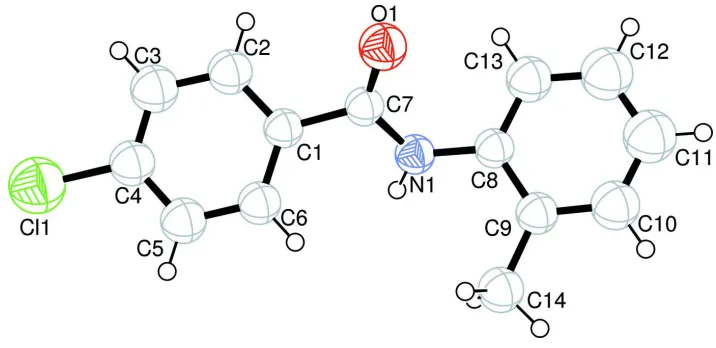
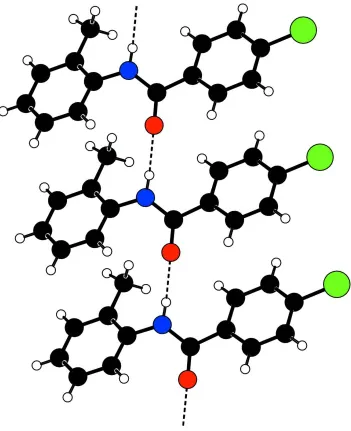
4 Chloro N o tolylbenzamide
8
0
0
Full text
Figure
Related documents