi, M.; Kraemer, A.; Bӧck, B. C.;
Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O. D.; Herold-Mende, C.; Roth, W.
Oncogene, 2008, 1 – 11.
19 Henson, J. D.; Neumann, A. A.; Yeager, T. R.; Reddel, R. R.
Oncogene, 2002, 21, 598 –
610.
20 Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.
Nature, 2008, 454, 436- 454.
21 Mantovani, A.; Romero, P.; Palucka, A. K.; Marincola, F. M.
Lancet, 2008, 371, 771 –
783.
22 Iwai, K.; Maeda, H.; Konno, T.
Cancer Research, 1984, 44, 2115 – 2121.
23 Wyld, D. K.; Selby, P.; Perren, T. J.; Jonas, S. K.; Allen-Mersh, T. G.; Wheeldon, J.;
Burchill, S. A. Int. J. Cancer, 1998, 79, 288 – 293.
24 Gosselin, M. V.; Rubin, G. D.; Leung, A. N.; Huang, J.; Rizk, N. W.
Radiology, 1998, 208(1), 209 – 215.
25 Kiesslich, R.; Burg, J.; Vieth, M; Gnaendiger, J.; Enders, M.; Delaney, P; Polgase, A.;
McLaren, W.; Janell, D.; Thomas, S.; Nafe, B.; Galle, P. R.; Neurath, M. F.
Gasteroenterology, 2004, 127, 706 – 713.
26 Pinkel, D.; Straume, T.; Gray, J. W.
Proc. Natl. Acad. Sci. USA, 1986, 83, 2934 – 2938.
27 Jiao, X.; Eslami, A.; Ioffe, O.; Kwong, K. F.; Henry, M.; Zeng, Q.; Refaely, Y.; Burrows,
W.; Gamliel, Z.; Krasna, M. J. The Annals of Thoracic Surgery, 2003, 76(4), 996- 1000.
28 Nelson, H.; Petrelli, N.; Carlin, A.; Couture, J.; Fleshman, J.; Guillem, J.; Miedema, B.;
Ota, D.; Sargent, D. J. Nat. Cancer Inst., 2001, 93(8), 583 – 596.
29 Spano, J.-P.; Chodkiewicz, C.; Maurel, J.; Wong, R.; Wasan, H.; Barone, C.; Létourneau,
R.; Bajetta, E.; Pithavala, Y.; Bycott, P.; Trask, P.; Liau, K.; Ricart, A. D.; Kim, S.; Rixe, O.
Lancet, 2008, 371, 2101 – 2108.
30 Bolla, M.; Gonzalez, D.; Warde, P.; Dubois, J. B.; Mirimanoff, R.-O.; Storme, G.; Bernier,
31 Bubendorf, L.; Kolmer, M.; Kononen, J.; Kovisto, P.; Mousses, S.; Chen, Y.; Mahlamäki,
E.; Schraml, P.; Moch, H.; Willi, N.; Elkahloun, A. G.; Pretlow, T. G.; Gasser, T. C.; Mihatsch, M. J.; Sauter, G.; Kallioniemi, O.-P. J. Nat. Cancer Inst., 1999, 91(20), 1758 –
1764.
32 Parmiani, G.; Castelli, C.; Dalerba, P.; Mortarini, R.; Rivoltini, L.; Marincola, F. M.;
Anichini, A. J. Nat. Cancer Inst., 2002, 94(11), 805 – 818.
33 Lehman, P.
British Journal of Dermatology, 2007, 156, 793 – 801.
34 Samuel, S. K.; Spencer, V. A.; Bajno, L.; Sun, J.-M.; Holth, L. T.; Oesterreich, S.; Davie,
J. R. Cancer Research, 1998, 58, 3004 – 3008.
35 Giannakakou, P.; Sackett, D. L.; Kang, Y.-K.; Zhan, Z.; Buters, J. T. M.; Fojo, T.;
Poruchynsky, M. S. J. Biol. Chem., 1997, 272(27), 17118 – 17125.
36 Blagosklonny, M. V.; Fojo, T.
Int. J. Cancer, 1999, 83, 151 – 156.
37 Gieschen, H. L.; Spiro, I. J.; Suit, H. D.; Ott, M. J.; Rattner, D. W.; Ancukiewicz, M.;
Willett, C. G. Int. J. Radiation Oncology Biol. Phys., 2001, 50 (1), 127 – 131.
38 Bush, D. A.; Hillebrand, D. J.; Slater, J. M.; Slater, J. D.
Gastroenterology, 2004, 127 (5),
S189 – S193.
39 Schwartz, D. L.; Einck, J.; Bellon, J.; Laramore, G. E.
Int. J. Radiation Oncology Biol. Phys., 2001, 50 (2), 449 – 456.
40 Krämer, M.; Jäkel, O.; Haberer, T.; Kraft, G.; Schardt, D.; Weber, U.
Phys. Med. Biol, 2000, 45, 3299 – 3317.
41 Huber, P. E.; Debus, J.; Latz, D.; Zierhut, D.; Bischof, M.; Wannenmacher, M.;
Engenhart-Cabillic, R. Radiotherapy and Oncology, 2001, 59, 161 – 167.
42 Hoskin, P. J.; Saunders, M. I.; Dische, S.
Cancer, 1999, 86 (7), 1322 – 1328.
43 Al-Sarraf, M.; Pajak, T. F.; Marcial, V. A.; Mowry, P.; Cooper, J. S.; Stetz, J.; Ensley, J.
F.; Velez-Garcia, E. Cancer, 1987, 59, 259 – 265.
44 Meresman, G. F.; Bilotas, M.; Buquet, R. A.; Barañao, R. I.; Sueldo, C.; Tesone, M.
45 Schneider, J.; Huh, M. M.; Bradlow, H. L.; Fishman, J.
J. Biol. Chem., 1984, 259 (8), 4840
– 4845.
46 Huggins, C.
Science, 1967, 156 (3778), 1050 – 1054.
47 Overgaard, M.; Jensen, M.-B.; Overgaard, J.; Hansen, P. S.; Rose, C.; Andersson, M.;
Kamby, C.; Kjær, M.; Gadeberg, C. C.; Rasmussen, B. B.; Blichert-Toft, M.; Mouridsen, H. T. Lancet, 1999, 353, 1641 – 1648.
48 Mouridsen, H.; Gershanovich, M.; Sun, Y.; Pérez-Carrión, R.; Boni, C.; Monnier, A.;
Apffelstaedt, J.; Smith, R.; Sleeboom, H. P.; Jänicke, F.; Pluzanska, A.; Dank, M.; Becquart, D.; Bapsy, P. P.; Salminen, E.; Snyder, R.; Lassus, M.; Verbeek, J. A.; Staffler, B.; Chaudri-Ross, H. A.; Dugan, M. J. Clin. Oncol., 2001, 19, 2596 – 2606.
49 Pejawar-Gaddy, S.; Finn, O. J.
Crit. Rev. in Oncology/Hematology, 2008, 67, 93 – 102.
50 von Mehren, M.; Adams, G. P.; Weiner, L. M.
Annu. Rev. Med., 2003, 54, 343 – 369.
51 Alberts, B.; Bray, D.; Hopkin, K.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P.
Essential Cell Biology, 2nd Edition, Garland Science, New York, NY, 2004.
52 Smith, A. E.; Helenius, A.
Science, 2004, 304, 237 – 242.
53 Decuzzi, P.; Ferrari, M.
Biomaterials, 2007, 28, 2915 – 2922.
54 Hoye, A. T.; Davoren, J. E.; Wipf, P.; Fink, M. P.; Kagan, V. E.
Acc. Chem. Res., 2008, 41 (1), 87 – 97.
55 Rosania, G. R.
Current Topics in Medicinal Chemistry, 2003, 3, 659 – 685.
56 Langer, R.; Tirrell, D. A.
Nature, 2004, 428, 487 – 492.
57 Zheng, G.; Patolsky, F.; Cui, Y.; Wang, W. U.; Lieber, C. M.
Nature Biotechnology, 2005, 23 (10), 1294 – 1301.
58 Hahm, J.; Lieber, C. M.
Nano Letters, 2004, 4 (1), 51 – 54.
59 Patolsky, F.; Zheng, G.; Hayden, O.; Lakadamyali, M.; Zhuang, X.; Lieber, C. M.
Proc. Nat. Acad. Sci., 2004, 101 (39), 14017 – 14022.
60 Pal, S.; Alocilja, E. C.; Downes, F. P.
Biosensors and Bioelectronics, 2007, 22, 2329 –
61 Kuo, C.-W.; Lai, J.-J.; Wei, K. H.; Chen, P.
Nanotechnology, 2008, 19, 1 – 7.
62 Li, C.; Curreli, M.; Lin, H.; Lei, B.; Ishikawa, F. N.; Datar, R.; Cote, R. J.; Thompson, M.
E.; Zhou, C. J. Am. Chem. Soc., 2005, 127, 12484 – 12485.
63 Kam, N. W. S.; O’Connell, M.; Wisdom, J. A; Dai, H.
Proc. Nat. Acad. Sci., 2005, 102 (33), 11600 – 11605.
64 James, J. T.; McCluskey, R.; Arepalli, S.; Hunter, R. L.
Critical Reviews in Toxicology, 2006, 36, 189 – 217.
65 Brigger, I.; Dubernet, C.; Couvreur, P.
Adv. Drug Del. Rev., 2002, 54, 631 – 651.
66 Hans, M. L.; Lowman, A. M.
Curr. Opin. Sol. State and Mat. Sci., 2002, 6, 319 – 327.
67 You, H. S.; Lee, K. H.; Oh, J. E.; Park, T. G.
Journal of Controlled Release, 2000, 68, 419
– 431.
68 Cherng, J.-Y.; van de Wetering, P.; Talsma, H.; Crommelin, D. J. A.; Hennik, W. E.
Pharm. Res., 1996, 13 (7), 1038 – 1042.
69 Huang, M.; Khor, E.; Lim, L.-Y.
Pharm. Res., 2004, 21 (2), 344 – 353.
70 Fischer, D.; Li, Y.; Ahlmeyer, B.; Krieglstein, J.; Kissel, T.
Biomaterials, 2003, 24, 1121 –
1131.
71 Kirchner, C.; Javier, A. M.; Susha, A. S.; Rogach, A. L.; Kreft, O.; Sukhorukov, G. B.;
Parak, W. J. Talanta, 2005, 67, 486 – 491.
72 Shenoy, D.; Little, S.; Langer, R.; Amiji, M.
Mol. Pharm., 2005, 2 (5), 357 – 366.
73 Pinaud, F.; King, D.; Moore, H.-P.; Weiss, S.
J. Am. Chem. Soc., 2004, 126, 6115 – 6123.
74 Ballou, B.; Lagerholm, B. C.; Ernst, L. A.; Bruchez, M. P.; Waggoner, A. S.
Bioconj. Chem., 2004, 15, 79 – 86.
75 Gao, X.; Cui, Y.; Levenson, R. M.; Chung, L. W.; Nie, S.
Nature Biotechnology, 2004, 22 (8), 969 – 976.
76 Jouet, R. J.; Warren, A. D.; Rosenberg, D. M.; Bellitto, V. J.; Park, K.; Zachariah, M. R.
77 Gupta, A. K.; Gupta, M.
Biomaterials, 2005, 26, 3995 – 4021.
78 Hrapovic, S.; Liu, Y.; Male, K. B.; Luong, J. H. T.
Anal. Chem., 2004, 76, 1083 – 1088.
79 Redmond, P. L.; Wu, X.; Brus, L.
J. Phys. Chem. C, 2007, 111, 8942 – 8947.
80 Murayama, H.; Narushima, T.; Negishi, Y.; Tsukuda, T.
J. Phys. Chem. B, 2004, 108 (11),
3496 – 3503.
81 Mornet, S.; Vasseur, S.; Grasset, F.; Duguet, E.
J. Mater. Chem., 2004, 14, 2161 – 2175.
82 Cserpán, I.; Udvardy, A.
J. Cell Sci., 1995, 108, 1849 – 1861.
83 Feldherr, C. M.; Akin, D.; Cohen, R. J.
J. Cell Sci., 2001, 114, 4621 – 4627.
84 Dworetzky, S. I.; Lanford, R. E; Feldherr, C. M.
J. Cell Biol., 1988, 107, 1279 – 1287.
85 Jen, C.-P.; Chen, Y.-H.; Fan, C.-S.; Yeh, C.-S.; Lin, Y.-C.; Shieh, D.-B.; Wu, C.-L.; Chen,
D.-H.; Chou, C.-H. Langmuir, 2004, 20, 1369 – 1374.
86 Faraday, M.
Philosophical Transactions of the Royal Society of London, 1857, 147, 145 –
181.
87 Daniel, M.-C.; Astruc, D.
Chem. Rev., 2004, 104, 293 – 346.
88 Bhattacharya, R.; Mukherjee, P.
Adv. Drug Del. Rev., 2008, 60, 1289 – 1306.
89 Templeton, A. C.; Wuelfing, W. P.; Murray, R. W.
Acc. Chem. Res., 2000, 33, 27 – 36.
90 Shenhar, R.; Norsten, T. B.; Rotello, V. M.
Adv. Mater., 2005, 17 (6), 657 – 669.
91 Joshi, H.; Shirude, P. S.; Bansal, V.; Ganesh, K. N.; Sastry, M.
J. Phys. Chem. B, 2004, 108, 11535 – 11540.
92 Quinten, M.
Appl. Phys. B, 2001, 73, 317 – 326.
93 Choi, H. S.; Liu, W.; Misra, P; Tnaka, E.; Zimmer, J. P.; Ipe, B. I.; Bawendi, M. G.;
Frangioni, J. V. Nature Biotechnology, 2007, 25 (10), 1165 – 1170.
94 Grabar, K. C.; Brown, K. R.; Keating, C. D.; Stranick, S. J.; Tang, S.-L.; Natan, M. J.
95 Franzen, S.; Folmer, J. C. W.; Glomm, W. R.; O’Neal, R.
J. Phys. Chem. A, 2002, 106,
6533 – 6540.
96 Simard, J.; Briggs, C.; Boal, A. K.; Rotello, V. M.
Chem. Commun., 2000, 1943 – 1944.
97 Storhoff, J. J.; Elghanian, R.; Mucic, R. C.; Mirkin, C. A.; Letsinger, R. L.
J. Am. Chem. Soc., 1998, 120, 1959 – 1964.
98 Gomez, S.; Philippot, K.; Collière, V.; Chaudret, B.; Senocq, F.; Lecante, P.
Chem. Commun., 2000, 1945 – 1946.
99 Hayat, M. A.
Colloidal Gold: Principles, Methods and Applications; Academic Press: New
York, 1989; Vol. 1.
100 Wang, G.; Zhang, J.; Murray, R. W.
Anal. Chem., 2002, 74 (17), 4320 – 4327.
101 Turkevich, J.; Garton, G.; Stevenson, P. C.
J. Colloid Science, 1954, 9, S26 – S35.
102 Hiemenz, P. C.; Rajagopalan, R.
Principles of Colloid and Surface Chemistry; 3rd ed.;
Marcel Dekker, Inc.: New York, 1997.
103 Zufferey, R.; Dull, T.; Mandel, R. J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D.
J. Virol, 1998, 72(12), 9873 – 9880.
104 Bouvet, M.; Ellis, L. M.; Nishizaki, M.; Fujiwara, T.; Liu, W.; Bucana, C. D.; Fang, B.;
Lee, J. J.; Roth, J. A. Cancer Research, 1998, 58, 2288 – 2292.
105 Wan, Y. Y.; Leon, R. P.; Marks, R.; Cham, C. M.; Schaack, J.; Gajewski, T. F.;
DeGregori, J. Proc. Nat. Acad. Sci., 2000, 97(25), 13784 – 13789.
106 Xiao, X.; Li, J.; McCown, T. J.; Samulski, R. J.
Experimental Neurology, 1997, 144, 113
– 124.
107 Monahan, P. E.; Samulski, R. J.
Mol. Med. Today, 2000, 6, 433 – 440.
108 Loo, L.; Guenther, R. H.; Lommel, S. A.; Franzen, S.
J. Am. Chem. Soc., 2007, 129,
11111 – 11117.
109 Ren, Y.; Wong, S. M.; Lim, L.-Y.
Bioconjugate Chem., 2007, 18, 836 – 843.
110 Thomas, C. E.; Ehrhardt, A.; Kay, M. A.
111 Liu, Y.; Liggit, D.; Zhong, W.; Tu, G.; Gaensler, K.; Debs, R.
J. Biol. Chem., 1995, 270 (42), 24864 – 24870.
112 Huang, H.; Pierstorff, E.; Osawa, E.; Ho, D.
Nano Letters, 2007, 7 (11), 3305 – 3314.
113 Dufes, C.; Keith, W. N.; Bilsland, A.; Proutski, I.; Uchegbu, I. F.; Schätzlein, A. G.
Cancer Researh, 2005, 65(18), 8079 – 8084.
114 Rakhmilevich, A. L.; Turner, J.; Ford, M. J.; McCabe, D.; Sun, W. H.; Sondel, P. M.;
Grota, K.; Yang, N.-S. Proc. Nat. Acad. Sci., 1996, 93, 6291 – 6296.
115 Dean, D. A.; Strong, D. D.; Zimmer, W. E.
Gene Therapy, 2005, 12, 881 – 890.
116 Padmanabhan, K.; Smith, T. J.
Pharm. Dev. Tech., 2002, 7 (1), 97 – 101.
117 Niidome, T.; Huang, L.
Gene Therapy, 2002, 9, 1647 – 1652.
118 Pack, D. W.; Hoffman, A. S.; Pun, S.; Stayton, P. S.
Nature Reviews: Drug Discovery, 2005, 4, 581 – 593.
119 Lundberg, P. Langel, Ü.
J. Mol. Recognit., 2003, 16, 227 – 233.
120 Sullivan, C. S.; Pipas, J. M.
Microbiology and Molecular Biology Reviews, 2002, 66 (2),
179 – 202.
121 Carbone, M.; Rizzo, P.; Grimley, P. M.; Procopio, A.; Mew, D. J. Y.; Shridhar, V.; de
Bartolomeis, A.; Esposito, V.; Guiliano, M. T.; Steinberg, S. M.; Levine, A. S.; Giordano, A.; Pass, H. I. Nature Medicine, 1997, 3 (8), 908 – 912.
122 DeCaprio, J. A.; Ludlow, J. W.; Figge, J.; Shew, J.-Y.; Huang, C.-H.; Lee, W.-H.;
Marsilio, E.; Paucha, E.; Livingston, D. M. Cell, 1988, 54, 275 – 283.
123 Robb, J.
J. Virol., 1973, 12 (5), 1187 – 1190.
124 Feldherr, C. M.; Akin, D.
Chapter 2
2.1. Introduction.
This dissertation chapter begins with a brief discussion of the constituent pieces of the
large T/BSA-gold nanoparticle complexes used in this research and the differences between
these complexes and similar constructs used in other research. Then, the processes involved
in synthesizing these complexes are described, including results from experiments performed
to define certain parameters of the assembly of these nanoparticle complexes. Finally, data
are presented concerning the stability of these complexes under varying conditions. Note
that all experimental results are shown and discussed first, followed by a section defining
materials used and a methodical description of experimental procedures which were
performed.
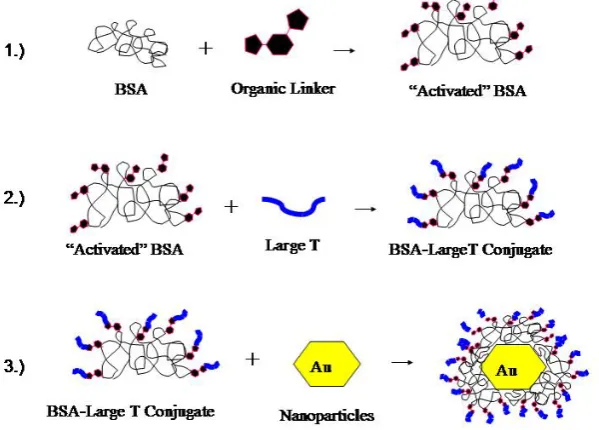
Briefly, the general three-step scheme utilized in this research for construction of the
large T/BSA-gold nanoparticle complexes is depicted in the cartoon in Figure 2.1.
Figure 2.1. Cartoon representing the general construction scheme of large
While the general idea of having large T NLS peptides conjugated to BSA as a
passivating layer for colloidal gold has been previously utilized – most notably by Feldherr,
et. al.1,2; see Chapter 1 for full discussion – there are differences between the individual parts of this cellular probe used in the original Feldherr research and what was used in the research
presented in this dissertation. The most ostensible difference is the organic
heterobifunctional linker employed in conjugating the large T peptides to BSA molecules.
The principle of using a heterobifunctional reagent is extremely useful for these types of
conjugations – one side of this linker preferentially reacts with terminal amines while the
other side preferentially binds with thiols. This enables one to use such molecular
“preferences” in sequence to give rise to 1) linkage of the heterobifunctional reagent to BSA
via its terminal amines, then 2) allow thiol-modified peptides to react with the other side of
the bound linkers. Previous research performed by Feldherr et. al.1,2 using these conjugates utilized m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS) as the organic linker
between large T peptide sequence and BSA. However, it is known3 that maleimides are subject to hydrolysis over short periods of time, which would potentially lower the amount of
peptides one might otherwise be able to attach to BSA. Consequently, the heterobifunctional
reagent chosen for use in this dissertation research was succinimidyl-4-(N
-maleimidomethyl)cyclohexane-1-carboxylate (SMCC), which does not have its maleimide
moiety directly connected to an extended -system as MBS does, thus minimizing
opportunity for hydrolysis. The structural differences between the two molecules can be seen
Figure 2.2. Structures of m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS) and
succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC).4
The large T NLS peptide sequence itself is a well-studied moiety which is known to
penetrate cell nuclei efficiently5,6,7 and is a fairly short peptide sequence compared to other known nuclear localization sequences; these are both good reasons to use this particular NLS
in the assembly of a novel cellular delivery vehicle. BSA is a well-studied protein,
biologically inert in its native form,8 theoretically has many attachment points for SMCC (54 available surface lysines),9 has many existing protocols for attaching various moieties of interest,10 and is also commonly used with colloidal gold.11,12
15 nm diameter gold nanoparticles were chosen as the nanoparticles to be used in the
cellular uptake experiments described in the next chapter. This decision was based upon the
following: 1) nuclear pores in human cervical cancer cells (HeLa, described further in
upper bound upon the overall nanoparticle complex size in order to allow for potential
nuclear localization during an experiment (moreover, dynamic light scattering experiments
performed during similar experiments involving BSA-20 nm diameter gold nanoparticle
complexes determined the overall complex size was increased by approximately 2 nm,14 potentially approaching the lower limit of what might pass through a nuclear pore); 2) 15 nm
diameter nanoparticles are readily visible using other microscopy methods – such as
VECDIC and confocal imaging; and 3) 15 nm particles can be seen as a compromise
between potential facile detection using optical microscopy and having a large enough
surface area to accommodate several conjugates/polypeptides for possible
multifunctionality.15
2.2. Results and Discussion
2.2.1. Large T-SMCC-BSA Synthesis.
As mentioned previously, the conjugation of large T to BSA is accomplished via an
organic linker, SMCC. An overview of the steps involved can be seen in Figure 2.1. Each of
these steps is described in detail below.
2.2.1.1. SMCC-BSA Conjugation.
The first step in the process of synthesizing these conjugates is attaching SMCC (in
organic solvent) to aqueous solution of BSA. There are existing protocols for this general


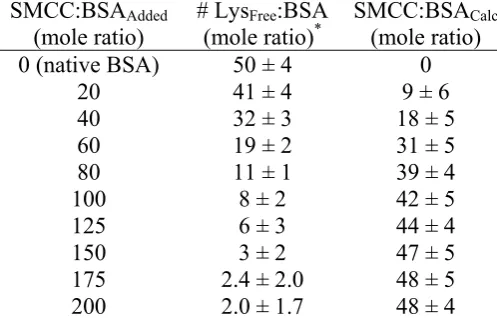
![Table 2.3. Mole ratio of SMCC molecules conjugated with BSA for samples to be used in HeLa delivery experiments; [Lys]Pre refers to the experimentally-determined concentration of lysine in the BSA samples prior to addition of SMCC, while [Lys]Post refers t](https://thumb-us.123doks.com/thumbv2/123dok_us/1662905.1208814/54.595.116.496.487.617/molecules-conjugated-delivery-experiments-experimentally-determined-concentration-addition.webp)