ARTICLE
New York State Cystic Fibrosis Consortium: The First
2.5 Years of Experience With Cystic Fibrosis Newborn
Screening in an Ethnically Diverse Population
Robert Giusti, MDa, Ashley Badgwell, MSb, Alejandro D. Iglesias, MDb,c, and the New York State Cystic Fibrosis Newborn Screening Consortium
Departments ofaPediatrics andbObstetrics and Gynecology, Long Island College Hospital, Brooklyn, New York;cDivision of Medical Genetics, Department of Pediatrics, Beth Israel Medical Center, New York, New York
The authors have indicated they have no financial relationships relevant to this article to disclose.
ABSTRACT
OBJECTIVE.The purpose of this work was to report on the first 2.5 years of newborn
screening for cystic fibrosis in New York.
METHODS.Directors of the 11 New York cystic fibrosis centers were asked to provide
mutation data, demographic data, and selected laboratory results for each patient diagnosed by newborn screening and followed at their center. Summary data were also submitted from the New York newborn screening laboratory on the total number of patients screened, the number of positive screens, and the number of patients that were lost to follow-up. A second survey was submitted by each center regarding the availability of genetic counseling services at the center.
RESULTS.A total of 106 patients with cystic fibrosis were diagnosed through newborn
screening in the first 2.5 years and followed at the 11 Cystic Fibrosis Foundation– sponsored cystic fibrosis care centers in New York. Two screen-negative infants were subsequently diagnosed with cystic fibrosis when symptoms developed. The allele frequency of⌬F508 was 57.4%, which is somewhat lower than the allele frequency of ⌬F508 in the US cystic fibrosis population of 70%. There were 90 non-Hispanic white (84%), 12 Hispanic, 2 Asian, and 1 black infants diagnosed with cystic fibrosis during this period. Five patients were diagnosed secondary to a positive screen based on a high immunoreactive trypsinogen and no mutations.
CONCLUSIONS.Newborn screening for cystic fibrosis has been effectively conducted in
New York using a unique screening algorithm that was designed to be inclusive of the diverse racial makeup of the state. However, this algorithm results in a high false-positive rate, and a large number of healthy newborns are referred for confirmatory sweat tests and genetic counseling. This experience indicates that it would be helpful to convene a working group of cystic fibrosis newborn screening specialists to evaluate which mutations should be included in a newborn screening panel.
www.pediatrics.org/cgi/doi/10.1542/ peds.2006-1415
doi:10.1542/peds.2006-1415
Key Words
newborn screening, cystic fibrosis, immunoreactive trypsinogen Abbreviations
CF— cystic fibrosis
CFTR— cystic fibrosis transmembrane regulator
NBS—newborn screening CDC—Centers for Disease Control and Prevention
IRT—immunoreactive trypsinogen PPV—positive predictive value Accepted for publication Sep 1, 2006
Address correspondence to Ashley Badgwell, MS, Long Island College Hospital, Department of Obstetrics and Gynecology, 97 Amity St, Brooklyn, New York 11201. E-mail: abadgwel@ chpnet.org
C
YSTIC FIBROSIS (CF)is an autosomal recessive genetic disorder caused by mutations of the CF transmem-brane regulator (CFTR) gene, located on chromosome 7, which encodes a chloride channel protein.1More than1100 mutations in the CFTR gene associated with dis-ease have been reported to the Cystic Fibrosis Genetic Analysis Consortium.2 The most common mutation,
⌬F508, accounts for 30% to 88% of CF chromosomes worldwide, depending on race/ethnicity.3In addition to
⌬F508,⬃25 mutations occur with a frequency ofⱖ0.1% in the non-Hispanic white population.2
Advocacy toward newborn screening (NBS) for CF is currently well established. Compelling data are currently available demonstrating the beneficial effect on the nu-tritional status, growth, and intellectual outcomes of infants diagnosed through screening.4In 2005, Wilfond
and Gollust5reported that NBS for CF had been
imple-mented in 15 states. Presently, 19 states have initiated NBS, andⱖ7 additional states are in the planning stag-es.6 Although data are mixed regarding the long-term
benefit on pulmonary outcomes, the general consensus is that the benefit of NBS for CF outweighs any risks. In 2003, the Centers for Disease Control and Prevention (CDC) convened an expert workshop that resulted in a report in 2004 that stated that NBS for CF is justified and recommending that it be considered in all states.7NBS is
intended to identify children at risk for a condition who are in need of confirmatory diagnostic testing.
The initial US CF NBS experimental trials used a single or repeat immunoreactive trypsinogen (IRT) pro-tocol.8,9To improve sensitivity, states that started
screen-ing between 1999 and 2002 chose to add mutation anal-ysis, initially using only the⌬F508 mutation. Published reports summarizing the experience of screening pro-grams in Wisconsin, Colorado, and Massachusetts indi-cate that using an IRT and 25 CFTR multimutation assay achieves improved sensitivity and postscreening predic-tion of CF at the cost of increased referrals for sweat testing and carrier identification.10–12
NBS for CF was implemented in New York in October 2002 as a result of legislation enacted by the state legis-lature. This legislation supplied funding to the state lab-oratory for the purchase of the equipment and staffing needs to implement these new screening protocols. There was no funding allocated for the 11 statewide Cystic Fibrosis Foundation-approved CF care centers where the screen-positive patients are to be referred for confirmatory sweat testing. Several planning meetings were held between the CF center directors and the di-rector of the state laboratory to aid in the implementa-tion of the screening protocol. No funding was allocated to support a statewide coordinator, similar to the posi-tion funded in the state of Massachusetts, to help imple-ment and analyze the effectiveness of the screening pro-gram.
It was concluded that using ⌬F508 as the sole
new-born CF mutation would potentially miss many patients in New York because of its diverse ethnic population. The population of New York State is more than 19 million and is comprised of a proverbial “melting pot” of racial and ethnically diverse groups. Three of every 10 persons belong to 1 of the state’s racial or ethnic minor-ity groups. According to the 2000 US Census, New York has the largest population of people of African/Carib-bean descent in the United States (3 014 385), is among the top 8 states with Hispanic residents (2 867 583), and includes⬎1 million Asian/Pacific Islanders (1 053 794).13
Therefore, in an attempt to minimize missed cases, it was decided that the New York state protocol would consist of a 2-tier algorithm consisting of a single IRT screen and expanded mutation panel. We present ana-lyzed data including mutation frequency, IRT cutoff ef-ficiency, sensitivity, specificity, positive predictive value (PPV), and availability of genetic counseling services from the first 2.5 years of NBS for CF in New York state. In addition, based on this experience, we present our recommendations to improve the protocol.
METHODS
Laboratory Screening Algorithm
A single IRT measurement was performed by the state NBS laboratory on all of the Guthrie card specimens.14
The top 5% of daily IRT results was the cutoff for per-forming a mutation analysis. The 32-mutation panel used was chosen by the state laboratory director because it was available as a panel for the automated equipment purchased for the laboratory (ABI PRISM 3100 Genetic Analyzer, automated capillary DNA sequencer; AME Bioscience, Toroed, Norway). To further improve the sensitivity of the screening program in this ethnically diverse state, very high IRT (top 0.2%) results. even when no mutations were detected. were considered screen positive and referred for a sweat test (Fig 1). Because infants with very high IRT levels are consider-ably more likely to have CF compared with those with lower values, this “fallback” group was intended to screen for infants with CF who do not carry common CF mutations.15 The inclusion of this group, however,
re-sults in an increased false-positive rate.12The birth
hos-pital, pediatrician of record, and the regional CF center are notified of all screen positive infants, and it is the responsibility of the pediatrician and the birth hospital to notify the family of a positive result and refer the infant for sweat testing.
Data Collection
the total number of patients screened, the number of positive screens, and the number of patients who were lost to follow-up. A second survey was submitted by each center regarding the availability of genetic counsel-ing services at their center. The surveys were developed through a joint effort of the CF center directors who comprise the New York Cystic Fibrosis Consortium. Con-ference calls were an essential tool in facilitating the planning of this study. EpiInfo software (CDC, Atlanta, GA) was used to create a database and to analyze data.
RESULTS
Detection of CF-Affected Infants
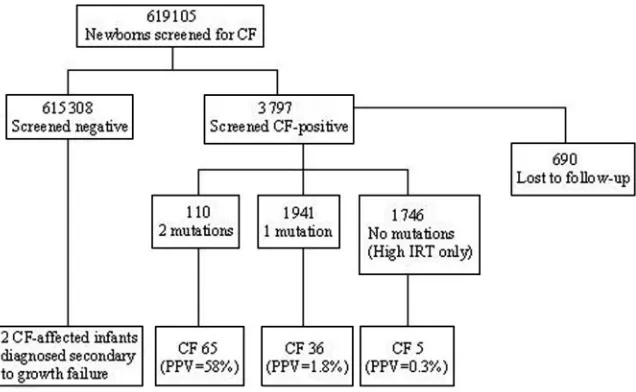
From October 2002 to August 2005, 619 105 infants were screened for CF. During this time period, 3797 were screen positive, 128 infants were diagnosed with CF, and 690 cases were “not closed” because of the inability to notify and arrange referral to regional CF center for a sweat test, per written communication with
the New York Cystic Fibrosis Newborn Screening Con-sortium and Ken Pass, director of the New York Cystic Fibrosis Newborn Screening Consortium (August 2005). Data from the 11 New York Cystic Fibrosis Foundation-sponsored care centers during the first 2.5 years of screening were obtained on 106 patients with CF diag-nosed through NBS and 2 screen-negative infants who, at ⬃24 to 30 months of age, were diagnosed with CF secondary to growth failure. There were 92 non-His-panic white (82%), 12 Hisnon-His-panic, 2 Asian, and 1 black patients diagnosed. Of the 1746 infants referred for sweat test secondary to very high IRT alone (no detected mutations), 5 infants were diagnosed with CF (Table 1). There were 14 screen-positive patients with a high risk of having CF based on the presence of 2 CF mutations who were found to have negative sweat test results. In addition, 2 siblings were referred for sweat testing and diagnosed with CF as a result of the birth of a screen-positive sibling. The 2 screen-negative patients who
FIGURE 1
New York State NBS protocol for CF.
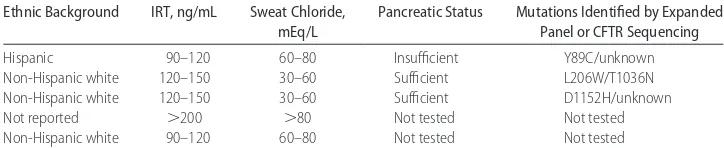
TABLE 1 Ethnic Background, IRT Values, Sweat Chloride Values, Pancreatic Status, and Mutation Status for 5 CF-Affected Infants Screened Positive by High IRT Only (No Mutations Detected by State Panel)
Ethnic Background IRT, ng/mL Sweat Chloride,
mEq/L
Pancreatic Status Mutations Identified by Expanded Panel or CFTR Sequencing
Hispanic 90–120 60–80 Insufficient Y89C/unknown
Non-Hispanic white 120–150 30–60 Sufficient L206W/T1036N
Non-Hispanic white 120–150 30–60 Sufficient D1152H/unknown
Not reported ⬎200 ⬎80 Not tested Not tested
were subsequently diagnosed with CF at ⬃24 to 30 months of age secondary to growth failure were non-Hispanic white. Neither carries a mutation included on the New York state panel, and the initial IRT is only known for 1 of the 2 patients (⬍60 ng/mL).
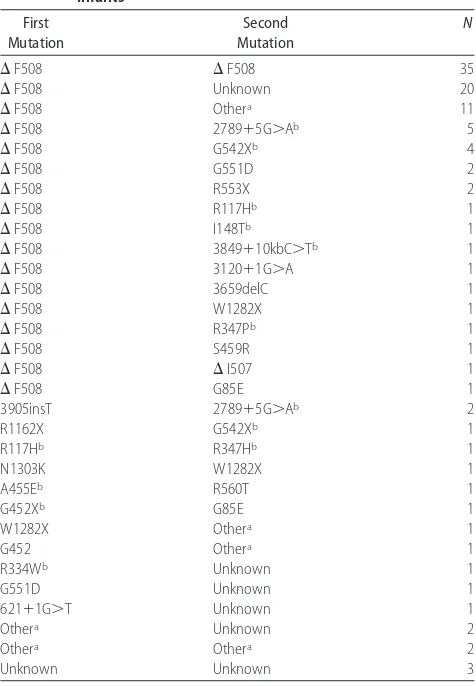
Mutation prevalence data are reported in Table 2. Of the 216 mutations, ⌬F508 was the most common mu-tation, accounting for 57.4% of disease alleles; 32% of patients were homozygous ⌬F508. This is indicative of the variability of CF mutations in this ethnically diverse population. (Among patients with CF living in the United States, the allele frequency of⌬F508 is 70%, and 50% of patients are homozygous for ⌬F508.)16 G542X
and 2789⫹G⬎A were the next frequent mutations (3.2%). G551D and W1282X were each present in 3 patients (1.4%). Four additional mutations were seen twice, 3905insT, R553X, G85E, and R117H (0.9%), and the majority of the other mutations were found in a single patient: N1303K, A455E, R1162X, R334W,⌬I507, 3120⫹1G⬎A, 3659delC, 3849⫹10KBC⬎T, R347D, R347H, R560T, I148T, S549R, and 621⫹1G⬎T. The screening
panel mutations found in Hispanics were ⌬F508,⌬I507, 3849⫹10KBC⬎T, 3120⫹1G⬎A, R334W, and R553X. Among the 12 Hispanic patients, the allele frequency of ⌬F508 was 45%, which is consistent with the reported incidence in this population.17,18 All of the identified
in-fants (n ⫽ 14) in the group with 2 mutations and a negative sweat test were found to have either the R117H or I148T mutations; 7 had the⌬F508/R117H genotype. Twelve mutations from the state panel were not found in any patients: 3905insT, 1717-1G⬎A, 1898⫹1G⬎A, 1078delT, 2184delA, 3876delA, 394delTT, 711⫹1G⬎T, S459N, R347H, I507V, and I506V. After being diagnosed with CF, many patients without 2 identified mutations underwent additional mutation analysis using an ex-panded CFTR mutation panel or CFTR gene sequencing. The decision of whether or not to perform gene sequenc-ing was made at each CF care center. Most commonly, gene sequencing is performed to help to confirm a CF diagnosis when an infant had a borderline sweat test or when only 1 or no mutations have been identified by NBS or an expanded mutation panel. However, some centers also requested genetic sequencing even when sweat testing was clearly diagnostic of CF. Of the addi-tional mutations detected through these means, D1152H was the most common, occurring in 3 patients.
A total of 690 screen-positive patients were lost to follow-up and never had a sweat test performed. The majority of lost cases occurred in the New York City metropolitan area, which has a large percentage of inner city, indigent, and non–English-speaking families. Al-though there were various reasons for the inability to follow-up a positive NBS, the most likely explanation is a lack of resources to support a statewide coordinator who could help to address the problem of the many patients who are lost to follow-up.
Based on these data, the NBS protocol for CF in New York state had a sensitivity of 98%, a specificity of 99.5%, and a PPV of 3.4% (Fig 2.) In the high IRT/no mutations group, the PPV was 0.3%. The 690 screen-positive infants who were lost to follow-up were not included in this calculation. Although the New York state NBS laboratory reported that 128 infants were screened positive for a presumptive diagnosis of CF as a result of NBS during this time period, the 11 CF care centers reported data from only 106 infants diagnosed as a result of NBS.
More than 50% of the positive patients were found to have sweat chloride levels⬎80 mEq/L. A decrease in the upper limit for normal sweat chloride has been sug-gested for infants on the basis of observations that in-fants with elevated IRT and 1 mutation detected and sweat chloride in the range of 30 to 59 mEq/L were at increased risk for having a second mutation present.9,19
Twenty-five percent of infants diagnosed with CF were found to have sweat chloride levels between 30 and 60 mEq/L, which is considered intermediate or
“border-TABLE 2 Genotypes and Frequencies Observed in 108 CF-Affected
Infants First Mutation Second Mutation N
⌬F508 ⌬F508 35
⌬F508 Unknown 20
⌬F508 Othera 11
⌬F508 2789⫹5G⬎Ab 5
⌬F508 G542Xb 4
⌬F508 G551D 2
⌬F508 R553X 2
⌬F508 R117Hb 1
⌬F508 I148Tb 1
⌬F508 3849⫹10kbC⬎Tb 1
⌬F508 3120⫹1G⬎A 1
⌬F508 3659delC 1
⌬F508 W1282X 1
⌬F508 R347Pb 1
⌬F508 S459R 1
⌬F508 ⌬I507 1
⌬F508 G85E 1
3905insT 2789⫹5G⬎Ab 2
R1162X G542Xb 1
R117Hb R347Hb 1
N1303K W1282X 1
A455Eb R560T 1
G452Xb G85E 1
W1282X Othera 1
G452 Othera 1
R334Wb Unknown 1
G551D Unknown 1
621⫹1G⬎T Unknown 1
Othera Unknown 2
Othera Othera 2
Unknown Unknown 3
aOther mutations identified by an expanded mutation panel or CFTR sequencing include: D1152H (n⫽3), 117P, R352W, P750L, 2622H, M470M, P67L, H607R, R74W, V201M, D1270N, 1716G⬎A, A309D, P5L, S492F, S1235R, L206W, T1036N, D110E, Y25X, and Y89C.
line,” and 16 (80%) of 20 patients with sweat chloride between 30 and 60 mEq/L who had a measurement of the fecal elastase were found to be pancreatic sufficient (elastase⬎250).
Genetic Counseling
The survey of genetic counseling services demonstrated that each center has a unique protocol regarding genetic counseling. Six of 11 centers report that a genetic coun-selor is available on the day that the sweat test is per-formed and that the majority of families with a positive screen are seen for genetic counseling on the day of the sweat test. All but 3 centers refer all patients for genetic counseling, 2 only refer those with ⱖ1 mutation, and the third gave no additional information. Regarding other staff, at 1 center, all of the patients seen for sweat testing meet with a doctor, at 3 centers the families speak with a nurse (by telephone at 1 center), and at the remaining 7 centers the patients only meet with the sweat test technician unless 2 mutations are found by screening. Seven centers routinely give out informa-tional handouts. When asked to estimate the percentage of families who used genetic services, 6 centers esti-mated ⬎50%, 4 estimated 25% to 50%, and 1 center estimated 10% to 25%. Nine centers reported that the genetic counseling staff had been able to handle the increased work load. Two centers wrote in unsolicited comments about the problematic lack of reimbursement for genetic counseling services.
DISCUSSION
This survey of CF center directors collected and analyzed data since the implementation of the CF NBS in New York during the first 2.5 years. The results validate and demonstrate that the program has been successful in terms of early detection of CF in an ethnically diverse state. However, despite the evident success of the
pro-gram, the inclusion of 32 mutations in the screening panel and the inclusion of the very high IRT group result in a low PPV; New York has the lowest PPV of any state offering screening.5
One approach to improving the PPV would be to raise the IRT cutoff, which results in a mutation analysis. Wisconsin is currently using the IRT cutoff at 96%.11We
had 2 non-Hispanic white infants who were screen neg-ative and were subsequently diagnosed with CF as a result of growth failure. Increasing the IRT cutoff will reduce the number of false-positive patients but also reduce the number of identified patients with CF. There needs to be ongoing risk benefit analysis that will permit adjustments to the screening algorithm.
The very high IRT group was established as a fallback group to attempt to diagnose infants with some of the ⬎1100 CF mutations not included in the panel. Because the false-positive rate in this group is unacceptably high, another approach to improving the PPV would be to permit a repeat IRT measurement in the very high IRT group.20 In most newborns with CF, the IRT remains
elevated for 2 to 3 weeks but then declines in some patients at 1 month.21In addition, perinatal asphyxia has
been found to correlate with an elevated IRT level pos-sibly related to pancreatic injury.22We have found that
infants hospitalized in NICUs frequently have a tran-siently elevated IRT value because of either a stressful delivery or intrauterine hypoxia, which may result in hypoxia of the pancreas and release of IRT into the bloodstream. Obtaining a repeat blood specimen at 2 weeks in the very high IRT group could help to improve the PPV of the screening algorithm. (Presently, the state NBS laboratory will not accept a repeat specimen for analysis and continues to notify the pediatrician until a sweat test is obtained.)
Another way to improve the sensitivity of the screen-ing protocol is to perform reflex testscreen-ing when the
“com-FIGURE 2
plex” mutations in the NBS panel, R117H and I148T, are found. These mutations are known to have a variable presentation. The I148T mutation is only expected to result in a CF phenotype when present on a gene incis location with the 3199del6 mutation.23In its 2004
revi-sion, the American College of Medical Genetics recom-mended removing the I148T mutation from the CF screening panel “because the frequency of I148T alone is 0.05% and I148T with 3199del6 is constantly lower than 0.1% and because I148T alone does not cause classic CF by itself.”24The R117H mutation is known to be
modi-fied by the 5/7/9T polythymidine tract; R117H/5T incis is associated with CF, but R117H/7T incisand R117H/5T in trans are associated only with congenital bilateral absence of the vas deferens. The ACMG also recom-mends that the 5/7/9T variant be included in CF screen-ing panels “to distscreen-inguish the genotypes of R117H asso-ciated with CF and those assoasso-ciated with [congenital bilateral absence of the vas deferens]”; however, the 5/7/9T variant is not a reflex test in the New York state protocol. We recommend that whenever R117H and I148T mutations are found, the laboratory will perform appropriate reflex testing and will not report a positive unless the modifiers are present. Of 14 infants who were presumptive positive subjects based on the presence of “2 CF mutations” but had negative sweat tests, all had either the I148T or R117H mutation as 1 or both of the detected mutations. When these mutations are included in an NBS panel, there will be a significant false-positive rate. Because of these concerns, the NBS program in the state of Massachusetts initially removed the RII7H mu-tation from the panel but recently decided to re-add it to their panel when patients with this mutation were found to be colonized withPseudomonas aeruginosaeven when the sweat test was negative.25,26
This New York NBS experience indicates that it would be helpful to convene a working group of CF NBS spe-cialists to evaluate which mutations should be included in panels for NBS and to make recommendations regard-ing mutation panel design for use in states that are contemplating a CF NBS. Mutations that should be in-cluded are those that occur with a prevalence of ⬎0.1%.27The top 20 mutations in frequency compiled in
the Cystic Fibrosis Foundation Registry were part of the New York screening panel, and these mutations encom-passed 96% of the mutations that were isolated in New York and 97.5% of recorded mutations in the registry. The additional 12 mutations in the New York panel resulted in finding an additional 6 mutations. A cost versus benefit analysis for the inclusion of these addi-tional rare CF mutations (⬍0.002% in the Cystic Fibrosis Foundation Patient Registry) in an NBS panel should be performed. We detected 20 additional mutations by gene sequencing. The cost of additional CF gene testing and sequencing should be included in this analysis.
Another approach in the development of a screening
panel would be to select CF mutations not just on the basis of relative frequency but to exclude most class IV and V mutations alleles, which are frequently found in pancreatic sufficient patients.19 The functional
conse-quences of CF mutations can be grouped into 5 classes. Class IV and V mutations that affect the transmembrane domain alter the chloride conductance properties of CFTR. Because the flow of ions across the membrane is reduced but not eliminated, some CFTR function is pre-served, which results in a milder phenotype or a non-classic form of CF with less severe lung disease and maintenance of pancreatic function.28,29 Seven
muta-tions in the New York panel are considered “mild” mu-tations, which might explain the high incidence of the infants diagnosed who were pancreatic sufficient (25% of patients who reported fecal elastase). Currently, pan-creatic sufficient patients are detected by NBS, and the benefit of early diagnosis in this group has yet to be documented. One would expect that the initiation of early therapy would improve outcomes in this relatively healthy group; however, currently there are no guide-lines for treating these patients who may not develop symptoms for 10 or 20 years. The experience of having diagnosed adults with CF who have had significant com-plications because of delayed diagnosis would argue for including these mutations and make the decision to eliminate class IV and V (mild) alleles exceedingly diffi-cult. An additional risk occurs for infants with positive NBS results who exhibit either borderline sweat test results or have 2 CF mutations documented in the set-ting of a normal sweat chloride result.30 It is clear that
psychosocial risks can occur in families with a child diagnosed with variant CF, and this diagnosis subjects the family to uncertainty, anxiety, and potentially un-necessary treatment without a clear compensating ben-efit.19 Modification of the mutation panel to exclude
pancreatic sufficient mutations not associated with clas-sic CF assumes that that the risk/benefit and cost analysis does not warrant identifying this group.
caretak-ers to ensure compliance. Our results suggest that the education process must be ongoing both at the state level and locally. We have found presentations at grand rounds and local pediatric society meetings to be the optimal venue for educating primary caretakers. The availability of a statewide coordinator to help in reaching out to these infants is strongly recommended.
Regarding genetic counseling, implementation of NBS screening for CF creates an expanded need for these services. Parents of infants who are carriers of a CF mutation and their extended families benefit from ge-netic counseling. Gege-netic counseling, whether per-formed by a certified genetic counselor or other qualified health care provider, is an effective means to alleviate the cognitive uncertainty and emotional distress that is frequently experienced by parents whose children had a positive newborn screen. Ciske et al31found that parents
who received genetic counseling had a significantly bet-ter understanding of the inheritance of CF, the signifi-cance of being a carrier, and the likelihood for 2 carriers to have an affected child compared with parents who did not have counseling. Tluczek et al32found that
informa-tion that parents received from a genetic counselor or pediatric nurse practitioner is more comforting and use-ful than information from any other source. The need for quality genetic counseling services only further in-creases when a state chooses to adopt an expanded mutation panel. In a state with a diverse ethnic popula-tion, the counselors must have the cultural sensitivity and skills to address the needs of a culturally diverse clientele. The availability of a translator and of printed genetic information materials in many languages is es-sential.
This study confirms that, in New York, although ge-netic counseling is available in at least some form to all parents of screen-positive newborns, each center has a unique counseling protocol. The majority of centers re-ported that their staff has been able to handle the in-creased workload. However, this likely reflects that the majority of parents of screen-positive infants are not receiving genetic counseling, because the existing staff presumably would not be able to meet the demand. Our impression is that although center directors agree that genetic counseling is imperative for the success of the screening program, most centers lack the funds for coun-seling services. When expanding the mutation panel, one of the consequences is the fact that many CF carriers will be found, and there is a significant need for coun-seling of families.7To maximize the efficacy and
mini-mize the psychosocial risks of CF screening, it is essential for all states to ensure equal accessibility to genetic coun-seling services as part of NBS for CF.
Based on this experience, our recommendations to improve the New York state CF NBS program are as follows: (1) consider raising the IRT cutoff, (2) imple-ment a repeat IRT test at 2 weeks for NICU patients and
for the very high IRT group, (3) remove I148T from the mutation panel or add 3199del6 as a reflex test and add polythymidine variant testing to distinguish phenotypes associated with R117H, (4) increase funds for genetic counseling and improve consistency in postscreening genetic counseling, and (5) establish a position for a statewide coordinator whose purpose would be to ad-dress the issue of screen-positive cases that are lost to follow-up and to assure that the data are analyzed and the program is reviewed annually to permit changes to the protocol.
CONCLUSIONS
The experience of New York confirms that a protocol consisting of a single IRT measurement and an expanded mutation panel is effective for CF NBS in an ethnically diverse state. However, the use of an overly sensitive protocol will result in the detection of a significant num-ber of healthy newborns who are carriers of CF. For this reason, states contemplating initiating NBS for CF must consider the need for genetic counseling services when planning their screening algorithm and ensure proper allocation of resources, as recommended by the CDC in their 2004 report.7In addition, states with large inner
city populations should anticipate difficulties in coordi-nating the follow-up of screen-positive infants and de-velop protocols to minimize cases that are lost to follow-up. It is incumbent on all state-mandated screening initiatives to work closely with the directors of CF refer-ral centers to assure adequate follow-up for screen-pos-itive infants and to implement an ongoing evaluation of the screening process so that adjustments to the program can be facilitated.7
ACKNOWLEDGMENTS
Additional authors from the New York State Cystic Fi-brosis Newborn Screening Consortium include the fol-lowing members: Robert Kaslovsky, MD, Albany Medi-cal College; Jack Sharp, MD, Children’s Hospital of Buffalo; Joan Germana, MD, Schneider’s Children’s Hospital; Andrew Ting, MD, Mount Sinai School of Med-icine; Lynne Quittell, MD, Children’s Hospital of New York, Columbia; Maria Berdella, MD, St Vincent’s Cath-olic Medical; Clement Ren, MD, Strong Memorial Hos-pital, Rochester; Cathy Kier, MD, University Medical Center at Stony Brook; Anbar Ran, MD, State University of New York Upstate Medical University; and Nikil Amin, MD, Maria Fareri Children’s Hospital, Westchester.
REFERENCES
1. Welsh MJ, Ramsy BW, Accurso F, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:5121–5188
3. Richards CS, Bardley LA, Amos J, et al. Technical standards and guidelines for CFTR mutation testing. Genet Med. 2002;4: 379 –391
4. Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics.2001;107:1–13
5. Wilfond BS, Gollust SE. Policy issues for expanding newborn screening programs: the cystic fibrosis newborn screening ex-perience in the United States.J Pediatr.2005;146:668 – 674 6. National Newborn Screening and Genetics Resource Center.
National newborn screen status report. Updated April 4, 2006. Available at: http://genes-r-us.uthscsa.edu/nbsdisorders.pdf. Accessed April 24, 2006
7. Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommen-dations for state newborn screening programs. MMWR Morb Mortal Wkly Rep.2004;53(RR-13):1–36
8. Crossley JR, Berryman CC, Elliott RB. Cystic-fibrosis screening in the newborn.Lancet.1979;2:1093–1095
9. Wilcken B, Towns SJ, Mellis CM. Diagnostic delay in cystic fibrosis: lessons from newborn screening.Arch Dis Child.1983; 58:863– 866
10. Comeau AM, Pared RB, Dorkin HL, et al. Population-based newborn screening for genetic disorders when multiple muta-tion DNA testing is incorporated: a cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections.Pediatrics.2004;113:1573–1581
11. Rock, MJ, Hoffman G, Laessig RH, et al. Newborn screening for cystic fibrosis in Wisconsin: nine-year experience with routine trypsinogen/DNA testing.J Pediatr.2005;147(3 suppl):S73–S77 12. Sontag MK, Hammond RB, Zielenski J, Wagener JS, Accurso FJ. Two-tiered immunoreactive trypsinogen-based newborn screening for cystic fibrosis in Colorado: screening efficacy and diagnostic outcomes.J Pediatr.2005;147(3 suppl):S83–S8 13. US Census Bureau. Census 2000 demographic profile
high-lights. Available at: http://factfinder.census.gov/servlet/ SaFFFacts. Accessed April 24, 2006
14. Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ. Laboratory standards and guidelines for pop-ulation-based cystic fibrosis carrier screening.Genet Med.2001; 3:149 –154
15. Gregg RG, Simantel A, Farrell PM, et al. Newborn screening for cystic fibrosis in wisconsin: comparison of biochemical and molecular methods.Pediatrics.1997;990:819 – 824
16. Cystic Fibrosis Foundation Patient Registry.2004 Annual Data Report to Center Directors. Bethesda, MD: Cystic Fibrosis Foun-dation Patient Registry; 2005
17. Sugarman EA, Rolhfs EM, Silverman LM, Allitto, BA. CFTR mutation distribution among U.S. Hispanic and African Amer-ican individuals: evaluation in cystic fibrosis patient and carrier screening populations.Genet Med.2004;6:392–399
18. Heim, RA, Sugarman, EA, Allitto, BA. Improved detection of
cystic fibrosis mutations in the heterogeneous U.S. population using an expanded, pan-ethnic mutation panel. Genet Med. 2001;3:168 –176
19. Lebecque P, Leal T, DeBoeck C, Jaspers M, Cuppers H, Cassi-man J. Mutations of the cystic fibrosis gene and intermediate sweat chloride levels in children.Am J Respir Crit Care Med. 2002;165:757–761
20. Therrell BL, Lloyd-Puryear MA, Mann MY. Understanding newborn screening system issues with emphasis on cystic fi-brosis screening.J Pediatr.2005;147(3 suppl):S6 –S10 21. Rock MJ, Mischler EH, Farrell PM, et al. Newborn screening for
cystic fibrosis is complicated by age-related decline in immu-noreactive trypsinogen levels.Pediatrics.1990;85:1001–1007 22. Rock MJ, Mischler EH, Farrell PM, Bruns WT, Hassemer DJ,
Laessig RH. Immunoreactive trypsinogen screening for cystic fibrosis characterization of infants with false-positive screening test.Pediatr Pulmonol.1989;6:42– 48
23. Rohlfs EM, Zhou Z, Sugarman EA, et al. The I148T allele occurs on multiplehaplotypes: a complex allele is associated with cys-tic fibrosis.Genet Med.2002;4:319 –323
24. Watson MS, Cutting GR, Desnick, RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American Col-lege of Medical Genetics mutation panel.Genet Med.2004;6: 387–391
25. Campbell PW, White TB. Newborn screening for cystic fibrosis: an opportunity to improve care and outcomes.J Pediatr.2005; 147(3 suppl):S2–S5
26. Parad RB, Anne CM, Soultan, Z, et al. Outcomes from a cohort of immunoreactive trypsinogen (IRT)/DNA cystic fibrosis new-born screen (CFNBS) positive infants who have R117H as one or two detected CFTR mutations: worrisome oropharyngeal cultures in infants with normal sweat chlorides.Pediatr Pulmo-nol.2005;(suppl 28):256
27. Rock MJ, Parrell PM. Neonatal screening for cystic fibrosis. In: Chernick V, Boat TF, Willmont RW, Bush A, eds. Kendig’s Disorders of the Respiratory Tract in Children. 7th ed. Philidelphia, PA: Saunders; 2006:861– 865
28. Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in Cystic Fibrosis. Cell. 1992;73: 1251–1254
29. Gallati, S. Genetics of cystic fibrosis.Semin Respir Crit Care Med. 2003;24:629 – 638
30. Parad RB, Comeau AM. Diagnostic dilemmas resulting from the immunoreactive trypsinogen/ DNA cystic fibrosis newborn screening algorithm.J Pediatr.2005;147(3 suppl):S78 –S82 31. Ciske DJ, Haavisto A, Laxova A, Rock LZ, Farrell PM. Genetic
counseling and neonatal screening for cystic fibrosis: an assess-ment of the communication process. Pediatrics. 2001;4: 699 –705
DOI: 10.1542/peds.2006-1415
2007;119;e460
Pediatrics
Robert Giusti, Ashley Badgwell and Alejandro D. Iglesias
With Cystic Fibrosis Newborn Screening in an Ethnically Diverse Population
New York State Cystic Fibrosis Consortium: The First 2.5 Years of Experience
Services
Updated Information &
http://pediatrics.aappublications.org/content/119/2/e460 including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/119/2/e460#BIBL This article cites 26 articles, 4 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/pulmonology_sub Pulmonology
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
DOI: 10.1542/peds.2006-1415
2007;119;e460
Pediatrics
Robert Giusti, Ashley Badgwell and Alejandro D. Iglesias
With Cystic Fibrosis Newborn Screening in an Ethnically Diverse Population
New York State Cystic Fibrosis Consortium: The First 2.5 Years of Experience
http://pediatrics.aappublications.org/content/119/2/e460
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.