organic papers
o1770
Liet al. C17H14O3 doi:10.1107/S1600536806012050 Acta Cryst.(2006). E62, o1770–o1771
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
2,7-Diacetylxanthene
Li Wu, Ai-Lin Liu , Yang Lu* and Guan-Hua Du
Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, 1 Xiannong tan street, Beijing 100050, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 296 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.048
wRfactor = 0.128 Data-to-parameter ratio = 8.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 5 March 2006 Accepted 3 April 2006
#2006 International Union of Crystallography
All rights reserved
In the crystal structure of the title compound, C17H14O3, the
asymmetric unit comprises one half-molecule; a mirror plane passes through the pyran O atom and thepara-carbon atom.
Comment
The title compound, (I), was synthesized from xanthene and acetyl chloride (Ng & Ng, 1952). Recently, we found that it exhibits anti-xanthine oxidase activity with an inhibition ratio of 71.28% at a concentration of 10 6g ml 1. In the light of this, we have synthesized this compound and determined its structure by X-ray analysis.
Compound (I) crystallizes in the space group Cmc21 with
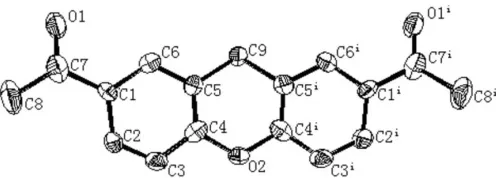
one half-molecule in the asymmetric unit (Fig. 1). The dihedral angle between the two benzene rings is 11.1 (1), and the
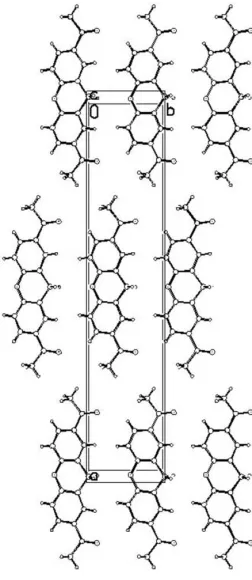
pyran ring adopts a boat conformation, in which atoms C4, C5, C4iand C5iform the bottom of the boat, O2 the prow and C9 the stern [deviations from the C4/C5/C4i/C5i mean plane = 0.132 (2) and 0.1921 (17) A˚ for O2 and C9, respectively; symmetry code: (i) x,y,z]. Atoms O2 and C9 are located on a mirror plane. No hydrogen-bond interactions are observed between molecules (Fig. 2)
Experimental
[image:1.610.243.422.314.392.2] [image:1.610.208.456.616.705.2]The title compound was prepared according to the procedure of Ng & Ng (1952). A single crystal was obtained by slow evaporation of a saturated methanol–hexane(1:1) solution at 283 K.
Figure 1
Crystal data
C17H14O3 Mr= 266.28
Orthorhombic,Cmc21 a= 29.875 (6) A˚ b= 6.0270 (12) A˚ c= 7.2560 (15) A˚ V= 1306.5 (5) A˚3
Z= 4
Dx= 1.354 Mg m 3
MoKradiation
= 0.09 mm 1 T= 296 (1) K Block, pale yellow 0.300.200.10 mm
Data collection
MAC DIP 2030K diffractometer
!scans
Absorption correction: none 1899 measured reflections
785 independent reflections 782 reflections withI> 2(I) Rint= 0.023
max= 27.3
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.048 wR(F2) = 0.128 S= 1.11 785 reflections 95 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0667P)2 + 0.4024P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.11 e A˚ 3
min= 0.11 e A˚ 3
[image:2.610.376.502.71.361.2]Extinction correction:SHELXL97 Extinction coefficient: 0.010 (3)
Table 1
Selected bond and torsion angles (). C4—O2—C4i
118.6 (2) C5—C9—C5i
112.0 (2)
C4i
—O2—C4—C3 168.35 (15)
C4i
—O2—C4—C5 12.8 (4)
C3—C4—C5—C6 0.2 (3)
O2—C4—C5—C6 179.0 (2)
C3—C4—C5—C9 176.8 (3)
O2—C4—C5—C9 2.0 (3)
C6—C5—C9—C5i
167.84 (16) C4—C5—C9—C5i
15.3 (4)
Symmetry code: (i) x;y;z.
In the absence of significant anomalous scattering, Friedel pairs were merged. Methyl H atoms were constrained to an ideal geometry, with C—H = 0.96 A˚ andUiso(H) = 1.5Ueq(C). All other H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms, with C—H = 0.93 A˚ andUiso(H) = 1.2Ueq(C).
Data collection: DENZO (Otwinowski & Minor, 1997); cell refinement: SCALEPACK (Otwinowski & Minor, 1997); data reduction: SCALEPACK; program(s) used to solve structure:
SHELXS97(Sheldrick, 1997); program(s) used to refine structure:
SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEPII
(Johnson, 1976) and PLATON (Spek, 2003); software used to prepare material for publication:SHELXL97.
The authors acknowledge the financial support of the International Centre for Diffraction Data.
References
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
Ng, D.-X. & Ng, P. B.-H. (1952).J. Chem. Soc.pp. 3741–3744.
Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Figure 2
supporting information
sup-1 Acta Cryst. (2006). E62, o1770–o1771
supporting information
Acta Cryst. (2006). E62, o1770–o1771 [https://doi.org/10.1107/S1600536806012050]
2,7-Diacetylxanthene
Li Wu, Ai-Lin Liu, Yang Lu and Guan-Hua Du
2,7-diacetylxanthene
Crystal data C17H14O3
Mr = 266.28
Orthorhombic, Cmc21
Hall symbol: C 2c -2 a = 29.875 (6) Å b = 6.0270 (12) Å c = 7.2560 (15) Å V = 1306.5 (5) Å3
Z = 4
F(000) = 560 Dx = 1.354 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1899 reflections θ = 2.7–27.3°
µ = 0.09 mm−1
T = 296 K
Block, pale yellow 0.30 × 0.20 × 0.10 mm
Data collection MAC DIP 2030K
diffractometer
Radiation source: rotating anode Graphite monochromator
Detector resolution: 0 pixels mm-1
ω scans
1899 measured reflections
785 independent reflections 782 reflections with I > 2σ(I) Rint = 0.023
θmax = 27.3°, θmin = 2.7°
h = −37→38 k = 0→7 l = −9→9
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.048
wR(F2) = 0.128
S = 1.11 785 reflections 95 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0667P)2 + 0.4024P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.11 e Å−3
Δρmin = −0.11 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.010 (3)
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.17114 (5) −0.1266 (3) 0.1160 (5) 0.0832 (8)
O2 0.0000 0.4390 (3) −0.1025 (3) 0.0520 (6)
C1 0.12308 (6) 0.1729 (3) 0.0456 (3) 0.0502 (6)
C2 0.11922 (7) 0.3845 (3) −0.0292 (3) 0.0548 (6)
H2B 0.1447 0.4704 −0.0472 0.066*
C3 0.07776 (7) 0.4680 (3) −0.0768 (3) 0.0520 (6)
H3A 0.0754 0.6091 −0.1277 0.062*
C4 0.03987 (6) 0.3413 (3) −0.0487 (3) 0.0457 (5)
C5 0.04197 (6) 0.1272 (3) 0.0262 (3) 0.0452 (5)
C6 0.08396 (6) 0.0465 (3) 0.0721 (3) 0.0478 (5)
H6A 0.0864 −0.0952 0.1219 0.057*
C7 0.16732 (7) 0.0715 (4) 0.0911 (4) 0.0617 (7)
C8 0.20751 (8) 0.2214 (5) 0.1045 (7) 0.0952 (12)
H8A 0.2337 0.1342 0.1304 0.143*
H8B 0.2030 0.3270 0.2017 0.143*
H8C 0.2115 0.2984 −0.0102 0.143*
C9 0.0000 −0.0111 (5) 0.0458 (5) 0.0518 (7)
H9A 0.0000 −0.0826 0.1656 0.062*
H9B 0.0000 −0.1264 −0.0475 0.062*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0560 (9) 0.0665 (12) 0.1270 (19) 0.0072 (7) 0.0005 (11) 0.0135 (11)
O2 0.0536 (11) 0.0395 (10) 0.0630 (15) 0.000 0.000 0.0093 (9)
C1 0.0451 (9) 0.0520 (10) 0.0535 (13) −0.0023 (7) 0.0033 (8) −0.0054 (10)
C2 0.0536 (11) 0.0511 (11) 0.0597 (13) −0.0108 (8) 0.0074 (10) −0.0019 (10)
C3 0.0607 (11) 0.0400 (9) 0.0554 (12) −0.0053 (8) 0.0045 (9) 0.0024 (8)
C4 0.0493 (10) 0.0395 (9) 0.0484 (12) 0.0005 (6) 0.0028 (8) 0.0001 (9)
C5 0.0481 (10) 0.0379 (9) 0.0495 (12) −0.0022 (6) 0.0023 (7) −0.0009 (8)
C6 0.0467 (9) 0.0435 (9) 0.0533 (12) −0.0003 (7) 0.0031 (8) 0.0003 (8)
C7 0.0476 (10) 0.0641 (13) 0.0736 (15) −0.0015 (9) 0.0054 (10) 0.0010 (12)
C8 0.0486 (12) 0.085 (2) 0.153 (3) −0.0077 (11) −0.0061 (17) −0.003 (2)
C9 0.0435 (13) 0.0400 (12) 0.0720 (19) 0.000 0.000 0.0099 (13)
Geometric parameters (Å, º)
O1—C7 1.213 (3) C5—C6 1.386 (3)
O2—C4 1.385 (2) C5—C9 1.512 (2)
supporting information
sup-3 Acta Cryst. (2006). E62, o1770–o1771
C1—C2 1.391 (3) C7—C8 1.506 (3)
C1—C6 1.408 (2) C8—H8A 0.9600
C1—C7 1.493 (3) C8—H8B 0.9600
C2—C3 1.381 (3) C8—H8C 0.9600
C2—H2B 0.9300 C9—C5i 1.512 (2)
C3—C4 1.381 (3) C9—H9A 0.9700
C3—H3A 0.9300 C9—H9B 0.9700
C4—C5 1.401 (3)
C4—O2—C4i 118.6 (2) C5—C6—H6A 119.1
C2—C1—C6 118.72 (18) C1—C6—H6A 119.1
C2—C1—C7 122.35 (18) O1—C7—C1 121.3 (2)
C6—C1—C7 118.87 (19) O1—C7—C8 120.4 (2)
C3—C2—C1 120.41 (18) C1—C7—C8 118.3 (2)
C3—C2—H2B 119.8 C7—C8—H8A 109.5
C1—C2—H2B 119.8 C7—C8—H8B 109.5
C2—C3—C4 119.79 (18) H8A—C8—H8B 109.5
C2—C3—H3A 120.1 C7—C8—H8C 109.5
C4—C3—H3A 120.1 H8A—C8—H8C 109.5
C3—C4—O2 115.35 (17) H8B—C8—H8C 109.5
C3—C4—C5 122.01 (17) C5—C9—C5i 112.0 (2)
O2—C4—C5 122.64 (16) C5—C9—H9A 109.2
C6—C5—C4 117.19 (16) C5i—C9—H9A 109.2
C6—C5—C9 122.28 (18) C5—C9—H9B 109.2
C4—C5—C9 120.46 (17) C5i—C9—H9B 109.2
C5—C6—C1 121.88 (18) H9A—C9—H9B 107.9
C6—C1—C2—C3 −0.2 (3) C4—C5—C6—C1 0.1 (3)
C7—C1—C2—C3 177.1 (2) C9—C5—C6—C1 177.1 (2)
C1—C2—C3—C4 0.5 (3) C2—C1—C6—C5 −0.1 (3)
C2—C3—C4—O2 −179.4 (2) C7—C1—C6—C5 −177.5 (2)
C2—C3—C4—C5 −0.5 (3) C2—C1—C7—O1 −163.1 (3)
C4i—O2—C4—C3 −168.35 (15) C6—C1—C7—O1 14.2 (4)
C4i—O2—C4—C5 12.8 (4) C2—C1—C7—C8 16.5 (4)
C3—C4—C5—C6 0.2 (3) C6—C1—C7—C8 −166.1 (3)
O2—C4—C5—C6 179.0 (2) C6—C5—C9—C5i 167.84 (16)
C3—C4—C5—C9 −176.8 (3) C4—C5—C9—C5i −15.3 (4)
O2—C4—C5—C9 2.0 (3)