INVESTIGATION
Inhibition of RNA Interference and Modulation
of Transposable Element Expression by Cell Death
in
Drosophila
Weiwu Xie,* Chengzhi Liang,†,1and James A. Birchler*,2 *Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211,yCold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724
ABSTRACT RNA interference (RNAi) regulates gene expression by sequence-specific destruction of RNA. It acts as a defense mechanism against viruses and represses the expression of transposable elements (TEs) and some endogenous genes. We report that mutations and transgene constructs that condition cell death suppress RNA interference in adjacent cells inDrosophila melanogaster. The reversal of RNAi is effective for both thewhite(w) eye color gene and greenfluorescent protein (GFP), indicating the generality of the inhibition. Antiapoptotic transgenes that reverse cell death will also reverse the inhibition of RNAi. Using GFP and a low level of cell death produced by a heat shock-head involution defective (hs-hid) transgene, the inhibition appears to occur by blocking the conversion of double-stranded RNA (dsRNA) to short interfering RNA (siRNA). We also demonstrate that the mus308 gene and endogenous transposable elements, which are both regularly silenced by RNAi, are increased in expression and accompanied by a reduced level of siRNA, when cell death occurs. Thefinding that chronic ectopic cell death affects RNAi is critical for an understanding of the application of the technique in basic and applied studies. These results also suggest that developmental perturbations, disease states, or environmental insults that cause ectopic cell death would alter transposon and gene expression patterns in the organism by the inhibition of small RNA silencing processes.
R
NA interference (RNAi) uses double-stranded RNA (dsRNA) to target the destruction of the homologous messenger RNAs via short interfering RNAs (siRNA) (Zamore and Haley 2005). In Drosophila, dsRNA can be generated by transcribing a sequence from both directions or from inverted repeats. The ribonuclease Dicer-2 processes the dsRNA into 21-nt siRNA. Single-stranded siRNA is then incorporated into the RNA-interference silencing com-plex (RISC) and the latter recognizes and cleaves a target RNA, which perfectly or nearly perfectly matches the siRNA. This targeted degradation mechanism has been demon-strated as an immunity defense against viruses that have dsRNA in their life cycle (Li and Ding 2005). Discovery of en-dogenous siRNAs homologous to transposable elements (TEs) also suggests this siRNA pathway plays a role in suppressingtransposition (Chung et al.2008; Czechet al. 2008; Ghildiyal et al.2008; Kawamuraet al.2008; Tamet al.2008; Watanabe et al. 2008) in addition to the PIWI-associated small RNA (piRNA) pathway (Aravinet al.2007; Brenneckeet al.2007). Cell death is a central part of the immune system of many multicellular organisms. Cells infected with pathogens can trigger the apoptotic pathway and cell death to prevent the pathogens from spreading (Postigo and Ferrer 2010). Such a mechanism is also used to remove damaged cells or extra cells unused in tissue formation. When diseased or damaged cells are removed, proliferation signals are generated from the dying cells to the adjacent cells to stimulate compensatory cell divisions (Huhet al.2004; Pérez-Garijoet al.2004; Ryooet al.2004).
Deep sequencing of endogenous siRNAs reveals that a significant proportion of them are derived from transcrip-tion of certain sequences from opposite directranscrip-tions, of hairpin-structured sequences, or of homologous sequences (e.g., genes and their pseudogenes) from which comple-mentary sense and antisense sequences are generated (Chung et al. 2008; Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008; Tam et al.2008; Watanabeet al.2008). These siRNAs have been Copyright © 2011 by the Genetics Society of America
doi: 10.1534/genetics.111.128470
Manuscript received March 5, 2011; accepted for publication May 15, 2011 Supporting information is available online at http://www.genetics.org/content/ suppl/2011/05/19/genetics.111.128470.DC1.
1Present address: IRRI, DAPO Box 7777, Metro Manila, Philippines. 2Corresponding author: Division of Biological Science, Tucker Hall, University of
demonstrated to match important genes and their expres-sion is repressed in oocytes of mice and somatic cells offlies, thus implying a genome-wide regulation role by RNAi. Among these genes, the endogenous mus308gene in Dro-sophila increases in expression when RNAi is compromised (Czech et al.2008; Okamura et al.2008).
Here we report that cell death in Drosophila, induced either by apoptotic genes or caused by developmental defects, inhibits RNAi. We also demonstrate that cell death suppresses the silencing of mus308 and transposable ele-ments. The increased expression is accompanied by siRNA reduction and dsRNA accumulation, suggesting that the pro-cessing of dsRNA to siRNA is impaired.
Materials and Methods
Strains, genetic tests, and microscopy
All eye images were obtained using a dissecting microscope with·4 magnification with an attached digital camera. Ten to 30 flies of the same genotype were observed and repre-sentativeflies photographed.
The RNAi strains with homozygous GMR-wIR insertions on chromosomes X or 3 (Lee et al.2004) were kindly pro-vided by R. Carthew, Northwestern University, Evanston, IL. These strains were crossed to the multiple balancer strain Basc/Basc; In(2LR)SM1, al2 Cy cn2 sp2/In(2LR)bwV1, ds33K
dpOVbwV1; In(3LR)Ubx130,Ubx130es/In(3LR)C,Sb; svspa-pol. The eye phenotype was recorded in the F1withBarand in
the F2withsvspa-pol/svspa-pol. Also a male withGMR-wIRand
B was recovered. A GMR-wIR B strain was generated and tested to confirm that the X chromosome carried w+, B, and GMR-wIRby recombination with a regularw2X chro-mosome. The Bareffect in the males was also observed by using another strainy/BsY;tra2ts2bw/CyO(From B. Taylor
at Oregon State University, Corvallis, OR), which carries the Barmutation on the Y chromosome.
TheGMR-wIR / Blarvae were treated with acetamine as described (Fristrom 1972).
The RNAi stocks were crossed to the following strainsPr Dr/TM3; Sco/In(2L)Cyt, Cy amosRoi-1; P{w+mc.hs = GawB}
elavc155w* P{ry+t7.2= neo FRT} 19A; Bc1 EgfrE1/CyOand
bic L2/CyO. The eye phenotype of the F
1withDr,Roi (amosRoi-1),
Egfr or L combined with GMR-wIR was documented. The strain w+; Gla/SM6a; TM3, Ser/MKRS was crossed to a
strain carryingGMR-wIRon the X to produce the strainw+
GMR-wIR; Gla/SM6a; TM3, Ser/MKRS. The wgGla-1 (sub-sequently referred to as Gla) effect on RNAi was examined in the F1. This new strain was crossed to the stocks ofroand
hhbar3and the F
2with heterozygous or homozygous
muta-tions andwIRwas examined with or withoutGla.
The GMR promoting cell death transgenes including the five inducers and the two inhibitors (Table S1) (Xu et al. 2003) were kindly provided by B. Hay, California Institute of Technology, Pasadena, CA. These strains,GMR-diap1on the X, GMR-p35on the X,GMR-hidon 2,GMR-grim/TM3,Sb,
GMR-rpr/TM3,Sb,GMR-strica/CyO, andGMR-ttk88-myc(19)/CyO, were crossed to the RNAi strains and the F1phenotype was
recorded (all the transgenes carry aw+mcmarker).
To examine the combination of cell death inducers and inhibitors, the multiple balancer stock with GMR-wIR de-scribed above was first mated to males of inducer strains. The F1males carrying the respective inducer transgene and
GMR-wIR were then crossed to virgins of the inhibitor strains. The phenotype of the female offspring of these crosses was then analyzed and documented.
The symmetrically transcribing w RNAi strains w1118; SympUAST-w#8 and w1118; SympUAST-w#23/CyO, and the long stem-loop ones w1118; pUAST-IRsp-w#32/CyO and w1118; pUAST-IRsp-w#41 were kindly provided by E. Giordano (Giordano et al. 2002), Università di Napoli, Naples, Italy. These strains were first crossed to w+; Gla/ SM6a;TM3,Ser/MKRSto replace thewmutant gene on the X chromosome and to balance the transgenes on the second chromosome and double balance the third chromosome. To test whether B affects RNAi in these strains, virgins of the multibalancer strainBasc/Basc;In(2LR)SM1,al2Cy cn2sp2/
In(2LR)bwV1,ds33K dpOV bwV1; In(3LR)Ubx130,Ubx130es/In
(3LR)C,Sb;spapolspapolwere crossed to males ofy w/y+w;
UAS-EGFP Tub-Gal4/TM6b, and then the Stubble F1 males
withTub-Gal4andBascwere crossed to the balanced trans-genic strains. To simplify the genetic tests,GMR- and act5c-Gal4 trangenes on the second chromosome were chosen to recombine together in one chromosome with the UAS w RNAi transgenes. The new strains were then crossed with the cell death strains and F1phenotypes were assayed.
The UAS-wRNAiDS (on the third chromosome) strain (Kalidas and Smith 2002) and the GMR-Gal4 or act5c-Gal4 (on the second chromosome) strain werefirst crossed to the multibalancer strainw+;Gla/SM6a;TM3,Ser/MKRS. Then the F1flies were crossed together to produce a strain
containing both transgenes balanced on the second and third chromosomes. This strain was used to cross with the cell death strains and offspring phenotype was documented. The EGFPRNAi strainw[1118];P{w[+mC]=UAS-EGFP. dsRNA.R}142 (Roignant et al. 2003) was crossed with the GFP strain y w/w+; UAS-EGFP Tub-Gal4/TM6b, and the F
1
females were then crossed to the multibalancer strain y w67c23; Gla/SM6a; TM3, Ser/MKRS. The flies with
for cell death genes on the second chromosome, they were recombined together with bw. We also balanced the third chromosome constructs withMKRSorTM3,Ser. Finally, the EGFPRNAi strain was crossed to the cell death strains, in-dividually, and the phenotype of the offspring was digitally photographed using the GFP dissecting microscope in the University of Missouri (MU) cytology facility.
The transgenic hs-hid stock strains from Bloomington Stock Center (y1w*;Bl1L2/CyO, P{w[+mC]=hs-hid}4 on
chromosome 2 and y1 w*; Pr1 Dr1/TM3,
P{w[+mC]=hs-hid}14, Sb1 on chromosome 3) were crossed to the strain GMR-wIRto observe color restoration in the eyes. TheEGFP RNAi strain v; bw; Tub-Gal4 UAS-EGFP UAS-EGFPir/MKRS was crossed to the line with the third chromosome insertion or first crossed to the multibalancer strain w+; Gla/SM6a; TM3, Ser/MKRS and then crossed to the second chromo-some insertion to generate thehs-hidandEGFPRNAi strains. The flies were raised at 18. The third instar larvae were collected for dissection. For midgut analysis, larvae were col-lected before emerging from the food to avoid degradation that occurs at later stages (Jianget al.1997). Larval GFP was observed with the GFP dissecting microscope in the MU cy-tology facility. Dissected tissues were immediately mounted on slides withfluorescence mounting medium without cover-ing and photographed under a Zeiss Universal microscope with a MagnaFire cooled charge-coupled device camera.
MultipleGMR-wIRstrains were produced as follows. Both theGMR-wIR Bstrain and theGMR-wIR(on chromosome 3) strain were mated to the strain w+ GMR-wIR; Gla/SM6a;
TM3,Ser/MKRSand the offspring was mated with each other to obtain males with the genotype GMR-wIR B; GMR-wIR/ TM3,Ser(orMKRS). These males were mated to females of GMR-wIR(on chromosome 3) and the phenotypes of 1 (from F1), 2 and 3 (from F2) copies of GMR-wIR with B were
photographed.
For mitotic recombination in the eyes, strains were generated with a cell death inducing transgene (GMR-hid, -ttk, -grim, or -rpr) on the second or third chromosome recombined in the same chromosome with a corresponding neoFRT transgene (Xu and Rubin 1993). The original neoFRTstrains were modified to maintainw+on the X
chro-mosome and ey . FLP1 on the other autosome. These strains were then crossed to female w+ GMR-wIR; Gla/
SM6a; TM3, Ser/MKRSflies and male offspring were col-lected to mate with theneoFRTcell death strains. The phe-notypes of the offspring were documented. For simplicity, GMR-ttk eyes with clear boundaries between GMR-ttk and wild-type cell clones were chosen for illustration.
Acridine orange staining, Northern blotting, and RT–PCR
The transgenic hs-hid strains from the Bloomington Stock Center (y w*;BlL2/CyO, P{w+mC=hs-hid}4 and y w*; Pr
Dr/TM3, P{w+mC=hs-hid}14, Sb) and Canton-S were used
for acridine orange (AO) staining. The larvae were raised at 18and collected between the second instar to early third instar. Late third instar larvae were avoided because of
nor-mal degradation of the midguts (Jianget al.1997). Midguts were dissected and stained with AO (Jianget al.1997) and observed for greenfluorescence under a·4 objective lens of a Zeiss Universal microscope with a MagnaFire cooled charge-coupled device camera. For positive controls, larvae at the same age were used after heat shock at 37in a water bath for 30 min and then allowed to recover at 18for.1 hr. More than 10 pairs of Canton-S and hs-hid larvae and three heat shockedhs-hidindividuals were analyzed.
A total of 50–80 late third instar larvae were collected for TRIzol RNA isolation according to the manufacturer’s pro-tocol (Gibco BRL Life Technologies). If the RNA was for siRNA preparation, the RNA precipitation was placed on dry ice for 20 min before centrifugation to ensure that the small RNA was precipitated. Isolated RNA was dissolved in formamide at 55–65and stored at220. RNA concentra-tion was measured by 10- to 100-fold water diluconcentra-tion with a NanoDrop ND-1000 spectrophotometer.
EGFPmRNA and dsRNA levels were analyzed by North-ern blotting according to Augeret al.(2005). Twenty micro-grams of total RNA ofTub-Gal4 UAS-EGFP/TM6b,Tb(EGFP) and Canton-S (CS) and 5, 10, and 20 mg of total RNA of Tub-Gal4 UAS-EGFP UAS-EGFPir/MKRS (EGFPir) and Tub-Gal4 UAS-EGFP UAS-EGFPir/TM3,hs-hid Sb(hs-hid; EGFPir) were loaded in the gel. EGFP coding sequence amplified by primers WXo28 (AAG GGC GAG GAG CTG TTC AC) and WXo29 (CCA TGT GAT CGC GCT TCT CG) was cloned into pGEM-T Easy Vector (Promega) and sense and antisense EGFPprobes were transcribed from the plasmid DNA using MaxiScript kit (Ambion). a-Tubulin exon 2 sequence was amplified by primers WXo67 (TCT ATC CAT GTT GGT CAG GC) and WXo68 (GGT AGT TGA TGC CAA CCT TG) and cloned into pGEM-T Easy Vector. Antisense probe was then transcribed to detect a-tubulin mRNA on the striped membranes as input controls.
EGFP siRNA detection by Northern analysis was per-formed according to Pal-Bhadra et al. (2002). Ten micro-grams of total small RNA ofTub-Gal4 UAS-EGFP/TM6b,Tb (EGFP) and 2.5, 5, and 10mg of total small RNA ofTub-Gal4 UAS-EGFP UAS-EGFPir/MKRS (EGFPir) and Tub-Gal4 UAS-EGFP UAS-UAS-EGFPir/TM3, hs-hid Sb (hs-hid; EGFPir) were loaded in the gel. Sense and antisense EGFP probes were fragmented before hybridization. The 5S rRNA sequence was amplified by WXo89 [TAA TAC GAC TCA CTA TAG GGc caa caa cac gcg gtg t, T7 promoter attached (upper case)] and WXo90 (gcc aac gac cat acc acg c) from genomic DNA and then purified to transcribe an antisense RNA probe, which was used to detect 5S rRNA level as input controls. Locked nucleic acid (LNA) probes against miRNA and endogenous siRNAs were designed and synthesized by Exiqon with optimal Tm for Northern blots.
or exposed and scanned by a phosphorimager (Fujifilm Global) and analyzed by Multi Gauge. The fold difference between the death and no death samples was calculated in the formula: (Sh/Lh)/(S/L), in which Sh and S are the detected strength from a Northern blot for hidand no hid samples, respectively; Lh and L are the corresponding load-ing controls. When a sample with the detected signal strength was significantly less than the double amount of the twofold diluted sample, it was discarded as overex-posed. Samples of death and no death were paired for com-parison with the same amount of RNA transferred on the same membrane.
Late third instar “hs-hid; EGFPir” larvae raised at 18 were collected and heat shocked at 37in a water bath for 30 min and allowed to recover at 18 for 1 hr and then frozen at 280. Total RNA isolated from “EGFPir” larvae and heat shocked and not shocked “hs-hid; EGFPir”larvae was first precipitated from the formamide solution (25– 40 mg RNA in 5 ml plus 15ml 100% ethanol and 0.5 ml 5 M NaCl). The RNA was dissolved in 100 ml of nuclease free water and the concentration was measured with a Nano-Drop spectrophotometer. A total of 10mg RNA was treated with RQ1 DNase (Promega). First-strand cDNA was synthe-sized with SuperScript III reverse transcriptase (Invitrogen) and poly(T) primer. Primer WXo93 (aac aag cgc agc tga aca ag) were designed matching the UTSflanking sequence sub-sequent to the hsp70 promoter in the hs-hid transgene (Grether et al. 1995). Primer WXo91 (ga tga act cga cgc tac gtc) matches the coding sequence of hid exon 1. This pair of primers was used to amplify specifically the hs-hid transgene cDNA. PCR was terminated after 25, 30, and 35 cycles, respectively. PCR products were analyzed after gel electrophoresis and ethidium bromide staining.
Semiquantitative and real-time PCR
To study whether the expression of endogenous genes and transposable elements was changed, RNAs and cDNAs were prepared as described above from the larvae of“EGFPir”and “hs-hid; EGFPir” or Canton-S and hs-hid strains from the BloomingtonDrosophilaStock Center.
At least two pairs of primers were designed for each mRNA matching close to the 59 end to avoid possible de-tection of degraded 39fragments with a poly(A) tail, which can be reverse transcribed by a poly(T) primer. For semi-quantitative PCR, a series of dilutions of the cDNA (one-, two-, and fourfold) was used as templates for PCR to ensure that the stop point was within the range of exponential amplification. Consensus results were observed for different primers and for technical and biological replications.
To confirm the mus308expression change, real-time PCR was performed in an ABI 7300 system using the manufactor’s supplied Syber Green reaction master mix and the results were analyzed by the provided software. Multiple primer pairs were tested for the amplification efficiency and the most efficient ones (59-CTTAATGGCGCTGGAGAAAG-39 and 59-GGCCTGAT TCACTTCGTAGG-39) were used. A cDNA concentration range
was used for detecting differences in a dilution series. Three biological replications were analyzed.
Sequencing and analysis of small RNAs
Total RNA from “EGFPir”and “EGFPir hs-hid”third instar larvae wasfirst size separated by polyacrymide gel electro-phoresis and 15–40 nt molecules were collected for con-structing a cDNA library and then sequenced by a Solexa system in the MU DNA core facility. Primers (59-CGA CAGGTTCAGAGTTCTACAGTCCGACGATC (N)n TCGTATG CCGTCTTCTGCTTG-39) from both ends in the raw reads were removed and only the sequences with sizes between 19 and 31 were selected. Trimming was performed at either or both ends of the read for all or part of the primer/adapter sequence above. If the 39end of the read matched all or part of the left end of the right primer then that matched part was removed from the read. Similarly, if the 59end of the read matched all or part of the right end of the left primer then that matched part was removed from the read. No mismatches were allowed in any of the trimming.
The reads for each sample are mapped to Drosophila known miRNAs (from miRBase, http://microrna.sanger.ac. uk/sequences/index.shtml), Drosophila transposons (from flybase, http://flybase.org/) using BLAT (James 2002) with which the parameters used allow maximum sensitivity. After BLAT search, however, more stringent criteria were used to parse the results to retain only the highly confident align-ments. For miRNAs, only exact matches (same query/target sequence length and 100% sequence alignment identity) were counted for normalization. For si/piRNAs mapped to transposons, only fully mapped sequences with 100% iden-tity were counted. To further verify the si/piRNAs, anno-tated pi/siRNAs were downloaded from GEO datasets (GPL4738 and GPL6664) (Brenneckeet al.2007; Ghildiyal et al. 2008) and all cleaned sequences were mapped on them with BLAT (only the exact matches were counted).
Results
Eye mutations producing cell death restore eye color otherwise reduced by white RNAi
from 24 to 36 hr after pupal formation to remove the extra accessory cells, namely, the secondary and tertiary pigment cells (Brachmann and Cagan 2003).
An RNAi construct (GMR-wIR) (supporting information, Figure S1) repressing the expression of thewhite(w) gene reduces the eye color from brick red to light yellow (Figure 1A) (Leeet al.2004). In the process of manipulation of this construct, we crossed the RNAi stock to laboratory strains carrying dominant or recessive marker genes causing eye morphological abnormalities. Surprisingly, the normal red eye color was partially restored in Bar (B) and Drop (Dr) flies, and weakly restored in the presence of the Gla and sparkling-poliert (svspa-pol) mutations (Figure 1A and Table S1). Because the eye color change occurs in living cells and is adjacent to the missing portions in theBmutant eyes, we hypothesized that the inhibition of RNAi is caused by adja-cent tissue that has undergone cell death.
Bar and Drop are gain-of-function mutants, ectopically expressing homeobox genes BarH1/BarH2 or muscle seg-ment homeobox(msh), respectively, in the eye disc.Bar pre-maturely stops the MF movement beginning from the dorsal–ventral center and spreading laterally (Heberlein et al. 1993). Cell death occurs anterior to the arrested MF (Fristrom 1969) causing the adult phenotype. In thewRNAi andBflies, the restored eye color gradually diminishes pos-teriorly and laterally (Figure 1A), suggesting it may be caused by cell death in the adjacent region.
WhenB mutant larvae are raised on food containing 1– 2% acetamine, the mutant effect can be largely (but not completely) reversed by prolonging development, which sig-nificantly reduces cell death in the late third instar (Fristrom 1972). When female animals withBandGMR-wIRon the X chromosomes were treated with acetamine, reversal of RNAi is diminished as expected (Figure 1A).
the initiation of differentiation (Singhet al.2006) and does not affectwsilencing, suggesting the duration of the signal from cell death is temporally limited.
To rule out the possibility that cell death affectsw expres-sion as the basis of the above results, we crossedDrorBto hypomorphicwmutants including point mutationswa2and wa3 and insertion mutations, w-blood (wbl) and w-apricot (wa) (Zachar and Bingham 1982) and found no impact ( Fig-ure S2). These results indicate that modulation of thewhite gene itself is not the basis for the suppression. Another GMR-wIRtransgene inserted in chromosome 3, was tested withB and showed similar inhibition of RNAi (data not shown), indicating that the location of the RNAi transgene also does not affect the outcome.
Proapoptotic transgenes inhibit w RNAi depending on cell death
To test further the role of cell death signaling for the inhibition of RNAi, cell death was induced by overexpression of apoptosis promoting genes. The proapoptotic genesgrim, head involution defective (hid, or Wrinkled, W) and reaper (rpr), the caspase genestrica(ordream) and the transcription factortramtrack(ttk) have been shown to cause extensive cell death and irregular eye phenotypes when ectopically expressed by the GMR promoter (Figure 1B) (for example, Xuet al.2003). WhenwRNAi was also introduced into these strains, the expression of the w gene can be assayed in the remaining cells and was partially restored, as indicated by dispersed red spots or a centered dorsal–ventral strip of in-creased color in femalettkflies, without exception, when cell death occurred (Figure 1B, second row).
The double homozygous mutations ofvermilion (v) and brown(bw) are epistatic tow+and show pale-yellow eyes, thus resembling thewRNAi phenotype. When the cell death inducer transgenes were combined with these mutations, no pigment accumulation was found (Figure S3). This control indicates that the increase in pigment with the cell death transgenes has white RNAi as a target rather than being a consequence of altered eye morphology (e.g., closer ar-rangement of pigment cells).
The possibility that the antiapoptotic transgenes restore RNAi directly can be excluded by the following experiments. The cell death phenotypes can be variably suppressed, except for the overexpression ofttk, by simultaneous expres-sion of the apoptosis inhibitors Drosophila inhibitor of apo-ptosis 1(diap1orthread,th) or baculovirusp35(Figure 1B) (Xuet al.2003; Hayet al.2004). When cell death is reversed fully (as indicated by the eye shape and ommatidia arrange-ment returning to normal), RNAi ofwis restored; when the cell death was reversed partially, inhibition still occurs (Fig-ure 1B,hid+diap1). The transgenes diap1and p35do not suppress the ttk induced cell death; in this combination, RNAi is still suppressed, excluding the possibility that these antiapoptotic genes themselves restore the RNAi silencing. Also, crosses that introduce only the antiapoptotic trans-genes show no impact onwRNAi.
It is known that cells overexpressing hid will produce a proliferation signal to neighboring cells, a process not af-fected by the suppression of cell death by p35 (Huh et al. 2004; Pérez-Garijo et al. 2004; Ryoo et al. 2004). Indeed, the expression of p35 suppressed both cell death and the RNAi inhibition, indicating that this proliferation signal is independent of any signal affecting RNAi. In contrast, an-other proliferation signal produced by dying cells is affected by p35 and diap1 (Fan and Bergmann 2008), and thus it cannot be ruled out as coincident with a potential RNAi suppression signal.
Cell death inhibits w RNAi induced by diverse constructs
Different RNAi constructs were used to generate dsRNAs of thewgene (Giordanoet al.2002; Kalidas and Smith 2002) (Figure S1). These transgenes utilize an upstream activating sequence (UAS) for expression and silence the w gene to varying degrees when the transcription factor Gal4, which targets the UAS, is expressed by the promoters GMR, actin5c (act5c), or a-tubulin (Tub) (Giordanoet al. 2002; Kalidas and Smith 2002) (Figure S4). To confirm that the RNAi inhibition is not limited to certain constructs, we crossed these strains to the eye mutations B, Dr, and Gla, as well as to cell death strains overexpressing grim,hid,rpr,strica, andttk. Eye color restoration was observed in all the tested strains withstrica, and in all but one construct withGla,Dr, and grim(Figure S4andTable S2). However, in some cell death and RNAi combinations, no clear restoration was detected, especially with the constructs that alone show stronger silencing. Weaker pigment restoration was also ob-served when the silencing is achieved by a stronger pro-moter for Gal4, and different degrees of restoration were observed with different transgenic strains from the same RNAi construct (Figure S4andTable S2). These results sug-gest that the strength of the silencing plays a role in the magnitude of RNAi reversal. Moreover, because different promoters were used in these experiments, they demon-strate that downregulation of the GMR promoter of GMR-wIR by cell death signaling is not the basis of the restored eye color in the original observations.
Cell death suppresses GFP RNAi
restoration of GFP was observed in the eyes ofgrim,strica, and ttk (Figure 1C and Figure S5). Compared to the cell death control, which did not cause detectable autofl uores-cence in this assay (Figure S5), weaker and area-restricted restoration occurred in theDr,hid, andrpreyes.
The results of cell death induced inhibition ofwandEGFP RNAi are summarized in Table S2. Greater cell death is caused by overexpression of hidandrpr than that ofgrim, strica, andttk (as shown by the eye size); yet there is less RNAi reversal for both w and EGFP. The strica construct restored EGFPexpression to the strongest level as was also the case with restoration of wexpression. Thus, in general, the relative strength of reversal of RNAi is related to the mode of cell death but is independent of the genes (w or EGFP) involved.
RNAi inhibition is quantitatively related to the level of RNAi
As noted above, a difference in expression of an RNAi transgene affects the amount of restoredwexpression. We also noticed a weaker restoration observable with lesser ex-pression of Gal4 (e.g.,GMR-Gal4) driving an RNAi construct but not with more strongly expressed cases (e.g.,Tub-Gal4). Thisfinding suggests that a stronger RNAi effect is above the threshold of suppression. To test this possibility, we crossed multiple GMR-wIR transgenes with B and Gla. B and Gla show an additive effect on RNAi suppression. The results indicate that the restored color is negatively related to the copy number of theGMR-wIRtransgene (Figure S6).
The RNAi inhibition can be nonautonomous
The reversal ofwhiteRNAi in cells adjacent to the cell death sectors ofBar,bar3, andDr(Figure 1A) provided an indica-tion of the nonautonomy of RNAi inhibiindica-tion. As noted above, all these mutants cause a defect of the movement of the MF (Heberleinet al. 1993). The MF determines the fate of the cells: those cells encompassed by it will differentiate and remain alive in the adult eye and the remaining cells will be removed by cell death and will not be present in the adult eye. The color reversal observed in the adult eyes must be located in living cells, which did not undergo cell death. Therefore, the inhibition ofwRNAi in adult eyes must have been induced nonautonomously by the death of adjacent cells.
To examine this feature of inhibition by a second method, we produced mosaic cell death in the eyes. Mitotic re-combination by FLP–FRT (Golic 1991; Xu and Rubin 1993; Stowers and Schwarz 1999) was conducted in the eye imag-inal discs to resolve the zygotic heterozygous genotype into homozygous sectors of proapoptotic transgenes and normal cells using a promoter for FLP that acts before cell death, which was induced by the GMR-driven expression of the death inducer genes in the late third instar (see Materials and Methods). This technique produces mosaic eyes com-posed largely of two kinds of homozygous lineages: those for the death inducer construct and those for wild-type
chro-mosomes (Stowers and Schwarz 1999). After the adults eclosed, cell death sectors in the eyes can be observed as an irregular arrangement of ommatidia. The most useful in-volves the overexpression ofttk, which has a dominant phe-notype and causes a smooth eye surface due to cell death of specific cell types within the ommatidia. Areas with smooth surfaces were observed in the mosaic eyes, which allow sectors homozygous or occasionally heterozygous for the ttk construct to be identified definitively. The adjacent regions are homozygous for normal chromosomes; other-wise they would show a smooth eye surface. Thettksectors were accompanied by color restoration spreading from two to several lines of ommatidia, depending on the size of the cell death containing areas (Figure 2A andFigure S7). Thus, the inhibition of RNAi is occurring in the homozygous nor-mal cells, consistent with the pattern of color restoration in theB,hhBar3, andDreyes. The color intensity in the normal
sectors progressively decreases with the distance from the death sectors. Collectively, these data suggest the existence of a signal to suppress RNAi initiates from the region of cell death and travels a limited distance into regions with nor-mal cells.
In contrast, cell death induced by thehidtransgene in an analogous mitotic recombination experiment did not invoke RNAi inhibition in most cases, potentially indicating cell death does not inhibit RNAi under certain conditions (Figure 2B). Given that the same transgene is able to inhibit RNAi when heterozygous (Figure 1B) and the mitotic recombina-tion is effective at homozygosis at .90% (Stowers and Schwarz 1999), we suggest this difference is due to the rapid elimination of the dying cells in the case of the homo-zygous transgene. Thus, there is possibly a required thresh-old of duration of the dying cells to provoke substantial inhibition of RNAi.
Mosaics ofrprorgrimproduced by the FLP–FRT method exhibit colored sectors with the size usually not over an ommatidium in all eyes examined (Figure 2, C and D). In this case, the eyes from experiments involving homozygosis ofrprandgrimtransgenes do exhibit RNAi inhibition, pos-sibly becauserprandgrimtransgenes cause less severe cell death effects thanhid.
Cell death inhibits RNAi in different tissues
by the hs-hid transgene was that suppression of RNAi was reversed by adding thep35transgene to the genotype (Figure S8C). Collectively, these results demonstrate that the trans-gene was expressed and caused sporadic cell death.
To analyze the transgenichideffect onEGFPRNAi, third instar larvae were directly examined for greenfluorescence or following dissection into tissues and organs. Stronger EGFPexpression was obvious in larvae over all and in midg-uts, salivary glands, brains, and imaginal discs (Figure 3, B– F). Similar results were observed when hs-hid was located on either chromosome 2 or 3 balancers (Figure S9). This result indicates that the phenotype is indeed caused by the hs-hid gene and not by other factors on the balancer chro-mosomes, which without the transgene have no effect on RNAi. Thus, we conclude that cell death inhibition of RNAi operates in different tissues and developmental stages.
RNAi is inhibited by blocking the processing of dsRNA into siRNA
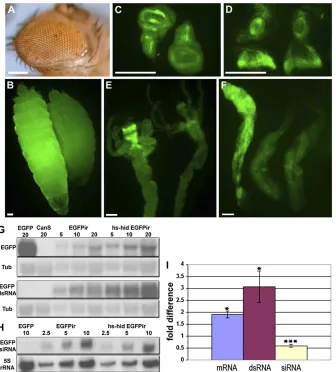
The expression change of EGFP in multiple larval tissues provides an opportunity to investigate mRNA, dsRNA, and siRNA changes during suppression of RNAi. As expected, the EGFP mRNA level in hs-hid transgenic animals increases about twofold (Figure 3, G and I). This change probably reflects a mixture of affected and unaffected cells, so the change in the affected cells is likely of much greater magni-tude. Interestingly, we detected more than a threefold
in-crease of the dsRNA in thehidtransgenic strain (Figure 3, G and I). This accumulation is likely caused by impairment of EGFPdsRNA processing. Consequently, we expected to see an siRNA decrease. Indeed,EGFPsiRNA is reduced40% in the transgenic strain (Figure 3, H and I). The results suggest the target step of RNAi suppression is the conversion of dsRNA to siRNA.
Adenosine deaminase acting on RNA (ADAR), an RNA editing enzyme, has been shown to compete for dsRNA as a substrate and thus inhibit RNAi (Scadden and Smith 2001; Wanget al.2005; Healeet al.2009). ADAR edits the dsRNA (Carpenter et al. 2009) and makes it unfavorable as a sub-strate for Dicer-2, the enzyme component of RNAi that processes dsRNA (Scadden and Smith 2001). Thus, we ex-amined the possibility that ADAR might be induced by cell death signaling and compete with RNAi for dsRNA substrates. However, this possibility was excluded because there is no increase in expression of ADAR accompanying cell death, and whenADARexpression is reduced by RNAi, cell death-mediated RNAi inhibition was not affected (Figure S10).
Cell death inhibition of RNAi regulates endogenous gene and transposable element expression
Recent work shows that siRNAs exist in somatic cells and match selected endogenous genes and transposable ele-ments. Among those genes, mus308, encoding an enzyme with DNA polymerase and helicase function, has been demonstrated to be repressed by RNAi (Czech et al.2008; Okamura et al. 2008). To investigate whether cell death upregulatesmus308by its inhibition of RNAi, RT–PCR was performed to detect the expression level of this gene in the hs-hid transgenic larvae. A doubling of the expression was observed by semiquantitative PCR, which was confirmed by real-time PCR (Figure 4A). This result is similar to theEGFP upregulation and suggests that endogenous genes under RNAi control can be affected in the same manner.
When RNAi is compromised, previous studies have shown that a severalfold increase of expression for some TEs was detected in S2 cells (Czech et al.2008; Ghildiyal et al.2008; Kawamuraet al.2008) and in adultflies (Chung et al.2008). We examinedfive of them, 1731, mdg1, 297, BEL, DOC, and S element to analyze the possible effect from cell death. It was demonstrated that dicer-2 knockout did not significantly affect 1731 and mdg1 expression in adult flies, whereas 297 and BEL mRNAs are increased, although both mdg1 and 297 siRNA levels are reduced (Chunget al. 2008). Our results also show BEL and 297 expression in-creased approximately twofold (P, 0.05) in the presence ofhs-hid–caused cell death, whereas mdg1 and 1731 expres-sion is not significantly changed, and DOC and S may be slightly upregulated (P,0.1) (Figure 4B). These results are consistent with the hypothesis that cell death reduces siRNA synthesis and thus impairs RNAi.
To further confirm the impact of hs-hid on siRNAs and miRNA, LNA probes were synthesized (Exiqon) for de-tecting let-7, miR-1, and esiRNA-sl-1 (the siRNA molecule Figure 2 Cell death caused RNAi inhibition in neighboring cells. Mosaic
complementary to mus308 mRNA; Kawamuraet al. 2008) by Northern blotting (Chung et al.2008). A change in the amount of a control miRNA was not detected (let-7 in Figure 4C). However, esiRNA-sl-1 was reduced to 45% of the control as also found for the EGFP siRNA (Figure 4C).
To determine whether any global change occurred for TE siRNAs, we sequenced the small RNAs (19–40 nt) from the hs-hid transgenic and control strains. The read numbers of sequenced RNAs matching TEs werefirst normalized by to-tal miRNA reads, then plotted on the basis of their size (Figure 4D, original data see Table S3). Peaks for siRNA (21 nt) and for piRNA (23–29 nt) were clearly evident. The siRNA peak is reduced to43% (averaged) in cell death samples. When the 21-mer set (excluding miRNAs) was compared for homology to the published siRNA/miRNA dataset fromfly heads (Ghildiyal et al.2008), we observed
40% reduction of matched siRNAs with cell death (620 to 372 reads). The 21-mer siRNA molecules derived from gene CG4068, which were shown to silence mus308 (Okamura et al.2008), are also reduced (35 to 13 reads) (seeTable S3 for total siRNA number derived from CG4068), matching the Northern results. There is also possibly a weaker reduc-tion in total piRNA amount (Figure 4D andTable S3).
How-ever, we were not able to detect a significant reduction of EGFP siRNA level in thehs-hidstrain by this method (Table S3) for unknown reasons, although this might be the case if the high expression ofEGFPis at saturating amounts. Over-all, our results indicate that the expression change of mus308 and TEs is caused by a genome-wide reduction of siRNAs.
Discussion
We demonstrate in this study that cells undergoing death inhibit RNAi. This process occurs in different stages of the life cycle, in different tissues, and for selected genes silenced by RNAi. Various constructs that generate dsRNA were all found to be effective, suggesting this step is not affected. Indeed, we observed accumulation of dsRNA and reduction of siRNA, indicating that the processing of dsRNA to siRNA is the step impaired. Either modification of dsRNA or Dicer-2 and/or its cofactors may be the target of the cell death signaling. However, we were not able to detect a change of Dicer-2 expression at either the transcriptional or protein level (Figure S11). The phenotype of the eye color suggests this is not an all-or-none regulation, but rather the restored Figure 3Cell death-related inhibition of RNAi occurs in different tissues and organs and the increased expression of the marker geneEGFP is caused by impaired processing of dsRNA into siRNA. (A–F). The default expression of the transgene hs-hid inhibits w silencing in the eye (A) andEGFPsilencing in larvae (B), wing disc (C), eye-antennal disc (D), midgut (E), and salivary glands (F). In each image except the eye, controls without hs-hid but with EGFP RNAi are located to the right. A salivary gland showing restored fluorescence and a control pair of glands is shown in the lower right. Bars, 0.1 mm. (G–I). Northern blotting detects altered levels of EGFP mRNA, dsRNA (G), and siRNA (H) in thehs-hidstrain. A dilution series (1X, 2X, and 4X) of total RNA or total small RNA was loaded and indicated by the numbers of micro-grams. Levels ofa-tubulin(Tub) mRNA (G), and 5S rRNA (H) were probed, respectively, as input controls. The patterns of siRNA were not differ-ent when probed with either sense or antisense EGFP probes (not shown). Statistical analysis of the EGFP mRNA, dsRNA, and siRNA levels in
color in the eye is quantitatively reversed relative to the silencing strength ofwRNAi.
We demonstrate that cell death inhibits RNAi nonauton-omously in neighboring normal cells. The inhibition is conditionally triggered in that when strong cell death was induced, little or no RNAi inhibition was observed. We assume that a threshold of duration of the cells undergoing cell death is required for the effect. This time-lag hypothesis is consistent with the fact that processing of dsRNA to siRNA is reduced and thus caused the RNAi inhibition. Therefore, the lag time may be related to the generation and perdurance of the siRNA molecules. This hypothesis also explains three other observations. First, we did not observe GFP RNAi inhibition during normal embryo development in which programmed cell death occurs. Second, when the hs-hid transgenic larvae were heat shocked, they died within 1 day. During this period of time, we were not able to detect any GFP expression difference in the GFP RNAi animals com-pared to the controls that were not heat shocked. Third, we also failed to detect a GFP expression change in the eye discs with EGFP RNAi in the late third instar when proapoptotic genes were expressed by the GMR promoter, that is, imme-diately after ectopic cell death was induced. However, it is important to note that when reversal of RNAi occurred, it did so in normal cells as evidenced from the results from eye mutations and the somatic recombination mosaic analysis.
One possibility for the existence of cell death induced inhibition of RNAi might be that RNAi acts as afirst line of defense against viruses (Li and Ding 2005). If this fails and
cell death results from viral proliferation, signaling might occur to neighboring cells to induce processes that modify dsRNA as a second line of defense, making them inaccessible for processing to siRNA, so that they cannot enter the RNAi pathway leaving the integrity of the target mRNA intact, analogous to the interferon response in mammals (Kawai and Akira 2007; Randall and Goodbourn 2008). This hy-pothesis can account for the collective data that indicate a quantitative negative relationship between the cell death signal and the effectiveness of RNAi. Such a second line of defense would be selected if RNAi regularly failed to stop viral infection, which is obviously the case, and would pro-vide a different means of attacking dsRNA.
Apoptosis is known to play a role in immunity to viruses in insects in that the apoptotic response can severely limit viral replication and thus many viruses encode antiapoptotic genes (reviewed in Clarke and Clem 2003; Clem 2005). Recent data on viral infection in Drosophilashow that long dsRNAs but not the siRNAs stimulate whole-body immunity to homologous virus infection (Salehet al.2009). Thus, the existence of cell death induced inhibition of RNAi causing local maintenance of dsRNA could possibly be beneficial for viral defense by RNAi throughout the body if localized path-ogen-induced cell death occurs.
et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008). Our findings suggest that chronic ectopic cell death inhibition of RNAi also elevates TE expres-sion. This finding suggests that chronic diseases or long-term exposure to pathogens, which might cause cell death in the stressed tissue, may cause activation of endogenous TEs in the adjacent normal cells. It has been documented in yeast cells under stress, that the retrotransposon Ty5 will randomize its integration, which otherwise mostly targets heterochromatin, and thus become mutagenic (Dai et al. 2007). Similarly, the activation of TEs in cells surrounding cell death may increase the somatic mutation frequency. Therefore, the inhibition of RNAi induced by chronic ectopic cell death may provide insight for the genesis of certain types of diseases, such as some cancers, in which persistent infl am-mation and somatic mutations are implicated (Cooper 1995; Coussens and Werb 2002; Copeland and Jenkins 2009).
Lastly, RNAi is widely used in genetic analysis to eliminate specific gene function and many practical appli-cations of this tool have been suggested (Aigner 2007). The finding that chronic ectopic cell death alters the pattern of RNAi is critical for an understanding of the application of the technique in basic and applied studies.
Acknowledgments
We thank R. Carthew, B. Hay, E. Giordano, C. Antoniewski, T. Zars, and C. Tan and the Bloomington DrosophilaStock Center for providingfly strains. We thank Ryan Donohue for performing some PCR experiments, Sean Blake for sequenc-ing small RNAs, and William G. Spollen for generatsequenc-ing and trimming the sequence data. We also thank Kyungju Chin, Robert Gaeta, and Louis Mega for advice.
Literature Cited
Aigner, A., 2007 Applications of RNA interference: current state and prospects for siRNA-based strategies in vivo. Appl. Micro-biol. Biotechnol. 76(1): 9–21.
Aravin, A. A., G. J. Hannon, and J. Brennecke, 2007 The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 318: 761–764.
Auger, D. L., A. D. Gray, T. S. Ream, A. Kato, E. H. Coe Jr.et al., 2005 Nonadditive gene expression in diploid and triploid hy-brids of maize. Genetics 169: 389–397.
Berghammer, A. J., M. Klingler, and E. A. Wimmer, 1999 A uni-versal marker for transgenic insects. Nature 402: 370–371. Brachmann, C. B., and R. L. Cagan, 2003 Patterning thefly eye:
the role of apoptosis. Trends Genet. 19: 91–96.
Brennecke, J., A. A. Aravin, A. Stark, M. Dus, M. Kellis et al., 2007 Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128(6): 1089–1103. Carpenter, J. A., L. P. Keegan, L. Wilfert, M. A. O’Connell, and F. M.
Jiggins, 2009 Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae). BMC Genet. 10: 75.
Chung, W.-J., K. Okamura, R. Martin, and E. C. Lai, 2008 Endo-genous RNA interference provides a somatic defense against Drosophila transposons. Curr. Biol. 18: 795–802.
Clarke, T. E., and R. J. Clem, 2003 Insect defenses against virus infection: the role of apoptosis. Int. Rev. Immunol. 22(5–6): 401–424.
Clem, R. J., 2005 The role of apoptosis in defense against bacu-lovirus infection in insects. Curr. Top. Microbiol. Immunol. 289: 113–129.
Cooper, G. M., 1995 Oncogenes. Jones and Bartlett Publishers, Boston.
Copeland, N. G., and N. A. Jenkins, 2009 Deciphering the genetic landscape of cancer-from genes to pathways. Trends Genet. 25: 455–482.
Coussens, L. M., and Z. Werb, 2002 Inflammation and cancer. Nature 420: 860–867.
Czech, B., C. D. Malone, R. Zhou, A. Stark, C. Schlingeheydeet al., 2008 An endogenous small interfering RNA pathway in Dro-sophila. Nature 453: 798–802.
Dai, J., W. Xie, T. L. Brady, J. Gao, and D. F. Voytas, 2007 Phos-phorylation regulates integration of the yeast Ty5 retrotranspo-son into heterochromatin. Mol. Cell 27(2): 289–299.
Fan, Y., and A. Bergmann, 2008 Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and dif-ferentiating tissues in the Drosophila eye. Dev. Cell 14(3): 399–410.
Fristrom, D., 1969 Cellular degeneration in the production of some mutant phenotypes in Drosophila melanogaster. Mol. Gen. Genet. 103: 363–379.
Fristrom, D., 1972 Chemical modification of cell death in the Bar eye of Drosophila. Mol. Gen. Genet. 115: 10–18.
Ghildiyal, M., H. Seitz, M. D. Horwich, C. Li, T. Duet al., 2008 Endo-genous siRNAs derived from transposons and mRNAs in Dro-sophila somatic cells. Science 320: 1077–1081.
Giordano, E., R. Rendina, I. Peluso, and M. Furia, 2002 RNAi triggered by symmetrically transcribed transgenes inDrosophila melanogaster. Genetics 160: 637–648.
Golic, K. G., 1991 Site-specific recombination between homolo-gous chromosomes in Drosophila. Science 252: 958–961. Grether, M. E., J. M. Abrams, J. Agapite, K. White, and H. Steller,
1995 The head involution defective gene of Drosophila mela-nogaster functions in programmed cell death. Genes Dev. 9(14): 1694–1708.
Hay, B. A., J. R. Huh, and M. Guo, 2004 The genetics of cell death: approaches, insights and opportunities inDrosophila. Nat. Rev. Genet. 5: 911–922.
Heale, B. S. E., L. P. Keegan, L. McGurk, G. Michlewski, J. Brindle
et al., 2009 Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 28: 3145–3156.
Heberlein, U., T. Wolff, and G. M. Rubin, 1993 The TGF beta homolog dpp and the segment polarity gene hedgehog are re-quired for propagation of a morphogenetic wave in the Drosoph-ila retina. Cell 75: 913–926.
Huh, J. R., M. Guo, and B. A. Hay, 2004 Compensatory prolifer-ation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr. Biol. 14(14): 1262–1266.
James, K. W., 2002 BLAT–the BLAST-like alignment tool. Genome Res. 12(4): 656–664.
Jiang, C., E. H. Baehrecke, and C. S. Thummel, 1997 Steroid regulated programmed cell death during Drosophila metamor-phosis. Development 124: 4673–4683.
Kalidas, S., and D. P. Smith, 2002 Novel genomic cDNA hybrids produce effective RNA interference in adultDrosophila. Neuron 33: 177–184.
Kawai, T., and S. Akira, 2007 Antiviral signaling through pattern recognition receptors. J. Biochem. 141: 137–145.
Lee, Y. S., K. Nakahara, J. W. Pham, K. Kim, Z. He et al., 2004 Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117: 69–81.
Li, H. W., and S. W. Ding, 2005 Antiviral silencing in animals. FEBS Lett. 579(26): 5965–5973.
Mozer, B. A., 2001 Dominant Dropmutants are gain-of-function alleles of themuscle segment homeoboxgene (msh) whose over-expression leads to the arrest of eye development. Dev. Biol. 233: 380–393.
Okamura, K., W.-J. Chung, J. G. Ruby, H. Guo, D. P. Bartelet al., 2008 The Drosophila hairpin RNA pathway generates endog-enous short interfering RNAs. Nature 453: 803–806.
Pal-Bhadra, M., U. Bhadra, and J. A. Birchler, 2002 RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing inDrosophila. Mol. Cell 9(2): 315–327. Pérez-Garijo, A., F. A. Martín, and G. Morata, 2004 Caspase
in-hibition during apoptosis causes abnormal signalling and devel-opmental aberrations in Drosophila. Development 131(22): 5591–5598.
Plautz, J. D., R. N. Day, G. M. Dailey, S. B. Welsh, J. C. Hallet al., 1996 Greenfluorescent protein and its derivatives as versatile markers for gene expression in living Drosophila melanogaster, plant and mammalian cells. Gene 173: 83–87.
Postigo, A., and P. E. Ferrer, 2010 Viral inhibitors reveal overlap-ping themes in regulation of cell death and innate immunity. Microbes Infect. 11(13): 1071–1078.
Randall, R. E., and S. Goodbourn, 2008 Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89(Pt 1): 1–47. Reifegerste, R., and K. Moses, 1999 Genetics of epithelial polarity
and pattern in the Drosophila retina. Bioessays 21: 275–285. Roignant, J.-Y., C. Carré, B. Mugat, D. Szymczak, J. A. Lepesant
et al., 2003 Absence of transitive and systemic pathways allows cell-specific and isoform-specific RNAi in Drosophila. RNA 9: 299–308.
Ryoo, H. D., T. Gorenc, and H. Steller, 2004 Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7(4): 491–501. Saleh, M. C., M. Tassetto, R. P. van Rij, B. Goic, V. Gaussonet al.,
2009 Antiviral immunity in Drosophila requires systemic RNA interference spread. Nature 458: 346–350.
Scadden, A. D., and C. W. Smith, 2001 RNAi is antagonized by A/I hyper-editing. EMBO Rep. 2(12): 1107–1111.
Singh, A., X. Shi, and K. W. Choi, 2006 Lobe and Serrate are required for cell survival during early eye development in Dro-sophila. Development 133(23): 4771–4781.
Stowers, R. S., and T. L. Schwarz, 1999 A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics 152: 1631–1639. Tam, O. H., A. A. Aravin, P. Stein, A. Girard, E. P. Murchisonet al.,
2008 Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453: 534–538. Wang, Q., Z. Zhang, K. Blackwell, and G. G. Carmichael, 2005 Vigilins
bind to promiscuously A-to-I-edited RNAs and are involved in the formation of heterochromatin. Curr. Biol. 15: 384–391.
Watanabe, T., Y. Totoki, A. Toyoda, M. Kaneda, S. Kuramochi-Miyagawa et al., 2008 Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453: 539–543.
Xu, P., S. Y. Vernooy, M. Guo, and B. A. Hay, 2003 The Drosophila microRNA Mir-14 suppresses cell death and is required for nor-mal fat metabolism. Curr. Biol. 13: 790–795.
Xu, T., and G. M. Rubin, 1993 Analysis of genetic mosaics in devel-oping and adult Drosophila tissues. Development 117: 1223–1237. Zachar, Z., and P. M. Bingham, 1982 Regulation of white locus expression: the structure of mutant alleles at thewhitelocus of
Drosophila melanogaster. Cell 30: 529–541.
Zamore, P. D., and B. Haley, 2005 Ribo-gnome: the big world of small RNAs. Science 309: 1519–1524.
GENETICS
Supporting Information http://www.genetics.org/content/suppl/2011/05/19/genetics.111.128470.DC1
Inhibition of RNA Interference and Modulation
of Transposable Element Expression by Cell Death
in
Drosophila
Weiwu Xie, Chengzhi Liang, and James A. Birchler
$#()'*)(*(#)(()*.023023#$#()'*)*(-$#8$'#+') '%)(/,((%'$#(()#$#)'$#72 755930230$#()'*)(#)$)'#(' (."")'!!.)-$#1''"#)24609 3#!*#-$#(719##)'$#(7182 755730230 ("'"#)'$"23*()$" #+')'%)(/,'(%').(%''"#)2 755730230#$)'$#()'*)$#)#(%')$-$#7/#)'$#7/-$#8##)'$#8/,'!# )$) $''(%$##$%%$()!.$'#)2# 755730230$#()'*)')!.!# () #+')'%)($)$#(&*#,)$*)(%'2 755830',#('#$))$(!0 !*''$,(#)))'#('%)$#$'#))$#$)$#()'*)(0! ''$,(($,)$'#!)'#('%)$# ')$#($)#'"#)(0
wRNAi#8 and
#23
wRNAi#32 and
#41
EGFPir
wRNAiDS
GMR-wIR
B
A
D
C
E
UAS w ~1.4 kb w ~1.4 kb SV40 Exon 3
GMR HSP70
Exon 3
UAS Exons 2-4
Exons 4-2 HSP7 0
UAS HSP7
0
EGFP
EGFP
SV40 UAS
UAS SV40
""%'(#" "'( &
"" '(% #" '
% %'(
"'( &"" '(% %
" " % '
! '% % )+1+./*
()+0-* ! )!-*#"%&'(
#$# ' ! # %%
% % ! +1&+./+0-!#' (
"#'$ *& !! "!)#"" #(#"*
!#!
" $! "# "$
&$$# $& &$%$ +-.+,1"&(!"##
$"#*'&"$"!$ "$'# #$"%$%"!" $"* # $$"#$ ") ")#+-.+, #$"$#$$$# #$"%$ '"#$ &'$ %$1(!"## +$$2,1*#"%& "+" "*$#"#%$ "#$$$ #$"%$#%$ +-.+$ "'$ %$$-#,.*(!"## # $
$"&+(!"## ## '#!%$ $" #+%"#/*01# '$
%##$!$ "+#$ #$"$$$ %$ $ %## $
FigureS11 ".5(!"## # $#$)$$#$"$$ "!" $&,
/0,".5" "$# $" "'###)).,4+56"&"#.
$"#"$ $'#%##$!$ "#!,& .%%'## ##)#!%$
$" ,/0,".5!" $&'###))#$" $, ) $ )#$".5/0$$
".5#1488,"#$""&'" $ *4!%#4+43; )" !$!" $#$ "/4!$-430, %$ $!" $'# $$"45+333
$"%$ "43%$#,45,7μ+57μ73μ $ $!" $'# "#!,#
"'#"%# "$$.%%!" $&,
$ +%* &% ##* (%
&$ %%* %6&6+%* &%2+'# * &%&' (& &$&&.%) *% +1('()) (%* * &% %%*( &('&(* &%&*/2+)
" %/6&((6# "/
) 4( )*(&$1''"'1?B?7=EBE84
4 ) ! $1"'1!$'1A<7=EE>84 4 (# %14+ %1 1E=?
7=EE?84 &$ %%* %6&6+%* &%2&,(.'()))#!
!"&7!8%1(()*)$&('&%* +((&-78$&,$%*1%+)),(/)$##/
) 4 (# %14+ %1 1E=?
7=EE?84
44&0(1$''1?D<7><<=84 &$ %%* %6&6+%* &%2*&' .'()) &%&
%!!+( %'+'#)*2+)))$&&*/
) 4(+%%(1"'1$''1=CD7=EEE84
, +*%*&%2&$&0/&*)+)
'%&*/') $ #(*&4 $ #(,#&'$%*# %/-)&)(,4
$'# 4 (# %14+ %1 1E=?
7=EE?84
&$$* #((/ ) )(+'*%*%+$(& '&*&('*&(##)&%&$$* +$ )&*%
(+4
$'# 4%*&144/)14#&0 "1'$4
7=1>81A=7=EED84
! +*%*&)&+%2+)).*(*&&%
%' $%*##)2)$&&*%(&-%&**/4 ) 44 ##1"'1$'!$'1=<C7><<?84
+ &$ %%*%* ,$+*%*%&)6)%) * ,2
$ *)") %# %2,%*(#(&-**% +)))$##/)
%(#/* ( %)*(&()&%
%)*(
44(%1464& 1$""1@<<A 7><<>84
4 %14 444& 1$"7>?81 @CC=7><<B84
&$ %%* %6&6+%* &%$+*%*&%!2
&+%*(*)% 2+)),( %*%+$(& '&*&('*&(%&%##) %%&$$* +$%
(&+/
&*# "#/ 4 (# %14+ %1 1E=?
7=EE?84
4%+*1"'1"!1B>?7><<>84
* #(&+/'%&*/'+*& )(+'*
&$$* #((/ &*# "#/ 44"(144+ %1"# 3=A<7=EDE84
) (&6'&'*&* %2.'()) %*/ ) 44 -" %)"41 ''' 1>C<D@
7><<<84
) (&6'&'*&* %2.'()) %*/ ) 44(*(1"41!$'"1=BE@7=EEA84
) (&6'&'*&* %2.'()) %*/ ) 44 /144))($%144+ %1!1
=>A?7=EEA84
)!" )')%2.'()) %*/ ) 4&+$% )1*#41" '!1?DC7><<=8
4+1"41# '41CE<7><<?84 )"".. (%)( '* &%*&((#**&('*-/2
.'()) %*/ ) 4 4%1*#41"1@AE7=EEC84
)* % *)'&'*&) )/( %)'))2.'()) %
*/
% *&( 44 /144))($%144+ %1!1
=>A?7=EEA84
),- % *)'&'*&) )2.'()) %*/ % *&( 44 /14+ %1$"1
>=>=7=EE@84 99&($&( %&($* &%1((*&#/)3**'355---4#/)4%*4
-( -( ,! -( -( -( -( (
' & ,! ,! -( +'-,! $005 ,! ,!
%' & $% ,! $% ,! $%
' & $%
' "# ,! -( ()'%$ ()'%$ -( ()'%$ ()'%$ -(
%' "# -( -( ()'%$ -( ()'%$ -(
' "# $%
' #" $% +'-,! +'-,! ,! $% $% -( $%1,!
%' #" $% +'-,! $% $% -( $%
' #" $%
' $! $% ,! ,! -( $% +'-,! -( $%1,!
%' $! $% -(
' $! $%
' $% +'-,! $% $% $% ,! $%
%' $% $% $% $% $% $%
' ,! ,! -( ,! ,! -( -(
%$()'*)(' )'' %'&'%#%)'0 '6@6 $ )())"=)'$($' +$-.)>%'*&'%#%)',('%# $)%)(#'%#%(%#, ))'$($07?.7;<.7<;.7=:$
' %$()'*)('%#0 %'$%03;99;4$0# )3$ ;99;40 '%$()'*)
('%#020% $$)3;99<40%#%# $) %$(,'$%))()., '"$! $))"0
+/ '+/ +0 '+0
0 24/2273 255/.36 2563317 220.710
//416/ //7//0 /24237 7//.3
! **
0. 43 44 16 16 61 45 00 07
0/ 471 5.2 074 072 /.11 612 06. 141
00 //2 //4 5. 5. /24 //6 14 25
01 53 54 60 6/ /1. /.3 41 60
02 /60 /63 /21 /20 0/5 /53 /00 /36
03 1.7 1/2 030 03. 23. 141 /6/ 013
04 107 112 061 06/ 217 132 0/0 053
05 0/. 0/1 /60 /6/ 1/7 036 /41 0/0
06 /17 /2/ /20 /2/ 0.0 /41 /.7 /2/
07 31 32 50 5/ //4 72 3/ 44
1. 2/ 20 16 16 66 5/ 12 22
1/ 04 04 /4 /4 26 17 10 20
!
0. 153 123 525 223
0/ 2470 272/ 7654 3574
00 4.2 437 /2.. 544
01 51 43 /40 53
02 04 04 2. 01
03 /6 0. 30 /6
04 /0 /3 01 /2
05 /3 // 00 /.
06 /0 01 12 /6
07 6 /2 23 6
1. 6 /4 05 0.
1/ 5 6 07 /1
! 2.46
0. . / / /
0/ /5 6 02 4
00 /1 7 0. 3
01 / 0 / 1
02 / . . .
03 / . . .
04 . . . .
05 / . . .
06 . . . .
07 . . 0 .
1. . . / .
1/ . . . .
! $( ) &"" 0/ &
13313133131311:88<#*+#&+)'$* '))'*'("#$#)59&#)5:#&+"
*#4%#*#$&#&!(+"./*3$$2=@5?93
131313&31:88:#+)#!!)/*/%%+)#$$/+)&*)#+)&*!&*#&
3 &+#*2=;>5=<?3
13&331:88:'-$!&'%#"/)#*()', +#-#&+) )&#&,$+3
,)'&29>>59?<3
13531313 13133&31:88;*&' +)&*#+#-&
*/*+%#(+"./*$$'.*$$5*(# #&#*' ')%5*(# ###&)'*'("#$3@2:@@5;8?3