research communications
Acta Cryst.(2014).E70, 203–206 doi:10.1107/S1600536814020169
203
Received 30 August 2014 Accepted 7 September 2014
Edited by W. T. A. Harrison, University of Aberdeen, Scotland
†
Keywords:crystal structure; pyrimidine–piper-azine heterocyclic salts; chloride salt; nitrate salt; bifurcated hydrogen bonds
CCDC references:1023201; 1023202
Supporting information:this article has supporting information at journals.iucr.org/e
Crystal structures of
4-(pyrimidin-2-yl)piperazin-1-ium chloride and 4-(pyrimidin-2-yl)piperazin-1-4-(pyrimidin-2-yl)piperazin-1-ium
nitrate
Thammarse S. Yamuna,aJerry P. Jasinski,b* Manpreet Kaur,aBrian J. Andersonband H. S. Yathirajana
aDepartment of Studies in Chemistry, University of Mysore, Manasagangotri, Mysore 570 006, India, andbDepartment of
Chemistry, Keene State College, 229 Main Street, Keene, NH 03435-2001, USA. *Correspondence e-mail: jjasinski@keene.edu
The title salts, C8H13N4 +
Cl, (I), and C8H13N4 +
NO3
, (II), contain linked pyridinium–piperazine heterocycles. In both salts, the piperazine ring adopts a chair conformation with protonation at the N atom not linked to the other ring. In the crystal of (I), weak N—H Cl interactions are observed, leading to zigzag chains along [100]. In the crystal of (II), both H atoms on the NH2
+
group form bifurcated N—H (O,O) hydrogen bonds. Weak C—H O interactions are also observed. These bonds collectively link the components into infinite chains along [100].
1. Chemical context
Pyrimidine-containing compounds exhibit various biological activities (Goldmann & Stoltefuss, 1991) and related fused heterocycles are unique classes of heterocyclic compounds that exhibit a broad spectrum of biological activities such as anticancer (Amin et al., 2009; Pandey et al., 2004), antiviral (Ibrahim & El-Metwally, 2010), antibacterial (Kuyper et al., 1996) and anti-oxidant (Padmajaet al., 2009), antidepressant (Kimet al., 2010) and possess anti-inflammatory effects (Clark
et al., 2007). In addition, several piperazine derivatives have reached the stage of clinical application; among the known drugs that are used to treat anxiety is a pyrimidinylpiperazinyl compound, buspirone (trade name BuSpar1) (Tollefsonet al., 1991). Our research group has published a number of papers on incorporated heterocyclic ring structures,viz.imatinibium dipicrate (Jasinski et al., 2010), 1-(2-hydroxyethyl)-4-[3-(2-trifluoromethyl-9H -thioxanthen-9-ylidene)propyl]piperazine-1,4-diium dichloride, which is the dihydrochloride salt of flupentixol (Siddegowda et al., 2011a) and opipramolium fumarate (Siddegowda et al., 2011b). Other related crystal structures are 4-(pyrimidin-2-yl)piperazin-1-ium (E )-3-carb-oxyprop-2-enoate (Yamunaet al., 2014a), flupentixol tartarate and enrofloxacinium oxalate (Yamunaet al., 2014b,c). As part of our ongoing studies in this area, we report herein the crystal structures of the title salts, (I) and (II).
2. Structural commentary
The structure of (I) and its atom numbering are shown in Fig. 1. It consists of a pyrimidylpiperazine cation joined by the C1/N3 atoms of each unit and a chloride anion. The C1—N3 bond is 1.373 (3) A˚ long, which compares favorably with similar ionic structures containing this cation [1.369 (3) (Yamunaet al., 2014a), and 1.36 (6) and 1.37 (1) A˚ (Dinget al., 2014)]. The N3/C5/C6/N4/C7/C8 piperazine unit adopts a slightly distorted chair conformation with protonation at the N4 nitrogen atom. The structure of (II) and its atom numbering are shown in Fig. 2. Similarly, it consists of a pyrimidylpiperazine cation joined by the C1/N3 atoms of each unit and a nitrate anion. The C1—N3 bond is 1.369 (3) A˚ , also in the range of the related structures described above. The N3/ C5/C6/N4/C7/C8 piperazine unit also adopts a slightly distorted chair conformation with protonation at the N4 atom.
3. Supramolecular features
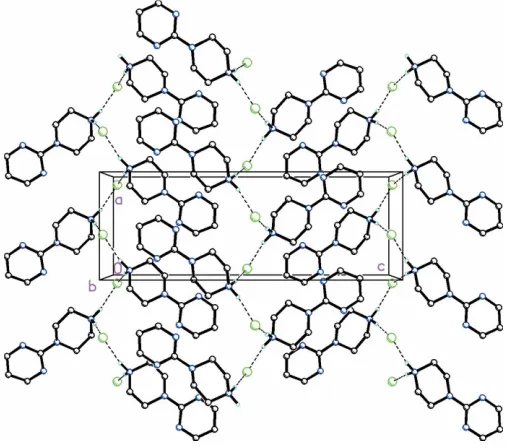
In the crystal of (I), N4—H4A Cl1 and N4—H4B Cl1 interactions are observed between pyrimidylpiperazine cations and chloride anions, forming zigzag chains along [100] (Fig. 3 and Table 1). In the crystal of (II), both of the H atoms on the N4 atom of the pyrimidylpiperazine cation are bifur-cated, forming N—H (O,O) hydrogen bonds (Fig. 4 and Table 2). Additional C—H O interactions between the pyrimidyl unit and the nitrate anion are present which, in concert with the N—H O hydrogen bonds between the
piperazine group and nitrate anions, form infinite chains along [100].
4. Database survey
A search of the Cambridge Structural Database (Version 5.35, last update May 2014: Allen 2002) revealed only three
struc-204
Yamunaet al. C8H13N4+Cland C8H13N4+NO3 Acta Cryst.(2014).E70, 203–206
[image:2.610.313.567.72.296.2]research communications
Figure 1
ORTEP drawing of C8H13N4+Cl, (I), showing 30% probability
displacement ellipsoids.
Figure 2
ORTEP drawing of C8H13N4 +
NO3
, (II), showing 30% probability displacement ellipsoids.
Figure 3
Molecular packing for C8H13N4+Cl, (I), viewed along thebaxis. Dashed
lines indicate N—H Cl interactions forming zigzag chains along thea
[image:2.610.46.296.72.174.2] [image:2.610.315.565.462.685.2]axis (see Table 1 for details). H atoms not involved in hydrogen bonding have been omitted for clarity.
Figure 4
Molecular packing for C8H13N4 +
NO3
[image:2.610.46.296.586.722.2]
tures containing the 4-(pyrimidin-2-yl)piperazin-1-ium cation similar to the structures reported here. These include the salts of 4-(pyrimidin-2-yl)piperazin-1-ium 3-carboxyprop-2-enoate (Yamuna et al. 2014a), 4-(pyrimidin-2-yl)piperazin-1-ium hydrogen d-tartrate monohydrate (Ding et al., 2014) and
4-(pyrimidin-2-yl)piperazin-1-ium hydrogen l-tartrate
mono-hydrate (Ding et al. 2014). The 3-carboxyprop-2-enoate
complex crystallizes in space group P21/c while the two
hydrogen (DandL)-tartrate monohydrate salts both crystal-lize in P212121. In comparison, title salt (I) crystallizes in
P212121 while (II) crystallizes in space group P21/c. In
addi-tion, as a related observaaddi-tion, 109 structures containing the pyrimidine–piperazine unit were also identified in this search. Some of these include, uniquely, the 4-(pyrimidin-2-yl)piper-azin-1-yl unit itself. These include 1-[4-(pyrimidin-2-yl)piper-azin-1-yl]ethanone, (1-methyl-1H -pyrrol-2-yl)[4-(pyrimidin-2-yl)piperazin-1-yl]methanone, [4-(pyrimidin-2-yl)piperazin-1-yl](2-thienyl)methanone, (4-fluorophenyl)[4-(pyrimidin-2-yl)-piperazin-1-yl]methanone (Spenceret al., 2011), (E )-1-phenyl-3-[4-(pyrimidin-2-yl)piperazin-1-yl]propan-1-one oxime (Kol-asa et al., 2006), N -(4-chlorophenyl)-4-(pyrimidin-2-yl)piper-azine-1-carboxamide (Li, 2011) and 6-{3-[4-(pyrimidin-2-yl)-piperazin-1-yl]propyl}-2,3-dihydro-5H-[1,4]dithiino[2,3-c ]pyr-role-5,7(6H)-dione (Bielenicaet al., 2011).
5. Synthesis and crystallization
For the preparation of title salt (I), a mixture of 1-(pyrimidin-2-yl)piperazine (0.2 g) and concentrated hydrochloric acid (5 ml) was stirred well over a magnetic stirrer at room temperature for 10 min and then warmed at 313 K for another 10 min. A white precipitate was obtained, which was dried in
research communications
Acta Cryst.(2014).E70, 203–206 Yamunaet al. C
[image:3.610.44.292.186.263.2]8H13N4+Cland C8H13N4+NO3
205
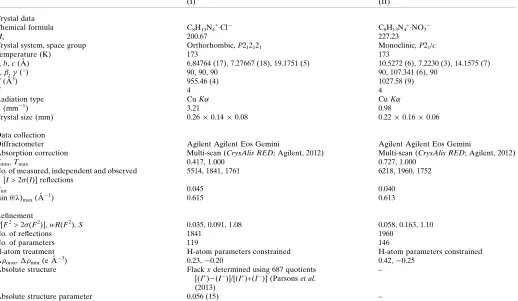
Table 3Experimental details.
(I) (II)
Crystal data
Chemical formula C8H13N4+Cl
C8H13N4+NO3
Mr 200.67 227.23
Crystal system, space group Orthorhombic,P212121 Monoclinic,P21/c
Temperature (K) 173 173
a,b,c(A˚ ) 6.84764 (17), 7.27667 (18), 19.1751 (5) 10.5272 (6), 7.2230 (3), 14.1575 (7) ,,(
) 90, 90, 90 90, 107.341 (6), 90
V(A˚3) 955.46 (4) 1027.58 (9)
Z 4 4
Radiation type CuK CuK
(mm1) 3.21 0.98
Crystal size (mm) 0.260.140.08 0.220.160.06
Data collection
Diffractometer Agilent Agilent Eos Gemini Agilent Agilent Eos Gemini
Absorption correction Multi-scan (CrysAlis RED; Agilent, 2012) Multi-scan (CrysAlis RED; Agilent, 2012)
Tmin,Tmax 0.417, 1.000 0.727, 1.000
No. of measured, independent and observed [I> 2(I)] reflections
5514, 1841, 1761 6218, 1960, 1752
Rint 0.045 0.040
(sin/)max(A˚
1) 0.615 0.613
Refinement
R[F2> 2(F2)],wR(F2),S 0.035, 0.091, 1.08 0.058, 0.163, 1.10
No. of reflections 1841 1960
No. of parameters 119 146
H-atom treatment H-atom parameters constrained H-atom parameters constrained
max, min(e A˚
3) 0.23,0.20 0.42,0.25
Absolute structure Flackxdetermined using 687 quotients
[(I+)(I
)]/[(I+)+(I
)] (Parsonset al. (2013)
–
Absolute structure parameter 0.056 (15) –
Computer programs:CrysAlis PROandCrysAlis RED(Agilent, 2012),SUPERFLIP(Palatinus & Chapuis, 2007),SHELXL2012(Sheldrick, 2008) andOLEX2(Dolomanovet al., 2009).
Table 1
Hydrogen-bond geometry (A˚ ,) for (I).
D—H A D—H H A D A D—H A
N4—H4A Cl1 0.91 2.21 3.102 (2) 167
N4—H4B Cl1i 0.91 2.21 3.114 (2) 175
Symmetry code: (i)xþ1 2;yþ
3 2;zþ1.
Table 2
Hydrogen-bond geometry (A˚ ,) for (II).
D—H A D—H H A D A D—H A
N4—H4A O2i 0.91 1.92 2.829 (3) 177
N4—H4A O3i 0.91 2.52 3.138 (3) 126
N4—H4B O1 0.91 2.35 3.197 (3) 155
N4—H4B O2 0.91 2.10 2.900 (3) 146
C3—H3 O1ii 0.95 2.46 3.240 (3) 140
C4—H4 O2iii 0.95 2.51 3.291 (3) 139
Symmetry codes: (i)xþ1;y1 2;zþ
1
[image:3.610.50.568.423.724.2]the open air overnight and then dissolved in hot dimethyl sulfoxide solvent. After few days, colourless blocks were obtained on slow evaporation (m.p. above 563 K).
For the preparation of title salt (II), a mixture of 1-(pyrimidin-2-yl)piperazine, from Sigma–Aldrich (0.2 g), and concentrated nitric acid (5 ml) was stirred well over a magnetic stirrer at room temperature for 10 min. A white precipitate was obtained immediately, which was dried in the open air overnight and then dissolved in water. After a few days, colourless blocks were obtained on slow evaporation (m.p. 463–470 K).
6. Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3. In both (I) and (II), all of the H atoms were placed in their calculated positions and then refined using a riding model with C—H bond lengths of 0.93 (CH) or 0.97 A˚ (CH2) and N—H bond lengths of 0.97 A˚ .
Isotropic displacement parameters for these atoms were set at 1.2Ueq(CH,CH2,NH).
Acknowledgements
TSY thanks the University of Mysore for research facilities and is also grateful to the Principal, Maharani’s Science College for Women, Mysore, for giving permission to under-take research. JPJ acknowledges the NSF–MRI program (grant No. CHE-1039027) for funds to purchase the X-ray diffractometer.
References
Agilent (2012).CrysAlis PROandCrysAlis RED. Agilent
Technol-ogies, Yarnton, England.
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Amin, K. M., Hanna, M. M., Abo-Youssef, H. E., Riham, F. &
George, R. F. (2009).Eur. J. Med. Chem.44, 4572–4584.
Bielenica, A., Kossakowski, J., Struga, M., Dybala, I., La Colla, P.,
Tamburini, E. & Loddo, R. (2011).Med. Chem. Res.20, 1411–1420.
Clark, M. P., George, K. M. & Bookland, R. G. (2007).Bioorg. Med.
Chem. Lett.17, 1250–1253.
Ding, X.-H., Li, Y.-H., Wang, S. & Huang, W. (2014).J. Mol. Struct.
1062, 61–67.
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. &
Puschmann, H. (2009).J. Appl. Cryst.42, 339–341.
Goldmann, S. & Stoltefuss, J. (1991).Angew. Chem. Int. Ed. Engl.30,
1559–1578.
Ibrahim, D. A. & El-Metwally, A. M. (2010).Eur. J. Med. Chem.45,
1158–1166.
Jasinski, J. P., Butcher, R. J., Hakim Al-Arique, Q. N. M., Yathirajan,
H. S. & Narayana, B. (2010).Acta Cryst.E66, o411–o412.
Kim, J. Y., Kim, D. & Kang, S. Y. (2010).Bioorg. Med. Chem. Lett.20,
6439–6442.
Kolasa, T.,et al.(2006).J. Med. Chem.49, 5093–5109.
Kuyper, L. F., Garvey, J. M., Baccanari, D. P., Champness, J. N.,
Stammers, D. K. & Beddell, C. R. (1996).Bioorg. Med. Chem. Lett.
4, 593–602.
Li, Y.-F. (2011).Acta Cryst.E67, o2575.
Padmaja, A., Payani, T., Reddy, G. D., Dinneswara Reddy, G. &
Padmavathi, V. (2009).Eur. J. Med. Chem.44, 4557–4566.
Palatinus, L. & Chapuis, G. (2007).J. Appl. Cryst.40, 786–790.
Pandey, S., Suryawanshi, S. N., Gupta, S. & Srivastava, V. M. L. (2004).
Eur. J. Med. Chem.39, 969–973.
Parsons, S., Flack, H. D. & Wagner, T. (2013).Acta Cryst.B69, 249–
259.
Sheldrick, G. M. (2008).Acta Cryst.A64, 112–122.
Siddegowda, M. S., Butcher, R. J., Akkurt, M., Yathirajan, H. S. &
Narayana, B. (2011a).Acta Cryst.E67, o2079–o2080.
Siddegowda, M. S., Jasinski, J. P., Golen, J. A., Yathirajan, H. S. &
Swamy, M. T. (2011b).Acta Cryst.E67, o2296.
Spencer, J., Patel, H., Callear, S. K., Coles, S. J. & Deadman, J. J.
(2011).Tetrahedron Lett.,52, 5905–5909.
Tollefson, G. D., Lancaster, S. P. & Montague-Clouse, J. (1991).
Psychopharmacol. Bull.27, 163–170.
Yamuna, T. S., Kaur, M., Anderson, B. J., Jasinski, J. P. & Yathirajan,
H. S. (2014b).Acta Cryst.E70, o206–o207.
Yamuna, T. S., Kaur, M., Anderson, B. J., Jasinski, J. P. & Yathirajan,
H. S. (2014c).Acta Cryst.E70, o200–o201.
Yamuna, T. S., Kaur, M., Jasinski, J. P. & Yathirajan, H. S. (2014a).
Acta Cryst.E70, o702–o703.
206
Yamunaet al. C8H13N4+Cland C8H13N4+NO3 Acta Cryst.(2014).E70, 203–206
supporting information
sup-1
Acta Cryst. (2014). E70, 203-206
supporting information
Acta Cryst. (2014). E70, 203-206 [doi:10.1107/S1600536814020169]
Crystal structures of 4-(pyrimidin-2-yl)piperazin-1-ium chloride and
4-(pyrimidin-2-yl)piperazin-1-ium nitrate
Thammarse S. Yamuna, Jerry P. Jasinski, Manpreet Kaur, Brian J. Anderson and H. S. Yathirajan
Computing details
For both compounds, data collection: CrysAlis PRO (Agilent, 2012); cell refinement: CrysAlis PRO (Agilent, 2012); data
reduction: CrysAlis RED (Agilent, 2012); program(s) used to solve structure: SUPERFLIP (Palatinus & Chapuis, 2007);
program(s) used to refine structure: SHELXL2012 (Sheldrick, 2008); molecular graphics: OLEX2 (Dolomanov et al.,
2009); software used to prepare material for publication: OLEX2 (Dolomanov et al., 2009).
(I) 4-(Pyrimidin-2-yl)piperazin-1-ium chloride
Crystal data
C8H13N4+·Cl−
Mr = 200.67
Orthorhombic, P212121
a = 6.84764 (17) Å
b = 7.27667 (18) Å
c = 19.1751 (5) Å
V = 955.46 (4) Å3
Z = 4
F(000) = 424
Dx = 1.395 Mg m−3
Cu Kα radiation, λ = 1.54184 Å Cell parameters from 2676 reflections
θ = 4.6–71.5°
µ = 3.21 mm−1
T = 173 K
Irregular, colourless 0.26 × 0.14 × 0.08 mm
Data collection
Agilent Agilent Eos Gemini diffractometer
Radiation source: Enhance (Cu) X-ray Source Detector resolution: 16.0416 pixels mm-1
ω scans
Absorption correction: multi-scan (CrysAlis RED; Agilent, 2012)
Tmin = 0.417, Tmax = 1.000
5514 measured reflections 1841 independent reflections 1761 reflections with I > 2σ(I)
Rint = 0.045
θmax = 71.4°, θmin = 4.6°
h = −8→8
k = −8→4
l = −23→23
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.035
wR(F2) = 0.091
S = 1.08 1841 reflections 119 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0504P)2 + 0.1163P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.23 e Å−3 Δρmin = −0.20 e Å−3
supporting information
sup-2
Acta Cryst. (2014). E70, 203-206
Absolute structure: Flack x determined using 687 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al. (2013)
Absolute structure parameter: 0.056 (15)
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Cl1 0.08383 (9) 0.49612 (9) 0.48653 (3) 0.0262 (2) N1 0.6948 (4) 0.6820 (3) 0.81551 (12) 0.0251 (5) N2 0.9664 (4) 0.5690 (3) 0.74930 (13) 0.0286 (6) N3 0.6688 (3) 0.6293 (3) 0.69659 (12) 0.0224 (5) N4 0.4359 (4) 0.6322 (3) 0.57422 (12) 0.0258 (5)
H4A 0.3467 0.5800 0.5451 0.031*
H4B 0.4718 0.7422 0.5556 0.031*
C1 0.7813 (4) 0.6281 (4) 0.75588 (14) 0.0208 (5) C2 0.8040 (4) 0.6746 (4) 0.87274 (15) 0.0269 (6)
H2 0.7471 0.7097 0.9159 0.032*
C3 0.9968 (5) 0.6181 (4) 0.87217 (16) 0.0318 (7)
H3 1.0742 0.6147 0.9133 0.038*
C4 1.0692 (5) 0.5668 (4) 0.80773 (17) 0.0330 (7)
H4 1.2013 0.5274 0.8052 0.040*
C5 0.7582 (4) 0.5944 (4) 0.62855 (14) 0.0245 (6)
H5A 0.8701 0.5096 0.6341 0.029*
H5B 0.8076 0.7111 0.6088 0.029*
C6 0.6103 (4) 0.5108 (4) 0.57949 (14) 0.0278 (6)
H6A 0.6694 0.4948 0.5328 0.033*
H6B 0.5705 0.3883 0.5969 0.033*
C7 0.3448 (4) 0.6631 (4) 0.64394 (14) 0.0246 (6)
H7A 0.2964 0.5449 0.6628 0.030*
H7B 0.2323 0.7475 0.6392 0.030*
C8 0.4936 (4) 0.7449 (4) 0.69357 (14) 0.0233 (6)
H8A 0.5297 0.8698 0.6777 0.028*
H8B 0.4359 0.7553 0.7407 0.028*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3
Acta Cryst. (2014). E70, 203-206
C1 0.0217 (13) 0.0157 (11) 0.0251 (13) 0.0000 (10) 0.0013 (11) 0.0022 (10) C2 0.0342 (16) 0.0238 (13) 0.0229 (13) −0.0013 (12) 0.0000 (12) −0.0001 (11) C3 0.0353 (16) 0.0287 (14) 0.0314 (14) −0.0017 (14) −0.0114 (14) 0.0056 (12) C4 0.0238 (14) 0.0339 (14) 0.0413 (17) 0.0072 (13) −0.0056 (14) 0.0062 (13) C5 0.0208 (13) 0.0292 (14) 0.0233 (13) 0.0030 (11) 0.0031 (11) −0.0032 (11) C6 0.0256 (14) 0.0316 (14) 0.0261 (13) 0.0000 (14) 0.0035 (10) −0.0058 (12) C7 0.0200 (13) 0.0272 (13) 0.0267 (14) −0.0009 (11) −0.0007 (11) 0.0007 (11) C8 0.0186 (13) 0.0244 (12) 0.0268 (13) 0.0046 (12) −0.0004 (12) −0.0027 (11)
Geometric parameters (Å, º)
N1—C1 1.346 (4) C3—H3 0.9500
N1—C2 1.329 (4) C3—C4 1.383 (4)
N2—C1 1.344 (4) C4—H4 0.9500
N2—C4 1.323 (4) C5—H5A 0.9900
N3—C1 1.373 (3) C5—H5B 0.9900
N3—C5 1.463 (3) C5—C6 1.510 (4)
N3—C8 1.466 (3) C6—H6A 0.9900
N4—H4A 0.9100 C6—H6B 0.9900
N4—H4B 0.9100 C7—H7A 0.9900
N4—C6 1.489 (4) C7—H7B 0.9900
N4—C7 1.492 (3) C7—C8 1.516 (4)
C2—H2 0.9500 C8—H8A 0.9900
C2—C3 1.383 (4) C8—H8B 0.9900
C2—N1—C1 116.2 (2) N3—C5—H5B 109.6
C4—N2—C1 115.2 (3) N3—C5—C6 110.2 (2)
C1—N3—C5 120.2 (2) H5A—C5—H5B 108.1
C1—N3—C8 119.7 (2) C6—C5—H5A 109.6
C5—N3—C8 114.0 (2) C6—C5—H5B 109.6
H4A—N4—H4B 108.0 N4—C6—C5 110.0 (2)
C6—N4—H4A 109.4 N4—C6—H6A 109.7
C6—N4—H4B 109.4 N4—C6—H6B 109.7
C6—N4—C7 111.3 (2) C5—C6—H6A 109.7
C7—N4—H4A 109.4 C5—C6—H6B 109.7
C7—N4—H4B 109.4 H6A—C6—H6B 108.2
N1—C1—N3 117.0 (2) N4—C7—H7A 109.7
N2—C1—N1 126.0 (2) N4—C7—H7B 109.7
N2—C1—N3 116.9 (2) N4—C7—C8 109.9 (2)
N1—C2—H2 118.6 H7A—C7—H7B 108.2
N1—C2—C3 122.9 (3) C8—C7—H7A 109.7
C3—C2—H2 118.6 C8—C7—H7B 109.7
C2—C3—H3 122.3 N3—C8—C7 110.4 (2)
C2—C3—C4 115.5 (3) N3—C8—H8A 109.6
C4—C3—H3 122.3 N3—C8—H8B 109.6
N2—C4—C3 124.2 (3) C7—C8—H8A 109.6
N2—C4—H4 117.9 C7—C8—H8B 109.6
supporting information
sup-4
Acta Cryst. (2014). E70, 203-206
N3—C5—H5A 109.6
N1—C2—C3—C4 −0.8 (4) C4—N2—C1—N1 −0.6 (4) N3—C5—C6—N4 55.8 (3) C4—N2—C1—N3 −178.9 (3) N4—C7—C8—N3 −54.5 (3) C5—N3—C1—N1 172.5 (2) C1—N1—C2—C3 1.0 (4) C5—N3—C1—N2 −9.0 (4) C1—N2—C4—C3 0.8 (4) C5—N3—C8—C7 55.2 (3) C1—N3—C5—C6 151.9 (2) C6—N4—C7—C8 57.4 (3) C1—N3—C8—C7 −152.4 (2) C7—N4—C6—C5 −58.1 (3) C2—N1—C1—N2 −0.3 (4) C8—N3—C1—N1 21.8 (4) C2—N1—C1—N3 178.0 (2) C8—N3—C1—N2 −159.8 (2) C2—C3—C4—N2 −0.2 (5) C8—N3—C5—C6 −55.8 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N4—H4A···Cl1 0.91 2.21 3.102 (2) 167 N4—H4B···Cl1i 0.91 2.21 3.114 (2) 175
Symmetry code: (i) x+1/2, −y+3/2, −z+1.
(II) 4-(Pyrimidin-2-yl)piperazin-1-ium nitrate
Crystal data
C8H13N4+·NO3−
Mr = 227.23 Monoclinic, P21/c
a = 10.5272 (6) Å
b = 7.2230 (3) Å
c = 14.1575 (7) Å
β = 107.341 (6)°
V = 1027.58 (9) Å3
Z = 4
F(000) = 480
Dx = 1.469 Mg m−3
Cu Kα radiation, λ = 1.54184 Å Cell parameters from 2763 reflections
θ = 6.2–71.4°
µ = 0.98 mm−1
T = 173 K
Irregular, colourless 0.22 × 0.16 × 0.06 mm
Data collection
Agilent Agilent Eos Gemini diffractometer
Radiation source: Cu Kα
Detector resolution: 16.0416 pixels mm-1
ω scans
Absorption correction: multi-scan (CrysAlis RED; Agilent, 2012)
Tmin = 0.727, Tmax = 1.000
6218 measured reflections 1960 independent reflections 1752 reflections with I > 2σ(I)
Rint = 0.040
θmax = 71.0°, θmin = 4.4°
h = −9→12
k = −8→8
l = −17→16
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.058
wR(F2) = 0.163
S = 1.10 1960 reflections 146 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0789P)2 + 0.9595P] where P = (Fo2 + 2Fc2)/3
supporting information
sup-5
Acta Cryst. (2014). E70, 203-206 Δρmax = 0.42 e Å−3
Δρmin = −0.25 e Å−3
Extinction correction: SHELXL2012 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 Extinction coefficient: 0.0099 (14)
Special details
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.4119 (2) 0.6964 (4) 0.41222 (17) 0.0615 (7) O2 0.50951 (18) 0.6257 (2) 0.30424 (14) 0.0381 (5) O3 0.55020 (17) 0.8884 (2) 0.37996 (13) 0.0323 (5) N5 0.49103 (17) 0.7390 (3) 0.36677 (13) 0.0238 (4) N1 0.00592 (19) 0.2396 (3) 0.48106 (14) 0.0291 (5) N2 −0.11846 (18) 0.3821 (3) 0.32856 (15) 0.0273 (5) N3 0.10930 (18) 0.3372 (3) 0.36702 (14) 0.0268 (5) N4 0.33344 (18) 0.3134 (3) 0.29632 (15) 0.0278 (5)
H4A 0.3814 0.2536 0.2617 0.033*
H4B 0.3777 0.4191 0.3216 0.033*
C1 −0.0049 (2) 0.3204 (3) 0.39365 (16) 0.0220 (5) C2 −0.1085 (3) 0.2126 (3) 0.50188 (19) 0.0346 (6)
H2 −0.1054 0.1544 0.5627 0.042*
C3 −0.2307 (2) 0.2647 (4) 0.4398 (2) 0.0362 (6)
H3 −0.3111 0.2420 0.4553 0.043*
C4 −0.2290 (2) 0.3519 (3) 0.3537 (2) 0.0329 (6)
H4 −0.3113 0.3927 0.3097 0.039*
C5 0.2387 (2) 0.2876 (4) 0.43489 (16) 0.0282 (5)
H5A 0.2266 0.2035 0.4867 0.034*
H5B 0.2848 0.4005 0.4676 0.034*
C6 0.3222 (2) 0.1932 (3) 0.37877 (17) 0.0270 (5)
H6A 0.4121 0.1674 0.4242 0.032*
H6B 0.2808 0.0738 0.3519 0.032*
C7 0.1993 (2) 0.3620 (3) 0.22801 (17) 0.0277 (5)
H7A 0.1537 0.2483 0.1960 0.033*
H7B 0.2095 0.4461 0.1755 0.033*
C8 0.1166 (2) 0.4552 (3) 0.28517 (17) 0.0276 (5)
H8A 0.1571 0.5756 0.3112 0.033*
H8B 0.0258 0.4789 0.2407 0.033*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-6
Acta Cryst. (2014). E70, 203-206
N5 0.0175 (9) 0.0301 (10) 0.0240 (9) −0.0015 (7) 0.0064 (7) 0.0012 (8) N1 0.0292 (10) 0.0327 (11) 0.0282 (10) 0.0020 (8) 0.0126 (8) 0.0038 (8) N2 0.0210 (9) 0.0270 (10) 0.0331 (11) 0.0037 (7) 0.0071 (8) 0.0001 (8) N3 0.0189 (9) 0.0380 (11) 0.0243 (9) 0.0068 (8) 0.0075 (7) 0.0087 (8) N4 0.0231 (9) 0.0278 (10) 0.0368 (11) −0.0042 (8) 0.0153 (8) −0.0045 (8) C1 0.0207 (10) 0.0220 (10) 0.0246 (11) 0.0023 (8) 0.0087 (8) −0.0035 (8) C2 0.0416 (14) 0.0328 (13) 0.0372 (13) −0.0021 (11) 0.0235 (11) −0.0014 (10) C3 0.0300 (13) 0.0340 (13) 0.0525 (15) −0.0049 (10) 0.0247 (11) −0.0130 (12) C4 0.0224 (11) 0.0304 (12) 0.0456 (15) 0.0020 (9) 0.0098 (10) −0.0063 (11) C5 0.0208 (11) 0.0395 (13) 0.0234 (11) 0.0087 (9) 0.0054 (9) 0.0023 (9) C6 0.0219 (10) 0.0296 (12) 0.0293 (11) 0.0038 (9) 0.0074 (9) 0.0014 (9) C7 0.0291 (11) 0.0305 (12) 0.0255 (11) −0.0013 (9) 0.0111 (9) 0.0039 (9) C8 0.0267 (11) 0.0290 (12) 0.0283 (11) 0.0033 (9) 0.0098 (9) 0.0080 (9)
Geometric parameters (Å, º)
O1—N5 1.233 (3) C2—C3 1.376 (4)
O2—N5 1.263 (2) C3—H3 0.9500
O3—N5 1.232 (2) C3—C4 1.377 (4)
N1—C1 1.342 (3) C4—H4 0.9500
N1—C2 1.337 (3) C5—H5A 0.9900
N2—C1 1.349 (3) C5—H5B 0.9900
N2—C4 1.333 (3) C5—C6 1.512 (3)
N3—C1 1.369 (3) C6—H6A 0.9900
N3—C5 1.459 (3) C6—H6B 0.9900
N3—C8 1.459 (3) C7—H7A 0.9900
N4—H4A 0.9100 C7—H7B 0.9900
N4—H4B 0.9100 C7—C8 1.512 (3)
N4—C6 1.487 (3) C8—H8A 0.9900
N4—C7 1.496 (3) C8—H8B 0.9900
C2—H2 0.9500
O1—N5—O2 118.2 (2) C3—C4—H4 118.2
O3—N5—O1 121.9 (2) N3—C5—H5A 109.7
O3—N5—O2 119.82 (18) N3—C5—H5B 109.7
C2—N1—C1 115.6 (2) N3—C5—C6 109.86 (18)
C4—N2—C1 115.5 (2) H5A—C5—H5B 108.2
C1—N3—C5 121.45 (19) C6—C5—H5A 109.7
C1—N3—C8 121.92 (18) C6—C5—H5B 109.7
C5—N3—C8 114.01 (18) N4—C6—C5 110.12 (18)
H4A—N4—H4B 108.0 N4—C6—H6A 109.6
C6—N4—H4A 109.4 N4—C6—H6B 109.6
C6—N4—H4B 109.4 C5—C6—H6A 109.6
C6—N4—C7 111.33 (17) C5—C6—H6B 109.6
C7—N4—H4A 109.4 H6A—C6—H6B 108.2
C7—N4—H4B 109.4 N4—C7—H7A 109.7
N1—C1—N2 126.0 (2) N4—C7—H7B 109.7
supporting information
sup-7
Acta Cryst. (2014). E70, 203-206
N2—C1—N3 117.06 (19) H7A—C7—H7B 108.2
N1—C2—H2 118.3 C8—C7—H7A 109.7
N1—C2—C3 123.4 (2) C8—C7—H7B 109.7
C3—C2—H2 118.3 N3—C8—C7 109.85 (18)
C2—C3—H3 122.1 N3—C8—H8A 109.7
C2—C3—C4 115.8 (2) N3—C8—H8B 109.7
C4—C3—H3 122.1 C7—C8—H8A 109.7
N2—C4—C3 123.6 (2) C7—C8—H8B 109.7
N2—C4—H4 118.2 H8A—C8—H8B 108.2
N1—C2—C3—C4 −1.3 (4) C4—N2—C1—N1 −2.7 (3) N3—C5—C6—N4 55.5 (3) C4—N2—C1—N3 175.4 (2) N4—C7—C8—N3 −55.3 (2) C5—N3—C1—N1 −6.3 (3) C1—N1—C2—C3 −0.7 (4) C5—N3—C1—N2 175.4 (2) C1—N2—C4—C3 0.2 (3) C5—N3—C8—C7 56.9 (3) C1—N3—C5—C6 141.1 (2) C6—N4—C7—C8 56.9 (2) C1—N3—C8—C7 −141.2 (2) C7—N4—C6—C5 −57.0 (2) C2—N1—C1—N2 2.9 (3) C8—N3—C1—N1 −166.9 (2) C2—N1—C1—N3 −175.2 (2) C8—N3—C1—N2 14.8 (3) C2—C3—C4—N2 1.6 (4) C8—N3—C5—C6 −57.0 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N4—H4A···O2i 0.91 1.92 2.829 (3) 177 N4—H4A···O3i 0.91 2.52 3.138 (3) 126
N4—H4B···O1 0.91 2.35 3.197 (3) 155
N4—H4B···O2 0.91 2.10 2.900 (3) 146
C3—H3···O1ii 0.95 2.46 3.240 (3) 140
C4—H4···O2iii 0.95 2.51 3.291 (3) 139