Clinical use of crizotinib for the treatment of non-small cell lung cancer
Full text
Figure
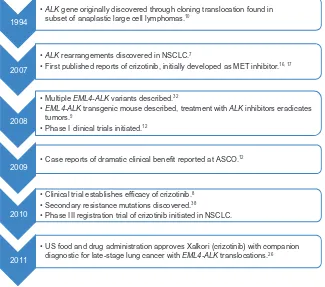

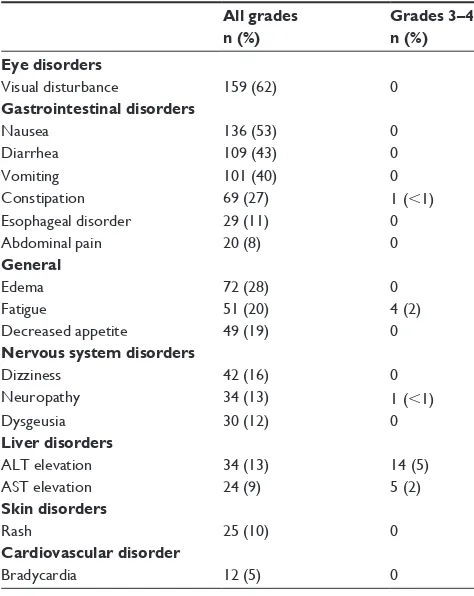

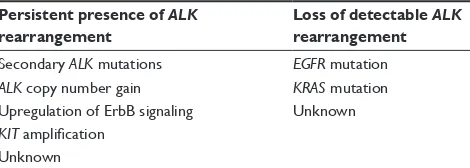
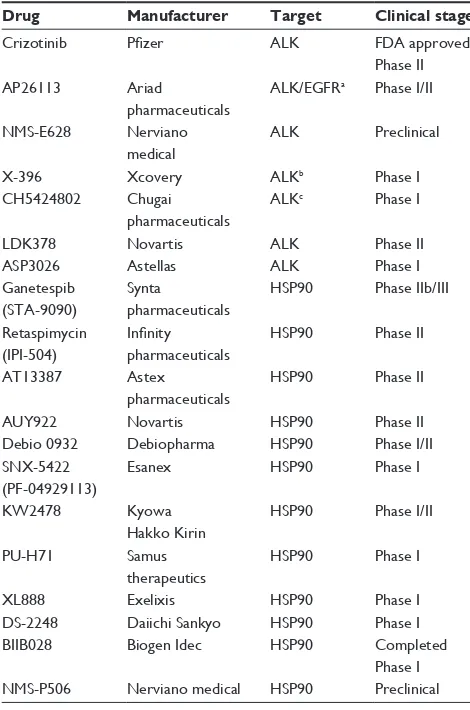
Related documents
Investigations of radon and CO 2 levels carried out at three geomorphologically different locations in the Postojna Cave uncovered significant differences in the spatial distribution
o Power oscillations and voltage decline cause cascading separations Blackout occurred in 3 minutes System restored in ~1-2 days o Voltage declines, causing cascading
The purpose of this study was to determine whether urban patterns of HR-HPV occurrence can be generalized to rural areas of the same developing country, using data from Mali,
PT100 evaluation unit for motor winding temperature, integrated in control cabinet for 3 × PT100/1000 or 3 × KTY83/84 sensors; temperature indication on evaluation unit, alarm
Its formation was considered to only occur in the night time and in the presence of water vapour.11 It is inadvertently formed when alkylamines, mainly dimethylamine DMA
We reported here a case of chronic disseminated histoplasmosis presenting with multiple mass lesions, weight loss, and hypercalcemia, mimicking metastatic cancer. The
The scheme combines the signal strength difference (SSD) and improved Signal Strength Difference (iSSD) to produce a hybrid fingerprint for the Wi-Fi based localization
An experimental study was conducted on a four stroke single cylinder compression ignition engine to determine the performance, combustion and exhaust emission characteristics
