Acta Cryst.(2003). E59, o825±o827 DOI: 10.1107/S1600536803010225 Itzia I. Padilla-MartõÂnezet al. C14H16N2O6
o825
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Diethyl
N
,
N
000-
m
-phenylenedioxamate
Itzia I. Padilla-MartõÂnez,aMarõÂa Chaparro-Huerta,aFrancisco J. MartõÂnez-MartõÂnez,aHerbert HoÈpflband EfreÂn V. GarcõÂa-BaÂeza*
aUnidad Profesional Interdisciplinaria de
BiotecnologõÂa, Instituto PoliteÂcnico Nacional, Avenida Acueducto s/n, Barrio La Laguna TicomaÂn, DF 07340, Mexico, andbCentro de Investigaciones QuõÂmicas, Universidad AutoÂnoma de Morelos, Cuernavaca Morelos, Mexico
Correspondence e-mail: vgarcia@acei.upibi.ipn.mx
Key indicators
Single-crystal X-ray study T= 100 K
Mean(C±C) = 0.003 AÊ Rfactor = 0.058 wRfactor = 0.124
Data-to-parameter ratio = 12.1
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, C14H16N2O6, crystallizes in the
mono-clinic space group, C2/c. C2 symmetry is imposed on the
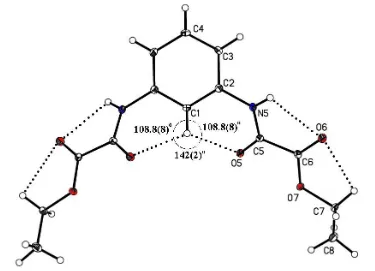
molecule. The ethyl oxamate groups are twisted out of the aromatic ring plane by 34.53 (6)and both carbonyl groups are antiperiplanar. The intramolecular hydrogen-bonding pattern is depicted by a soft CÐH O/O0 three-centered hydrogen
bond, and NÐH O and CÐH O hydrogen-bonding
interactions which form anS(5)S(5)S(6)S(6)0S(5)0S(5)0motif. The molecules are linked into C(3) and C(4) chain motifs, forming supramolecular layers in theacplane.
Comment
The title compound, (I), forms monoclinic crystals (C2/c, Z= 4).C2 symmetry is crystallographically imposed on the
mol-ecule; thus only one half molecule is present in the asymmetric unit. The bond lengths and angles are in the standard ranges, with a mean CÐC bond length of 1.384 (3) AÊ (Table 1). The
ethyl oxamate ±N(H)ÐC(O)ÐC(O)ÐOCH2CH3 group is
almost planar, with an N5ÐC5ÐC6ÐO7 torsion angle of 170.2 (2). Both carbonyl groups are antiperiplanar, with an O5ÐC5ÐC6ÐO6 torsion angle of 170.8 (2). The C5ÐC6 bond distance is almost exactly 1.541 (1) AÊ (Dewar & Schmeizing, 1968), the value usually assumed for a Csp3ÐCsp3
single bond, while for the Csp2ÐCsp2single bond, the average
value from the literature (Allenet al., 1987) is 1.478 (12) AÊ. This may be indicative of the absence of conjugation, in spite of the planarity of the oxamate system, but can be justi®ed by steric hindrance; at 2.640 (2) AÊ, the O5 O7 contact is markedly shorter than the sum of the van der Waals radii (3.04 AÊ; Bondi, 1964). The two ethyl oxamate groups are
Received 16 April 2003 Accepted 9 May 2003 Online 16 May 2003
Figure 1
The molecular structure of the title compound, showing the atom-labelling scheme, the three-centered hydrogen-bonding interaction CÐ H O(O0) and the H5 O6 H7A hydrogen-bonding interactions.
organic papers
o826
Itzia I. Padilla-MartõÂnezet al. C14H16N2O6 Acta Cryst.(2003). E59, o825±o827 twisted by 34.53 (6)away from the aromatic ring mean plane(Fig. 2). These results contrast with those recently reported for the 1,2-isomer,viz. N,N0-o-phenylenedioxamate (Martinet al., 2002). The analysis of this latter compound reveals a molecule with one ethyl oxamate group synperiplanar and the other antiperiplanar, both twisted with respect to the benzene ring as a result of steric interactions. The above-mentioned conformation allows the formation of the intramolecular three-centered hydrogen-bonding interaction (THB) C1Ð H O5(O50). Selected bond distances and torsion angles are listed in Table 2.
According to graph-set notation (Bernsteinet al., 1995), two adjacent S(6) rings are formed. A particular feature of this interaction is the O5 H1 O50 angle of 142 (2), and the sum of the angles at the donor is 360, as required by the crystallographic symmetry (Fig. 1). The H1 O5 distance [2.45 (1) AÊ] is shorter than the mean value reported for similar systems [2.553 (4) AÊ; Steiner, 2002]; however, it is larger than the values found for other intramolecular THB involving NÐ H as donor [2.09 (2) AÊ; Padilla-MartõÂnez et al., 2001]. These results reveal the relative weakness of the soft CÐH O C interaction (Vargaset al., 2000), compared with the hard NÐ H O C interaction (Desiraju, 1996) involving THB systems. The intramolecular hydrogen-bonding interactions N5ÐH5 O6 and C7ÐH7A O6 (Table 2) complete the overall hydrogen-bonding scheme, which can be depicted as a
set of six hydrogen-bonded rings forming an
S(5)S(5)S(6)S(6)0S(5)0S(5)0 motif. It is noteworthy that this last interaction should have some importance in determining
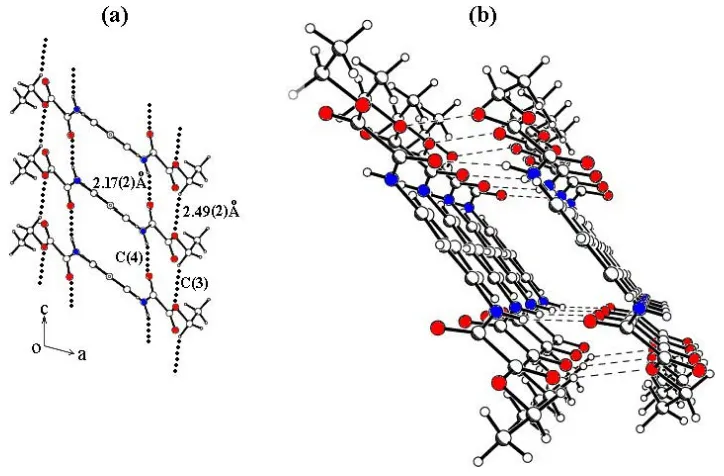
the location of the ethyl group in the oxamate plane. Supra-molecular layers of the title molecule are formed in the ac
plane due to N5ÐH O5i[2.17 (2) AÊ] and C7ÐH7B O7ii
[2.49 (2) AÊ] intermolecular hydrogen bonds (see Table 2 for symmetry codes), which lead toC(4) andC(3) chains motifs, respectively (Fig. 3a), whose topological motif corresponds to a ring graph-set descriptorR22(11)[C(3)C(4)] (Bernsteinet al.,
1995). These parallel chains are generated by translation along the [010] direction (Fig. 3b) and are formed because of the close proximity of alternate acceptors. There are no hydrogen bonds between these layers.
Experimental
The compound was prepared from 2.3 ml (20.4 mmol) of ethyl chlorooxoacetate and 1.0 g (9.3 mmol) of 1,3-diaminobenzene, according to reported procedures (MartõÂnez-MartõÂnezet al., 1998), to yield 20.0 g (70%) of a pale yellow solid (m.p. 425±427 K). IR (KBr, cmÿ1): 3349 (NH), 1699 (CO); 1H NMR (300.08 MHz, DMSO-d
6, p.p.m.): 10.8 (s, 2H, NH), 8.2 (s, 1H), 7.5 (d, 2H), 7.3 (t, 1H), 4.3 (q, 4H, CH2), 1.3 (t, 6H, CH3);13C NMR (75.46 MHz, DMSO-d6, p.p.m.): 161.3 (COO), 156.4 (CON), 138.3 (Ci), 129.6 (Cm), 117.7 (2Co), 113.5
(Co), 63.0 (CH2), 14.5 (CH3). Crystals suitable for X-ray analysis were obtained after slow crystallization from acetone.
Crystal data
C14H16N2O6 Mr= 308.29
Monoclinic,C2=c a= 25.086 (5) AÊ b= 7.1225 (14) AÊ c= 8.2529 (17) AÊ
= 104.870 (3) V= 1425.2 (5) AÊ3 Z= 4
Dx= 1.437 Mg mÿ3
MoKradiation Cell parameters from 600
re¯ections
= 20.0±1.0
= 0.11 mmÿ1 T= 100 (2) K Prism, colourless 0.320.210.20 mm
Data collection
Bruker SMART area-detector diffractometer
'and!scans
5869 measured re¯ections 1607 independent re¯ections 1464 re¯ections withI> 2(I)
Rint= 0.032
max= 27.5 h=ÿ31!32 k=ÿ8!9 l=ÿ10!10
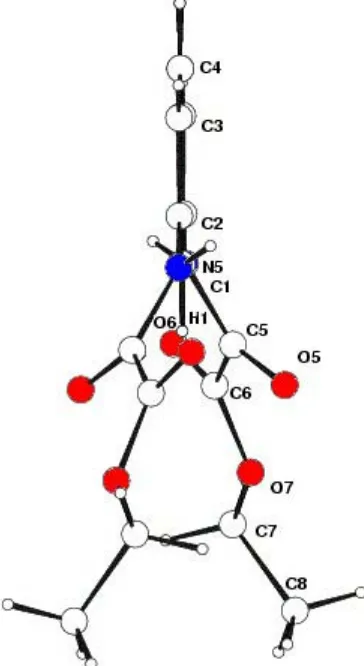
Figure 2
Projection of the molecular structure, showing the twist.
Figure 3
(a) Part of the packing of the title compound, showing the formation of
C(3) andC(4) chain motifs along the [010] direction (R22(11)[C(3)C(4)]);
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.058 wR(F2) = 0.124 S= 1.22 1607 re¯ections 133 parameters
All H-atom parameters re®ned
w= 1/[2(F
o2) + (0.0415P)2
+ 2.0276P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.36 e AÊÿ3
min=ÿ0.22 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
C1ÐC2 1.385 (2)
C2ÐC3 1.386 (3)
C2ÐN5 1.425 (2)
C3ÐC4 1.382 (3)
C5ÐO5 1.223 (2)
C5ÐN5 1.343 (2)
C5ÐC6 1.543 (3)
C6ÐO6 1.204 (2)
C6ÐO7 1.325 (2)
C7ÐO7 1.468 (2)
C7ÐC8 1.499 (3)
C1ÐC2ÐC3ÐC4 ÿ0.4 (3) N5ÐC2ÐC3ÐC4 ÿ179.55 (18) O5ÐC5ÐC6ÐO6 170.82 (19) N5ÐC5ÐC6ÐO6 ÿ9.6 (3) O5ÐC5ÐC6ÐO7 ÿ9.3 (2) N5ÐC5ÐC6ÐO7 170.22 (15) O5ÐC5ÐN5ÐC2 4.1 (3)
C6ÐC5ÐN5ÐC2 ÿ175.42 (16) C1ÐC2ÐN5ÐC5 37.0 (3) C3ÐC2ÐN5ÐC5 ÿ143.9 (2) O6ÐC6ÐO7ÐC7 4.6 (3) C5ÐC6ÐO7ÐC7 ÿ175.28 (15) C8ÐC7ÐO7ÐC6 ÿ169.18 (17)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
C1ÐH1 O5 0.94 (4) 2.450 (12) 2.896 (2) 108.8 (8) N5ÐH5 O6 0.85 (2) 2.35 (2) 2.734 (2) 108 (2) C7ÐH7A O6 0.95 (2) 2.49 (2) 2.717 (3) 93 (1) N5ÐH5 O5i 0.85 (2) 2.17 (2) 3.001 (2) 165 (2) C7ÐH7B O7ii 0.97 (2) 2.49 (2) 3.361 (3) 149 (2) Symmetry codes: (i)x;ÿy;1
2z; (ii)x;1ÿy;12z.
Data collection:SMART(Bruker, 2000); cell re®nement:SMART; data reduction: SAINT (Bruker, 2000); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 2000); software used to prepare material for publication:SHELXL97 andWinGX(Farrugia, 1999).
The authors gratefully acknowledge ®nancial support from CGPI±IPN (grant 5201) and CONACYT±MeÂxico (grant 33438-E).
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans.2, pp. S1±19.
Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995).Angew. Chem. Int. Ed. Engl.34, 1555±1573.
Bondi, A. (1964).J. Phys. Chem.68, 441±451.
Bruker (2000).SMART,SAINTandSHELXTL.Bruker AXS Inc., Madison, Winsconsin, USA.
Desiraju, G. R. (1996).Acc. Chem. Res.29, 441±449.
Dewar, M. J. S. & Schmeizing, H. N. (1968).Tetrahedron,11, 96±120. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837±838.
Martin, S., Beitia, J., Ugalde, M., Vitoria, P. & Cortes, R. (2002).Acta Cryst. E58, o913±o915.
MartõÂnez-MartõÂnez, F. J., Padilla-MartõÂnez, I. I., Brito, M. A., Geniz, E. D., Rojas, R. C., Saavedra, J. B. R., HoÈp¯, H., Tlahuex, M. & Contreras, R. (1998).J. Chem. Soc. Perkin Trans.2, pp. 401±406.
Padilla-MartõÂnez, I. I., MartõÂnez-MartõÂnez, F. J., GarcõÂa±BaÂez, E. V., Torres-Valencia, J. M., Rojas-Lima, S. & HoÈp¯, H. (2001).J. Chem. Soc. Perkin Trans.2, pp. 1817±1823.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Steiner, T. (2002).Angew. Chem. Int. Ed. Engl.41, 48±76.
Vargas, R., Garza, J., Dixon, D. A. & Hay, B. P. (2000).J. Am. Chem. Soc.122, 4750±4755.
supporting information
sup-1
Acta Cryst. (2003). E59, o825–o827supporting information
Acta Cryst. (2003). E59, o825–o827 [doi:10.1107/S1600536803010225]
Diethyl
N
,
N
′
-
m
-phenylenedioxamate
Itzia I. Padilla-Mart
í
nez, Mar
í
a Chaparro-Huerta, Francisco J. Mart
í
nez-Mart
í
nez, Herbert
H
ö
pfl and Efr
é
n V. Garc
í
a-B
á
ez
S1. Comment
The title compound, (I), forms monoclinic crystals (C2/c, Z = 4). A C2 symmetry is crystallographically imposed on the
molecule, thus only one half is present in the asymmetric unit. The bond lengths and angles present the usual features,
with a mean C—C bond length of 1.384 (3) Å (Table 1). The ethyl oxamate –N(H)—C(O)—C(O)—OCH2CH3 group is
almost planar, with an N5—C5—C6—O7 torsion angle of 170.2 (2)°. Both carbonyl groups are antiperiplanar, with an
O5—C5—C6—O6 torsion angle of 170.8 (2)°. The C5—C6 bond distance is coincident with that of 1.541 (1) Å (Dewar
& Schmeizing, 1968) usually assumed for a Csp3—Csp3 single bond, while for the Csp2—Csp2 single bond, the average
value from the literature (Allen et al., 1987) is 1.478 (12) Å. This can be indicative of absence of conjugation, in contrast
with the planarity of the oxamate system, but can be justified by steric hindrance indicated by the O5···O7 = 2.640 (2) Å
contact that is remarkably shorter than the sum of the van der Waals radii (2.84 Å; Bondi, 1964). The two ethyl oxamate
groups are twisted by 34.53 (6)° away from the aromatic ring mean plane (Fig. 2). These results contrast with those
recently reported for its 1,2-isomer, viz. N,N′-o-phenylenedioxamate (Martin et al., 2002). The analysis of this last
compound reveals a molecule with one ethyl oxamate group synperiplanar and other antiperiplanar, both twisted with
respect to the benzene ring as result of steric interactions. The above-mentioned conformation allows the formation of the
intramolecular three-centered hydrogen-bonding interaction (THB) C1—H···O5(O5′). Selected bonds and dihedral angles
are listed in Yable 2.
According to graph-set notation (Bernstein et al., 1995), two adjacent S(6) rings are formed. A particular feature of this
interaction is the O5···H1···O5′ angle of 142 (2)°, and the sum of the angles measured at the donor is 360 (2)° (Fig. 1).
The H1···O5 distance [2.45 (1) Å] is shorter than the mean value reported for similar systems [2.553 (4) Å; Steiner,
2002]; however, it is larger than the values found for other intramolecular THB involving N—H as donor [2.09 (2) Å;
Padilla-Martínez et al., 2001]. These results reveal the relative weakness of the soft C—H···O═C interaction (Vargas et
al., 2000) respect to the hard N—H···O═C interaction (Desiraju, 1996) involving THB systems. The intramolecular
hydrogen-bonding interactions N5—H5···O6 and C7—H7A···O6 (Table 2) complete the overall hydrogen-bonding
scheme which can be depicted as a set of six hydrogen-bonded rings forming an S(5)S(5)S(6)S(6)′S(5)′S(5)′ motif. It is
noteworthy that this last interaction should have some importance in determining the lying of ethyl in the oxamate plane.
Supramolecular layers of the title molecule are formed on the ac plane due to N5—H···O5i [2.17 (2) Å] and C7—
H7B···O7ii [2.49 (2) Å] intermolecular hydrogen bonds (see Table 2 for symmetry codes), which lead to C(4) and C(3)
chains motifs, respectively (Fig. 3a), whose topological motif corresponds to a ring graph-set descriptor R22(11)[C(3)
C(4)] (Bernstein et al., 1995). These parallel chains are generated by translation along [010] direction (Fig. 3 b) and
supporting information
sup-2
Acta Cryst. (2003). E59, o825–o827S2. Experimental
The compound was prepared from 2.3 ml (20.4 mmol) of ethyl chlorooxoacetate and 1.0 g (9.3 mmol) of
1,3-diamino-benzene, according to reported procedures (Martínez-Martínez et al., 1998), to yield 20.0 g (70%) of a pale yellow solid
(m.p. 425–427 K). IR (KBr, cm−1): 3349 (NH), 699 (CO); 1H NMR (300.08 MHz, DMSO-d6, p.p.m.): 10.8 (s, 2H, NH),
8.2 (s, 1H), 7.5 (d, 2H), 7.3 (t, 1H), 4.3 (q, 4H, CH2), 1.3 (t, 6H, CH3); 13C NMR (75.46 MHz, DMSO-d6, p.p.m.): 161.3
(COO), 156.4 (CON), 138.3 (Ci), 129.6 (Cm), 117.7 (2Co), 113.5 (Co), 63.0 (CH2), 14.5 (CH3). Crystals suitable for X-ray
[image:5.610.119.494.182.453.2]analysis were obtained after slow crystallization from acetone.
Figure 1
The molecular structure of the title compound, showing the atom-labelling scheme, the three-centered hydrogen-bonding
interaction C—H···O(O′) and the H5···O6···H7A hydrogen-bonding interactions. Displacement ellipsoids are drawn at the
supporting information
[image:6.610.215.397.79.412.2]sup-3
Acta Cryst. (2003). E59, o825–o827Figure 2
supporting information
[image:7.610.124.486.69.307.2]sup-4
Acta Cryst. (2003). E59, o825–o827Figure 3
(a) Part of the packing cell of the title compound, showing the formation of C(3) and C(4) chain motifs growing along the
[010] directions (R22(11)[C(3) C(4)]); (b) three-dimensional view of the supramolecular layers of the title compound.
(I)
Crystal data
C14H16N2O6 Mr = 308.29
Monoclinic, C2/c a = 25.086 (5) Å b = 7.1225 (14) Å c = 8.2529 (17) Å β = 104.870 (3)° V = 1425.2 (5) Å3 Z = 4
F(000) = 648 Dx = 1.437 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 600 reflections θ = 20.0–1.0°
µ = 0.11 mm−1 T = 100 K Prism, colourless 0.32 × 0.21 × 0.20 mm
Data collection
Bruker SMART area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
5869 measured reflections 1607 independent reflections
1464 reflections with I > 2σ(I) Rint = 0.032
θmax = 27.5°, θmin = 1.7° h = −31→32
k = −8→9 l = −10→10
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.058 wR(F2) = 0.124 S = 1.22 1607 reflections
133 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
supporting information
sup-5
Acta Cryst. (2003). E59, o825–o827Hydrogen site location: inferred from neighbouring sites
All H-atom parameters refined
w = 1/[σ2(Fo2) + (0.0415P)2 + 2.0276P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.36 e Å−3 Δρmin = −0.22 e Å−3
Special details
Experimental. Diffractometer operator H. Höpfl scanspeed_seconds/frame 10 1200 frames measured in phi (0–360) with chi=0 and om=2t h=25 65 frames measured in om (15–35) with chi=280, 2t h=29 and phi=0 Crystal mounted in
perfluorpolyetheroil
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
H1 0.5000 0.105 (5) 0.7500 0.014 (7)*
C1 0.5000 −0.0276 (4) 0.7500 0.0137 (5)
C2 0.46019 (8) −0.1249 (3) 0.8052 (2) 0.0162 (4)
C3 0.46013 (10) −0.3195 (3) 0.8059 (4) 0.0349 (6)
C4 0.5000 −0.4146 (5) 0.7500 0.0548 (13)
C5 0.39661 (8) 0.1366 (3) 0.7962 (2) 0.0142 (4)
C6 0.35751 (8) 0.2201 (3) 0.8944 (2) 0.0147 (4)
C7 0.30890 (9) 0.4968 (3) 0.9306 (3) 0.0183 (4)
C8 0.28844 (10) 0.6681 (3) 0.8280 (3) 0.0254 (5)
N5 0.41957 (6) −0.0250 (2) 0.8636 (2) 0.0149 (4)
O5 0.40436 (6) 0.21590 (19) 0.67286 (16) 0.0168 (3) O6 0.34333 (6) 0.1378 (2) 1.00371 (18) 0.0220 (4) O7 0.34272 (6) 0.39224 (18) 0.84013 (16) 0.0164 (3)
H3 0.4342 (12) −0.385 (4) 0.844 (3) 0.039 (7)*
H4 0.5000 −0.544 (8) 0.7500 0.075 (16)*
H5 0.4097 (9) −0.068 (3) 0.947 (3) 0.019 (6)*
H7A 0.2805 (9) 0.414 (3) 0.942 (3) 0.014 (5)*
H7B 0.3325 (10) 0.527 (3) 1.040 (3) 0.025 (6)*
H8A 0.2629 (11) 0.634 (4) 0.723 (3) 0.033 (7)*
H8B 0.3183 (11) 0.733 (4) 0.801 (3) 0.034 (7)*
H8C 0.2693 (11) 0.751 (4) 0.893 (3) 0.040 (7)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-6
Acta Cryst. (2003). E59, o825–o827C4 0.048 (2) 0.0098 (16) 0.123 (4) 0.000 0.052 (3) 0.000 C5 0.0139 (9) 0.0151 (9) 0.0125 (8) −0.0009 (7) 0.0016 (7) −0.0017 (7) C6 0.0120 (9) 0.0173 (9) 0.0137 (9) −0.0017 (7) 0.0015 (7) −0.0009 (7) C7 0.0219 (10) 0.0166 (10) 0.0193 (10) 0.0019 (8) 0.0109 (8) −0.0039 (7) C8 0.0321 (13) 0.0226 (11) 0.0250 (11) 0.0087 (9) 0.0136 (10) 0.0023 (9) N5 0.0137 (8) 0.0172 (8) 0.0144 (8) −0.0005 (6) 0.0047 (6) 0.0015 (6) O5 0.0188 (7) 0.0175 (7) 0.0154 (7) 0.0021 (5) 0.0066 (5) 0.0016 (5) O6 0.0250 (8) 0.0219 (8) 0.0223 (7) 0.0047 (6) 0.0118 (6) 0.0056 (6) O7 0.0195 (7) 0.0143 (7) 0.0172 (7) 0.0028 (5) 0.0077 (5) 0.0001 (5)
Geometric parameters (Å, º)
C1—C2i 1.385 (2) C5—C6 1.543 (3)
C1—C2 1.385 (2) C6—O6 1.204 (2)
C1—H1 0.94 (3) C6—O7 1.325 (2)
C2—C3 1.386 (3) C7—O7 1.468 (2)
C2—N5 1.425 (2) C7—C8 1.499 (3)
C3—C4 1.382 (3) C7—H7A 0.95 (2)
C3—H3 0.92 (3) C7—H7B 0.97 (2)
C4—C3i 1.382 (3) C8—H8A 0.97 (3)
C4—H4 0.92 (5) C8—H8B 0.96 (3)
C5—O5 1.223 (2) C8—H8C 1.00 (3)
C5—N5 1.343 (2) N5—H5 0.85 (2)
C2i—C1—C2 119.9 (2) O7—C6—C5 109.87 (15)
C2i—C1—H1 120.04 (12) O7—C7—C8 106.54 (15)
C2—C1—H1 120.04 (12) O7—C7—H7A 106.5 (13)
C1—C2—C3 120.25 (19) C8—C7—H7A 113.8 (13)
C1—C2—N5 119.99 (18) O7—C7—H7B 107.1 (14)
C3—C2—N5 119.75 (18) C8—C7—H7B 112.5 (14)
C4—C3—C2 119.1 (2) H7A—C7—H7B 109.9 (19)
C4—C3—H3 120.1 (17) C7—C8—H8A 110.5 (16)
C2—C3—H3 120.7 (17) C7—C8—H8B 110.7 (16)
C3i—C4—C3 121.3 (3) H8A—C8—H8B 107 (2)
C3i—C4—H4 119.36 (16) C7—C8—H8C 108.9 (16)
C3—C4—H4 119.36 (15) H8A—C8—H8C 109 (2)
O5—C5—N5 126.74 (17) H8B—C8—H8C 110 (2)
O5—C5—C6 121.17 (17) C5—N5—C2 123.78 (16)
N5—C5—C6 112.08 (15) C5—N5—H5 117.7 (16)
O6—C6—O7 126.36 (17) C2—N5—H5 118.5 (16)
O6—C6—C5 123.76 (17) C6—O7—C7 116.45 (14)
C2i—C1—C2—C3 0.22 (18) N5—C5—C6—O7 170.22 (15)
C2i—C1—C2—N5 179.3 (2) O5—C5—N5—C2 4.1 (3)
C1—C2—C3—C4 −0.4 (3) C6—C5—N5—C2 −175.42 (16)
N5—C2—C3—C4 −179.55 (18) C1—C2—N5—C5 37.0 (3)
C2—C3—C4—C3i 0.22 (17) C3—C2—N5—C5 −143.9 (2)
supporting information
sup-7
Acta Cryst. (2003). E59, o825–o827N5—C5—C6—O6 −9.6 (3) C5—C6—O7—C7 −175.28 (15)
O5—C5—C6—O7 −9.3 (2) C8—C7—O7—C6 −169.18 (17)
Symmetry code: (i) −x+1, y, −z+3/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C1—H1···O5 0.94 (4) 2.45 (1) 2.896 (2) 109 (1)
N5—H5···O6 0.85 (2) 2.35 (2) 2.734 (2) 108 (2)
C7—H7A···O6 0.95 (2) 2.49 (2) 2.717 (3) 93 (1)
N5—H5···O5ii 0.85 (2) 2.17 (2) 3.001 (2) 165 (2)
C7—H7B···O7iii 0.97 (2) 2.49 (2) 3.361 (3) 149 (2)