S. Pra nathi Reddy et al., Jour. of Sci. Res. in Phar. 2012, 1(1), 1-9
J
ournal of
S
cientific
R
esearch in
P
harmacy
Review Article
Available online thr oug h
www.jsrponline.com
Detecti on Tec hni ques for Eli minating Phar maceutical Impurities: Focus on Ge notoxic Impurities
S. Pranathi Reddy1*, E. Swathi1, Jyothisri S1, T. Siddartha Reddy2, P. Monica3, T. Sowjanya21St. Mary’s College of Pharmacy, St. Francis Street, Secundrabad, India-500025. 2
CMR College of Pharmacy, Kompally, Hyderabad, India.
3
Bharat College of Pharmacy, Ibrahimpatnam, Hyderabad, India.
Received on: 04-02-2012; Revised on: 09-02-2012; Accepted on: 14-03-2012
ABSTRACT
The elimination of organic impurities to produce highly pure drug substan ces is an important goal o f process chemistry. For t he detection o f gen eral impurities, hyphenated techniques(such as HPLC,Electrophoresis) play a critical role in rapid structural identi fication (qualitative detection) and in understanding the mech anisms o f fo rmation o f the impurities, enabling in form ed decisions to control and eliminate the impurities resulting from the chemical process wh ere possible. Drug impurity profiling, i.e. identification, structure elu cidation and quantitative determination o f impurities an d degrad ation products in bulk drug materials and pharmaceutical formulations is one o f the most important fields o f activities in modern pharmaceutical an alysis. The reason fo r the increased importance o f this area is that unidentified, potentially toxic impurities are health hazards and in order to increas e the sa fety o f drug therapy, impurities should be identified and d etermined by selective methods. Genotoxic impurities (GTIs) in pharmaceuticals at trace levels are o f increasing concerns to both pharmaceutical industries and regulatory agenci es due to their potentials for human carcinogenesis. Determination o f these impurities at ppm levels requires highly sensitive analytical methodologies, which poses tremendous ch allenges on an alytical communities in pharmaceutical R&D. This article pro vides an industrial perspective with regard to the analysis o f various structural classes o f GTIs that are commonly encount ered during ch emical dev elopment. The primary aim of this revi ew is to highlight recent advances in qualitative and quantitative detection of impurities at trace levels, with a particular fo cus on GTIs.
Key Words: Geotropic impurities, pharmaceuticals, electrophoresis.
INTRODUCTION
Aim and Scope:
The primary aim of this review is to highlight recent adv ances in qualitative and quantitative detection o f impurities at trace levels, mainly based on the papers published in the previous years. This paper deals with the sources o f pharm aceutical impurities along with qu antitative determination by speci fic methods fo r eliminating pharmaceutical impurities, with a particular focus on GTI‟S (genotoxic impurities).
According to the d efinition o f ICH (Intern ational Con ference on Harmonization) impurity profile o f a drug materi al is "A description of the identified and unidenti fied impurities, present in a n ew d rug substan ce" [1]
.The impurities in drug are unwanted chemicals that remains with the active pharmaceutical ingredients (API), dev elop during formulation and upon aging o f API/drug produ cts. The presence o f these un wanted chemicals even in trace amount may in fluen ce the efficacy and s afety o f pharmaceutical products. These impurities often posses unwanted pharmacological or toxicological effects by which any ben efits from thei r administration may be outweighed [2]. The composition of impurities allows one to draw conclusions regarding the manu facturing o f th e produ cts and its adulteration, which is becoming widespread in all countries of the world therefore, it is necessary to strictly control the quality of pharmaceutical products and to determine the concentration o f foreign impurities at all stages o f produ ction from raw materials to finished medicinal fo rms [2, 3].
Impurities in drugs are originated from v arious sources and ph ases o f the synthetic process and preparation o f pharmaceutical dosages fo rms. However majority of the impurities are characteristics of the synthetic route of the manu factu ring process. Since there are sev eral possibilities of synthesizing a drug, it is possible that the same product o f different sources may give rise to different impurities. According to the international conference on harmonization (ICH) o f technical requirem ents fo r registration of pharmaceuticals fo r human use, impurities are classi fied as org anic impurities, inorganic impurities and residual solvents. Organic impurities may arise from starting materi als, by products, synthetic intermediates and degrad ation products. Inorgani c impurities may derived from the manu factu ring process and are no rmally known and id enti fied as reagents, ligands, inorganic salts, heavy metal, catalysts, filter aids and charcoal et c. Residual solvents are the impurities introduced with solvents [4 - 7].
*Corresponding author:
S. Pranathi Re ddy
St. Mary’s College of Pharmacy, St. Francis Street, Secundrabad, India-500025.
*E-Mail: [email protected]
Mobile No.: +91-9866198327
A general scheme is set for the estimation of the impurity o f bulk drug substances by the rational use o f chrom atographic, spectroscopi c and analytical techniqu es. The various parameters to be ful filled in an impurity pro file o f drug substances are discussed. Impurity is defined as any substance coexisting with the original drug, such as starting material or intermediates or that is fo rmed, due to any sid e reactions. The presen ce o f these unwanted chemicals, even in small amount, may influence the efficacy and safety of the pharmaceutical produ cts. Impurity profiling (i.e., the identity as well as the quantity of impurity in the pharmaceuticals), is now gaining critical attention from regulatory authorities. The different Pharmacopoei as, such as the British Pharmacopoei a (BP), Unite States Pharmacopeia (USP), and Indian Pharmacopoei a (IP) are slowly incorporating limits to allowable levels of impurities present in the API‟s or formulations.
The International Conferen ce on Harmonization of Technical Requirements for Registration o f Pharm aceuticals for Human Us e (ICH) has also published guidelines for validation o f methods fo r analyzing impurities in new drug substances, products, residual solvents and microbiological impurities [8, 9].
Types of Impurities
Organic medi cinal substances are contaminated in exactly the same manner as inorg anic substan ce during th eir manu facturing p rocesses. Since the organic substances belong to a very wide rang e o f ch emical groups and at the s ame time the cont aminating impurities being o f varied nature the task of det ecting the impurities becomes a difficult job. Therefore, the contaminating impurities for o rganic m edicinal compounds can be classi fied into –
(1) Inorgani c impurities. (2) Organic impurities.
(3)Contamination by chemical intermediates [10]
Impurities closely related to the product and coming from the chemical o r from th e biosynthetic route itsel f, Impurities form ed due to spontaneous decomposition of th e drug du ring the storage o r on exposu re to extreme conditions, or the precu rsors which may b e present in the final product as impurities. Impurities pres ent in excess o f 0.1% should be identified and quanti fied by sel ective methods. The suggested structures o f the impurities can be synthesized and will provide the fin al evidence for their structures, previously determined by spectroscopi c methods. Therefo re it is essential to know the structure o f these impurities in the bulk drug in order to alter the reaction condition and to redu ce the quantity o f impurity to an acceptable level. Isolation, identification and quanti fication of impurities help us in various ways, to obtain a pure substance with less toxicity and, safety in drug therapy.
pro filing o f drugs. Impurities in new drug substan ces can b e address ed from two perspectives,
1) The chemical aspect which includes classi fication and
identification of impurities, report generation, listing of impurities in specifications, and a brief discussion o f analytical procedu res.
2) The safety aspect which includes sp eci fic guid ance for
quantifying impurities, pres ent, substantially at lower lev els, in a drug substance used in clinical studies.
Organic Impurities:
The actual and potential impurities most likely to arise during the synthesis, purification, and storag e o f the drug substan ce should be summarized ,based on sound s cienti fic appraisal o f the ch emical reactions involved in the synthesis, impurities associated with raw m aterials that could contribute to the impurity pro file o f the drug substan ce. The labo ratory studies conducted to detect impurities in the drug substance, which include test results o f materials manu factured during the dev elopment process and batches from the commercial pro cesses. The impurity profile of the drug lots, intended for marketing should be compared with those used in dev elopment. The spectroscopic studies (NMR, IR, MS etc. ) conducted to characteri ze the structure o f actual impurities present in the drug substance abov e an app arent level of 0.1% (e.g., calculated using the response factor o f the drug substance) should be d escribed. All recurring impurities above an app arent level o f 0.1% in b atches manu factured by the p roposed commercial pro cess should be identified of these studies.
Inorganic Impurities: Inorganic impurities are norm ally detect ed and quantifi ed using Pharm acopei al or oth er appropri ate stand ards. Carryover o f catalysts to the drug substance should be evaluated during development.
Residual Solvents: The control o f residues o f solvents used in the manu factu ring process for the drug substance should be discuss ed. Acceptance criteria should be based on Pharmacopeial standards, or ICH guidelines or known safety d ata, depends on the dose, duration o f treatment, and route of administration.
Sources of Pharmaceutical Impurities:
Impurities associated in with APIs Organic impurities, Inorg anic impurities, Solvent residues Impurities rel ated to formulation Impurity forms during fo rmulation, Formation of impurities on aging. Medicines are the fo rmulated forms o f active ph armaceutical ingredi ents. There are 2 types o f impurities in medicines:
1) Impurities associated in with active pharmaceutical ingredients. 2) Impurities that form are creat ed during fo rmulation and or with
aging or that are related to the formulated fo rms.
Degradation Products: Impurities can also b e formed by deg radation o f the end product during manu facturing o f bulk drugs. However, degrad ation products resulting from storage or formulation to different dosage forms or aging are common impurities in the medicines. The degradation of penicillin‟s and cephalosporins is a well-known example of degradation products. The presence o f a ß-lactam ring as well as that of an a-amino group in the C6/C7 side chain plays a critical role in their degradation [11].
Reagents, ligands, and catalysts – These chemicals are less commonly found in APIs; however, in some cases they may pose a problem as impurities.
In gen eral, an individual API may contain all o f th e abov e-mentioned types o f org anic impurities at l evels varying from n egligible to significant. A det ailed investigation o f impurities in semi-synthetic penicillin was perform ed both by th e manu factu rers and th e di fferent research g roups. A review paper on penicillin‟s and cephalosporin‟s [11]
describes methods o f isolation, detection, and quanti fication o f degradation p roducts, and antigenic polymeric by-produ cts. Studies show the pres ence o f traces o f ampicillin polymers and hydrolyzed products in the API.7
It has also been found that the presen ce o f cert ain chemicals such as triethylamine has a degradative effect on the product. Ampicillin trihydrate samples having triethylamine content o f 2000 ppm to 4000 ppm (determined by visual color method dev eloped by Gist- Bro cad es, Del ft, Holland)7 were found to be stable under accelerated stability testing. However, the product showed appreciable d egrad ation when triethylamine content became 7000 ppm. Recent pharmacopo eia8 included the limit tests fo r the traces o f impurities present in ampicillin and amoxicillin bulk raw mat erials. The residual solvents associated with these APIs have also been determined [12].
As the organi c impurities are the most common product- as well as process-related impurities, it is the responsibility of both the manufacturers of APIs and the users (ie, fo rmulators) to t ake care o f these impurities acco rding to ICH guidelines or compendia.
In addition, for an optically active single isomer drug there could be enantiomeric impurities present in the API.
Enantiomeric Impurities - The single enantiomeri c fo rm o f a chiral drug is now considered as an improv ed ch emical entity that may o ffer a b etter pharmacological pro fil e and an in creased th erap eutic index with a more favorable advers e reaction pro file [13]. However, the ph armacokinetic p ro file
of levo floxacin (S-isomeric form) and o flox acin (R-isomeric fo rm) are Comparable, suggesting the lack of adv antages o f single isomer in this regard [13]. In any case, cost benefits as well as the patient‟s compliance need to be
considered in selecting drugs. For the manufacturers o f single enantiomeric drug (eutomer), the undesirable stereoisomers in d rug control are considered in the same manner as other org anic impurities. The prominent single isomer drugs, which are being marketed, include levo floxacin (S-o floxacin),
levalbuterol (R-albuterol), esomeprazole (S-omeprazole). Inorg anic
impurities may also derive from the manu facturing pro cesses used fo r bulk drugs. They are normally known and identified and include the following:
Reagents, ligands, and catalysts- The chances o f having thes e impurities are rare: howev er, in some processes, these could creat e a p roblem unless the manu factu rers take prop er care during production.
Heavy metals- The main sources of heavy metals are the wat er used in the processes and the reactors (i f stainless steel reactors are us ed), where acidi fication or acid hydrolysis takes place. These impurities of heavy metals can easily be avoided using demineralized wat er and glass-lined reactors.
Other materials (eg: filter aids, charcoal)- The filters or filtering aids such as centri fug e bags are routinely used in the bulk drugs m anu facturing plants, and, in many cas es, activated carbon is also used. The regul ar monitoring o f fib ers and black particles in the bulk drugs is essential to avoid these contaminations.
Residual solvents are o rganic volatile chemicals used du ring the manu factu ring process or generated du ring the production. It is very di ffi cult to remove these solvents completely by the work-up process; however, efforts should be taken to the extent possible to meet the safety data. Some solvents that are known to cause toxicity should be avoided in the production of bulk drugs. Dep ending on the possible risk to human health, residual solvents are divided into 3 classes.10 Solvents such as benzen e (Class I, 2 ppm limit) and carbon tetrachlorid e (Class I, 4 ppm limit) are to be avoid ed. On the other hand, the most commonly used solvents such as methylene chloride (600 ppm), methanol (3000 ppm), pyridine (200ppm), toluene (890 ppm), N, N-dimethylformamide (880 ppm), and acetonitrile (410 ppm) are o f Class II. Class III solvents (acetic acid, aceton e, isopropyl alcohol, butanol, ethanol, and ethyl acetate) hav e permitted daily exposures o f 50 mg or less per d ay. In this regard, ICH guidelines [14] for limits should be strictly followed.
MATERIALS AND METHODS
High-Performance Liquid Chromatographic (HPLC):
In the majority of cas es the use o f traditional reversed phas e (RP) HPLC conditions and UV detection has been report ed. S everal gen eral studies support the selection of the optimal chromatographic conditions. Van Gyseghem et al. [15, 16], investigated the orthogonality of 38 chromatographic systems with different stationary and mobile phases with di fferent org anic modifiers, pH and column temperature using68 drug substances as model compounds. The same group des cribed a method fo r s creening RP -HPLC columns (27 di fferent brands) o ft en used in impurity pro filing studies b ased on selectivity di fferen ces and overall sep aration p erfo rmances [17]. The widely known Dry lab system is o ften us ed for th e optimization o f HPLC systems fo r impurity pro filing. A further d evelopment o f this using the Plackett–Burman exp erimental design is described by Li and R asmussen [18]. The use of exp erimental design to optimize the separation o f drugs and its impurities is described by M edenica el al[19] taking imanitib mesylate as the example.
Chemometric methods are time-to-time published not only for separation optimization but also fo r improving the quantitative analysis. For example, Vial et al. [20] described the validation o f internal normalization for impurity assays by using the statistical tool “ analysis of covariance”. In a HPLC/diode array UV chemometri c study Wiberg [21] demonstrated on the example o f the impurities o f prilocaine and other d rugs the usefuln ess o f principal compon ent an alysisin selecting suitable wav elengths fo r the quantifi cation. Gavin and Olsen [22] demonstrated on the ex ample o f the impurities of atomoxetine ho w HPLC system optimization can contribute to the “ Quality by design” principle. The same group used HPLC for the impurity profiling o f ph enylmethylamino-propanol (a common starting material in the synthesis o f atomoxetine and fluox etine) to find co rrel ation between th e impurity pro files o f th e drugs and the starting mat erial in the
course o f a quality evaluation strategy formulti-sourced active
pharmaceutical ingredi ent starting materials with the aim of selecting the most suitable vendor fo rthe starting material [23].
Some applications of HPLC with UV detection for the separation
separation and quanti fication o f sev en impurities in paracet amol. The determination o f sev eral process-related impurities and degradants in the steroidal drug lot eprednoletabon ate and its fo rmulations was des cribed by Yasuedaet al. [26]. Medenica et al. [27] developed a method for the sep aration and quantifi cation of the impurities of perindopriltert-butylamine in a tablet fo rmulation.
Drug stability studies require th e use o f stability indicating methods enabling the simultaneous determination of the undecomposed drug and its degradants. The most frequ ently used method is HPLC with UV detection as fo r example in the stability study of amiodarone [28], estradiol [29], gabapentin [30]pipamperone [31] and zolmitriptan [32].
In all cases discussed so far 150-250 x 4.0-4.6 mm HPLC columns packed with RP-stationary phase with particle si ze o f 3.5-5.0 μm was used and the retention times o f the analytes were in the rang e o f 10-20 min. The recently introduced ultra -performan ce liquid chrom atography (UPLC) uses short and narrow-bore columns with small particle size packing. For example, in the course o f the separation and quanti fication o f the impurities of primaquine phosphate by Dongre et al. [48] the dimensions of the column and the C18-silica packing were 50 x 2.1mm and 1.7 μm, respectively, and the separation was achieved within 2 minutes.
Another d evelopment in HPLC stationary phases has been the introduction of monoliths. In addition to many applications, attempts have been mad e to mak e use o f this in drug impurity pro filing as well. For
example, Tsanavaras and Themelis [33] ed a 100 x 4.6 mm Chromolith column
to separat e within 3 minutes acyclovir and its impurity guanine. Using the same column, Liu et al [34] parated and quanti ficated ri fampicine and its four related impurities within 10 mins, while the run time with conventional column was 60 minutes. Monolith column was successfully used by Rocheleau et al. [35] the impurity profiling of taxanes.
A new d evelopment in RP-HPLC is the introdu ction o f oil-in-water emulsion into the mobile phase. This method, named micro emulsion liquid chromatography (MELC) [36, 37] (not to be con fused with micro emulsion electrokinetic chromatography (MEEKC), to be discussed later!) is especially us eful wh en water -soluble pharmaceutical compounds are p resent in non-polar matrices such as creams, ointments or suppositories. Malenovic
et al. [38] success fully used this technique fo r the determination o f the impurities in simvastatin, while a recent application described by M cEvoy et al. [39] is the separation and determination o f five impurities and degradants in paracetamol within 7 minutes in a suppository formulation without the n eed of sample prep aration.
As already mentioned, in the overwh elming majority of cases (in all cases discussed so far) UV det ection was us ed after the HPLC separation. In the case o f poorly UV active drugs sensitive electroch emical det ection can be a useful altern ative. For example, Hanko and Rohrer [40] used integrated pulsed amperom etric d etection for s canning chromatograms in the course o f the separation o f neomy cin sulphate and its impurities by anion-exch ange chromatography. Pulsed electroch emical d etection was used also by Zawilla [41]
et.al for the analysis of amikacin and its 16 impurities.
Another (rather scarcely used) possibility for the separation and determination o f UV-in active impurities in UVinactive drugs by UV det ector is derivatisation prior to the analysis. For example, the Europ ean Pharmacopoei a [42] uses this method in the related impurities test of the above mentioned amikacin and its impurities the derivatising agent being 2,4,6-trinitro-benzen e sulphonic acid. Another example is the work of Medvedovici
et al.[43] who determined an UV-in active impurity
2-[(dimethylamino)methyl]cy clohexanon e in the UV -aciv e drug Tramadol, using a precolumn derivatisation reaction with 2,4-dinitrophenylhydrazine.
A special application fi eld of impurity profiling by HPLC is high-speed counter-current chromatography (HSCCC), a (semi)preparative method where the collected fractions are g enerally an alyzed by HPLC -DAD to determine the relative purities o f each fraction in the course of the preparative isolation of drugs from e.g. medicinal plant matrices [44].
Finally, it is worth mentioning that the literature o f drug impurity pro filing by HPLC until 2003 is summarised in the revi ew o f R ao and Nagaraju while the review o f Olsen et al. deals with method development for impurity profiling.
Thin-Layer Chromatographic (TLC) Methods
Thin-layer chromatography (TLC) is a widely used separation technique becaus e o f its ease o f use, cost-effectiveness, good sensitivity, speed o f s eparation, as well as its cap acity to an alyze multiple samples simultaneously. TLC plays an essential rol e in the early stag e o f drug development when kno wledge about the impurities and degradants in drug substance and drug product is limited.
As a consequen ce o f the dev elopment in the fi eld o f TLC plates (introduction o f high-performan ce thin-lay er chromatography – HPTLC) and the instrumentation (versatile densitometers) TLC plays an import ant role in the quantitative analytical aspects o f d rug impurity pro filing: it is a complementary technique to HPLC. The state-of-art o f TLC in drug impurity pro filing was reviewed by Ferenczi-Fodor et al. [14d].
A few o f the sev eral applications are as follows. Krzek etal [45] determined diclo fen ac and its impurities and Degradants using UV 248 nm to scan the chromatograms. UC scanning without spraying was used by the same group for the d etermination o f cipro floxacin [46] , chlorpromazin e, trifluoperazine, promazin e and doxepin [46] , The HPTLC method of Anjaneyulu et al. [47] is suitable for the quantitation of the impurities of alprazol am. Another HPTLC method was developed by Agbab a et al. [48] for the determination of impurities in omeprazole and pantoprazole. In a study by Naidong et al. [49] the layer was impregnated by sodium-EDTA to increase the selectivity. The separated impurities o f t etracycline and chlort etracycline were measu red by fluorimetric scanning at 400 nm. An SPE/TLC method with fluorescen ce s canning was d eveloped by Ko chan a et al. [50] for the impurity profiling o f 3,4- methylenedioxymethamph etamine in Ecstasy tablets.
Overpressured liquid chromatography (OPLC) a forced flow variant o f HPTLC was used by Bagócsi et al. [51] for the d etermination o f the impurities of norethisterone by fluo rimetric scanning after a sulphuric acid spray. Better selectivity and effi ciency were found with this method than with the pharmacopo eial TLC test.
Electrophoreti c and Related Methods
Capillary Electrophoresis [CE] Since CE is primarily suitable for the separation and qu anti fication o f charged impurities, the first example for its use is related to ammonium ion. This is an important point since ammonium salts as impurities do not contribute to the residue o f ignition and are usu ally not detected by TLC or HPLC methods. Gong et al. [52] determined this cation by non-aqueous CE with indirect UV-detection the UV-probe being imidazole. Schöftner et al. [53] also used indirect UV detection (probe creatinine) for the sep aration and det ermination o f tetra alkyl ammonium salts as impurities in colesev elam hyd rochloride and compared the perform ance of this technique with the other alternative: ion chromatography. Ibandron ate and its impurities are also salt-type materials. Phosphate and phosphate impurities were det ermined by indirect UV d etection (p robe sodium chromate) [53].
Basic materials are proton ated at low pH v alues and become therefo re suitable for impurity profiling by CE. For example Quaglia et al. [54] separated and qu antifi cated five impurities in fenticonazole using aqueous buffer o f pH 2.5 containing trimethyl-�-cyclod extrin as the running bu ffer using direct UV det ection. Similarly low pH and UV detection were used by Sabbah and Scriba [55] in the impurity pro filing o f 3, 4-diaminopyridine and 4-aminopyridine. Saaved ra et al. [56] separated by CE a d egradants o f alprazol am which was not separable by HPLC [57].
The Box-Behnken experimental design was applied in the optimization of the CE parameters o f the separation of ethambutol hydrochloride and it‟s degradants 2-amino-1- Butanol [58]
. Generally speaking it can be stat ed, that – although in certain cases CE can be a good complementary method to chromatographi c techniques – due to its inherent limitation in investigating uncharged molecules it is in the majo rity o f cases not a real altern ative to HPLC in separating and d etermining impurities in drugs.
Electrophoresis-related Chromatographic Techniques The above mentioned inherent limitations of CE in the separation an d det ermination o f impurities in drugs can be overcome by using el ectropho resis-related chromatographic techniques such as micellar electrokinetic chrom atography (MEKC), micro emulsion el ectrokinetic ch romatography (MEEKC) and capillary electroch romatography (CEC). Several pap ers deal with the application o f these techniques for the sep aration and determination o f impurities in drugs. These are good complement ary methods to HPLC but their importance in this field is not comparable with the latter.
The pharmaceutical application o f CEC (among oth ers to impurity profiling) was reviewed by Eeltink and Kok [59]. The advantages o f the method (fl at flow p ro file resulting in sharp er p eaks and b etter effici ency than with HPLC) are success fully exploited by Orlandidni et al. [60] and Quaglia et al. [61] in the separation and determination o f impurities in ketorolac and ibupro fen, respectively. The method was employed in drug research, too, for the impurity pro filing o f exp erimental drug m aterials [62, 63] The paper by Lurie et al. [64] is especially interesting. Making use of the high effici ency o f th e applied columns (non -porous ODS column and sulphonic acid C-12 polymer monolith column) as well as the high sensitivity of the laser-induced fluorimet ric detection even th e geog raphic o rigin o f heroin samples was distinguishable.
MEKC is also a g enerally applicable method in the sep aration and quantitative d etermination o f impurities in drugs as shown on sev eral examples by Hilhorst et al. [65] a further example is the determination of process-relat ed impurities is pioglitazone by Radhakrishna et al. [66].
Of the el ectrically-driven chromatographic sep aration methods
by Furlanetto et al. [67], in atropine sulphate by Bitar and Holzgrabe[68] and in various corticosteroid drugs by Pomponio et al. [69] .
Especially interesting are the papers where the performan ces o f electrically-driven and pressure-d riven ch romatographic and electropho retic methods are compared. A pap er o f this kind was published by Quaglia et al. [70]
on the determination o f the extrem ely toxic N-methyl-4 -phenyl-1, 2, 3, 6 tetrahydropyridine impurity/degrad ants in pethidine. The perfo rmances o f HPLC, CE and MEKC were compared. The shortest run time (less than 5 min) was achi eved by CE while the b est sensitivity by MEKC. In another study Hansen and Sheribah [71] compared aqueous and non-aqu eous CE, MEKC and MEEKC on the ex ample o f impurities in bromazepam. Due mainly to the poor solubility o f brom azep am in water, non-aqueous CE was selected as the most suitable method.
SELECTED TOPICS IN IMPURITY PROFILINGOF DRUGS
Determination of Related Impurities in Pharmacopoeias and ICH Guidelines
As a consequ ence o f th e key role o f impurities in drug s afety issues and the increasing demands regarding the qu ality/purity of drug materials, impurity-related methods hav e becom e the most important parts of monographs o f drug m aterials in pharmacopo eias. The principal ones, the European Pharmacopoeia [72] the United States Pharm acopoei a [73] and the Japanes e Pharmacopo eia [74] contain in the overwhelming majority of cases purity tests based almost ex clusively on TLC and HPLC. In the m ajority o f cases these are g eneral tests: the pharmacopo eias usu ally do not speci fy the impurities in the purity tests; these are expressed as the main component neglecting the di fferences in their responses. Of course this can be source o f uncertain, moreov er sometimes erron eous results. This is why structure elucidation o f impurities is a key issue in modern pharmaceutical analysis. On the b asis o f this, speci fic methods can b e d eveloped and applied for the determination o f individual impurities. This creates the b asis for the success ful application o f the principles outlined in the FDA/ ICH documents [75]
for controlling the limits of individual impurities taking into consideration the toxicity of th e impurity and the daily dose o f the d rug thus greatly contributing to the safety o f drug th erapy. Drug autho rities require that the drug master files o f (new) drug materials cont ain the stru cture o f impurities above 0.1% (in certain cases 0.05%) and in poss ession o f this, speci fic methods for their det ermination. This tendency is slowly gaining ground in the pharmacopo eias, too.
As for the limits for related impurities, these vary from drug to drug and monograph to monograph. In the case o f classical drug mat erials usually 1.0-1.5% of total impurities and 0.5% of individual impurities is permitted but in the case o f new monographs the tendency is to increase the severity of the requirem ents down to about 0.5% of total impurities.
DETERMINATION OF GENOTOXIC IMPURITIES [76-81]
One o f the most important developments in the field of regulatory aspects o f the drug impurity issue is related to genotoxic impurities. Due to the extrem ely high genotoxicity /carcinog enicity o f a certain g roup o f compounds these require mu ch stricter limits to ful fill the requirements o f drug safety.
Decisions to approve, prescribe and consume medicin es involve risk/benefit assessments by regulatory ag enci es, health care p ro fessionals and consumers. For serious or life threatening conditions, drugs with higher risks fo r adverse effects or for serious advers e effects are sometimes acceptabl e. For example, some li fe-saving can cer ch emotherapi es are kno wn human carcinogens. However, if one is su ffering from a li fe threatening tumor, a 5% risk of a secondary, treatment-related tumor is generally considered acceptable. Arguably, the sam e is not true for impurities found in drug substances and drug produ cts; impurities convey only risk with no associated benefit. Drug impurities might be viewed as “ pollutants” in the pharmaceutical world. Much like pollutants in the environment, few people believe that they can be entirely eliminated. The challenge for regulatory agenci es is to promulgat e stand ards th at assure that unavoidable drug impurities impart no or acceptable levels o f risk.
Residual impurities resulting from manu facturing and
fo rmulation, or from deg rad ation o f the active ph armaceutical ingredient (API) and ex cipients, may be present in pharmaceutical products. A subset of these impurities may present a potential for genotoxicity and therefore pose an additional safety con cern to clinical subjects and patients. The pharmaceutical industry and those that regulate it recognize their resp ective obligation to limit genotoxic impurities. Therefore, substantial efforts are made during development to control all impurities at safe concentrations. However, the effo rt made to limit impurities must be commensurate with the risk assessed at each phase o f clinical development, taking into account the extent o f the hazard, the diseas e indication, the size and ch aracteristics o f the exposed population, and the duration of that exposure, as well as the likely delay in the availability of beneficial medicines i f the burden o f limiting or controlling impurity levels is disproportionate. A balance o f these considerations can be d escribed b est as the „„as low as reasonably practicable‟‟ (ALARP) principle. It follows that the presence of impurities with genotoxic (mutagenic) potential may be un avoidable in clinical trial and ultimately in approved and marketed materials. Control of impurities in the
drug substance and deg rad ants in drug product are addressed in ICH Qu ality Guidelines Q3A(R) and Q3B(R), respectively, and the Q3C guideline that deals with residual solvents. However, no speci fic guidan ce for determining acceptable levels for genotoxic impurities is provided in these documents other than to recognize the fact that unusually toxic impurities may require tighter limits of control.
Limiting Genotoxic Impurities:
A major chang e in impurity testing involves a new regulatory guidance design ed to tightly limit substances possessing potential for genotoxicity. The European Agency fo r the Evaluation of Medi cinal Products (EMEA) guidance on genotoxic impurities, which became effective on Jan. 1, 2007, now applies a 1.5 μg daily exposure limit for such substances in most pharmaceuticals based on a precedent application of the threshold o f toxicological concern (TTC) concept to food additives and food contact materials. Befo re the EMEA d raft guidan ce, genotoxic impurities had been addressed only as a footnote in ICH Q3A (R2) " Impurities in New Drug Substances".
In 2004, the Pharmaceutical Research and Manu facturers o f America (PhRMA) form ed a task force to discuss genotoxic impurity limits. Concerned the 1.5 μg/day limit would be applied to drugs synthesi zed in the United States, even while thes e drugs were still in clinical d evelopment, the PhRMA group proposed a staged TTC approach that ties permissible impurity levels to the stage of dev elopment. Because clinical studies are conducted with limited duration o f dosing, the group reasoned th at total exposure is very low, and thus higher intake lev els should be allowable during early clinical studies without a net increase in risk. PhRMA's staged TTC approach applies to all clinical routes and to compounds at all stages o f development, for identi fied and p redict ed impurities. The TTC limits would not apply to already marketed produ cts.
US FDA has considered both the guidelines in setting limits on genotoxic impurities.
Emea Guideline on Limits of Genotoxic Impurities:
The European Medicines Ag ency's (EMEA) Committee for Medicinal Products fo r Human Use (CHMP) published a guid eline on the limits of genotoxic impurities. This guideline recommends dichotomizing genotoxic impurities into those for which there is “ sufficient (experimental) evidence fo r a threshold-related mech anism” and those “ without sufficient (experimental) evidence for a threshold-related mech anism.
“Those genotoxic compounds with suffi cient evidence would be regulated using methods outlined in ICH Q3C, for class 2 solvents. This approach calculat es a “ permitted daily exposure” (PDE) which is cal culated using the NOEL o r LOEL from th e most rel evant animal study plus incorporation o f safety factors. Examples o f genotoxins that may fall into this class include chemicals that induce aneuploidy by interfering with the mitotic spindle, chemicals interfering with activity of topoisomerase or chemicals that inhibit DNA synthesis.
For genotoxic compounds without su ffici ent eviden ce for a threshold-rel ated mech anism, the guideline proposes a policy of controlling levels to “ as low as reasonably practicable” (ALARP principal). This approach speci fies that ev ery effo rt should be made to p rev ent the form ation of such compounds during drug substance synthesis and, if not possible, efforts should b e made to redu ce su ch impurities through techni cal efforts (e.g. purification steps). Compounds falling into this class are generally those that interact with DNA either directly or indirectly such as alkylating agents, intercalating agents or agents gen erating free radicals. Since all exposures to such ag ents theoretically convey som e lev el o f carcinogeni c risk, regulatory agenci es gen erally p erfo rm quantitative risk assessments to calculat e the increas ed levels o f adverse ev ents, such as cancers, that result from particular exposures and set exposure levels which result in “ acceptable” risks; often 1 in 105 or 1 in 106 additional cancers from lifetime exposures.
While the approach described abov e has sound scienti fic support, in most instances sufficient mech anistic data will be lacking with which to decide wh ether a threshold mechanism is applicable for genotoxic impurities. Furthermore, it is also unlikely that data will exist on which quantitative risk assessments can be perfo rmed. The guideline recognizes these limitations and therefore proposes the use of a “ threshold of toxicological concern” (TTC) fo r genotoxic impurities. The TTC refers to a threshold exposure level to compounds that will not pose a significant risk o f carcinog enicity or other toxic effects and was originally dev eloped as a “ threshold of regulation” for food co ntact materi als by the FDA. The draft guideline proposes a TTC of 1.5 µg/day. This threshold corresponds to a 10-5 lifetime risk of cancer, a risk level that the EMEA considers justified due to the benefits derived from pharmaceuticals. Import antly, however, this draft guideline only add resses levels of g enotoxic impurities in marketed products; the guideline is silent on what might constitute acceptable TTCs for drugs du ring dev elopment, especially for trials o f short duration.
1). Genotoxic Compounds with Sufficient Evidence for a Threshold-Related Mechanism
aneuploidy, topoisomerase inhibition, inhibition of DNA synthesis, overloading o f defence mechanisms, metabolic ov erload and physiological perturbations (e.g. induction of erythropo eisis, hyper- or hypothermia). For (classes o f) compounds with clear evid ence for a th reshold genotoxicity, exposure l evels which are without app reciable risk o f g enotoxicity can be established according to the procedur e as outlined for class 2 solvents in the Q3C Note fo r Guidance on Impurities: Residual Solvents. This approach calculat es a “ Permitted Daily Exposure” (PDE), which is derived from the NOEL, or the lowest observ ed effect level (LOEL) in the most relevant (animal) study using “uncertainty factors” (UF).
2). Genotoxic Compounds without Sufficient Evidence for a Threshold-Related Mechanism
The assessment of acceptability of genotoxic impurities for which no threshold mechanisms are identi fied should include both pharmaceutical and toxicological evalu ations. In gen eral, pharmaceutical measurements should be guided by a policy of controlling levels to “ as low as reasonably practicable” (ALARP principle), where avoiding is not possible. Levels considered b eing consistent with the ALARP principle following pharmaceutical assessment should be assess ed for accept ability from a toxicological point of view.
2.1). Pharmaceutical Assessment:
A rationale o f the p roposed fo rmulation/manu factu ring strategy should be provided bas ed on av ailable formulation options and technologies. The applicant should highlight, within the chemical process and impurity pro file o f active substance, all chemical substances, used as reag ents or present as intermediat es, or side-products, known as genotoxic and/or carcinogenic (e.g. alkylating agents). More generally, reacting substances and substances which show “ alerting structure” in terms of genotoxicity which are not shared with the active substan ce should b e considered. Potential alternatives which do not lead to genotoxic residues in the final product should be used if available. A justification needs to be provided that no viable alternative exists, including alternative rout es o f synthesis or formulations, different starting materials. This might for instance include cases wh ere the structure, which is responsible for the genotoxic and/or carcinogenic potential, is equivalent to that needed in chemical synthesis (e.g. alkylation reactions).
If a genotoxic impurity is considered to be unavoid able in a drug substance, technical efforts (e.g. purification steps) should be undertaken to reduce the content o f the genotoxic residues in the fin al product in compliance with safety needs or to a level as low as reason ably practicabl e. Data on ch emical stability o f reactive intermediat es, reactants, and other components should be included in this ass essment. Detection and/or quantifi cation o f these residues should b e done by stat e-o f-the-art analytical techniques.
2.2). Toxicological Assessment:
The impossibility of defining a safe exposure level (zero risk concept) for g enotoxic carcinogens without a th reshold and the realization that complete elimination o f genotoxic impurities from drug substan ces is oft en unachievable, requires implementation o f a concept o f an acceptable risk level, i.e. an estimate of daily human exposu re at and b elow which there is a negligible risk to human health. However, thes e app roach es require availability of adequ ate d ata from long-term carcinogenicity studies. In most cases o f toxicological assessment o f genotoxic impurities only limited data from in vitro studies with the impurity (e.g. Ames test, chromosomal aberration test) are availabl e and thus established app roaches to d etermine acceptable intake lev els cannot be applied. Calculation o f “ safety multiples” from in vitro data (e.g. Ames test) are considered inapp ropriate for justification o f accept able limits. Moreover, neg ative carcinog enicity and genotoxicity data with the drug substance containing the impurity at low ppm levels do not provide sufficient assurance for setting accept able limits for the impurity due to the lack of sensitivity of this testing approach. Even potent mutagens and carcinogens are most likely to remain und etected when tested as part of the drug substance, i.e. at very low exposure levels. A pragmatic approach is therefore needed whi ch recognizes th at the presence o f v ery low levels of genotoxic impurities is not associated with an unacceptable risk.
Benefits of the use of the TTC approach:
The establishment of more widely accepted TTC values would benefit consumers, industry and regulators. By avoiding unnecessary extensive toxicity testing and safety ev aluations when human intakes are below the relev ant TTC value, it will allow limited resources o f time, animal use, cost and expertise to be devoted to the testing and evaluation of those substances with g reat er pot ential to pose risks to human health. In consequen ce, its application will contribute to a considerable reduction in the number of animals used for safety testing.
Considerations on Testing Of Impurities for Genotoxic Potential:
The general framewo rk for genotoxicity testing o f pharm aceuticals is given in two internationally agreed ICH s afety guid elines (ICH S2A, ICH S2B). One of these guidelines (ICH S2B) describes the standard b attery o f tests for genotoxicity fo r drug substance, which consists of:
i. A test for gene mutation in bacteria.
ii. An in vitro t est with cytog enetic ev aluation o f
chromosomal damage in mammalian cells.
iii. An in vivo test for chromosomal d amage in rodent
hematopoietic cells.
The ICH safety guidelines (S2A and S2B) state: „„for compounds giving negative results, the completion of this 3-test battery, performed and evaluated in accord ance with cu rrent recommendations, will usually provide a sufficient level of safety to demonstrate the absence of genotoxic activity.‟‟ In this context, genotoxicity is a b road t erm en compassing effects from mutagenicity through DNA reactivity, DNA damag e, and chromosomal damage, both structural chromosome b reak age and an euploidy. Any compound that produce a positive result in one or more assays in the standard battery has historically been regarded as genotoxic, which may require fu rther testing fo r risk assessment. Thus, the standard battery o f g enotoxicity assays used for testing the API p rovides important in formation abo ut a diversity o f mech anisms o f genotoxicity, both directly and indirectly associated with effects on DNA. Genotoxicants that do not act directly on DNA are typically associated with threshold-rel ated mech anisms, while those that directly t arget DNA (typically detected in assays measuring the reverse or fo rward mutations in a specific gen e with a selection agent) are considered by regulato ry autho rities not to h ave threshold- rel ated mechanisms. Requirements for control o f g enotoxic impurities in pharmaceutical products are di fferent depending upon whether or not there is evidence for a threshold-related mechanism. DNA-reactive g enotoxic impurities for which th ere is no evidence o f a threshold-relat ed mechanism are reg ard ed to be potentially trans-species and multi-organ carcinogens that may require control at relatively low levels. In contrast, it is accepted that impurities acting via threshold-rel ated mechanisms do not require control at similarly low levels. Since the main concern that should drive control o f impurities to relatively low levels is direct DNA reactivity, the primary endpoint o f relevan ce for genotoxic impurities is mutagenicity. DNA-reactive carcinog ens can be identified with a low in cidence o f false n egative results by a procedure that combines the assessment o f chemical structu ral features that in fer DNA reactivity (such as electrophilicity) with a single biological h azard identification test such as a bacterial rev erse mutation test, known as the „„Ames test‟‟. A flexible use of this approach is sometimes advisable since genotoxicity assessment o f impurities in mammalian cells may be need ed for speci fic stru ctural groups, such as carbam ates, which are known carcinogens and that are known to be ineffici ently detected in bacterial genotoxicity tests. A clearly n egative result in an approp riate genotoxicity test (i.e., a bacterial reverse mutation test or mammalian cell assay ) usually indicat es a su fficient level o f s afety to conclude the abs ence o f g enotoxicity for the purpos e o f controlling impurities.
Impurity Classification With Respect To Genotoxic Potential:
It is proposed here that impurities be classified into one o f five classes using data (either published in the literature or from genotoxicity testing) and comparative stru ctural an alysis to identify chemical functional moieties correlated with mutagenicity. The five classes are:
Class 1—Impurities known to be both genotoxic (mutagenic) and carcinogenic:
This group includes known animal carcinogens with reliabl e data fo r a g enotoxic mech anism and human carcinogens. Published data on the chemical structure exist demonstrating the genotoxic nature of the impurity.
Class 2—Impurities known to be genotoxic (mutagenic), but with unknown carcinogenic potential:
This group includes impurities with demonstrated mutageni city based on testing o f th e impurity in conv entional g enotoxicity tests, but with unknown carcinog enic potential.
Class 3— Alerting structure, unrelated to the structure of the API and of unknown genotoxic (mutagenic) potential:
This group includes impurities with functional moieties that can be linked to genotoxicity b ased on stru cture, but which hav e not been tested as isolated compounds. T hey are identi fied bas ed on ch emistry and using knowledge b ased expert systems fo r stru cture– activity rel ationships. The alerting functional moiety is not present in the structure o f the parent API. Some widely recognized alerts for DNA reactivity, i.e., mutagenic activity.
Some generic rule-b ased alerts may b e quite unsp eci fic (e.g., the general alerts for arom atic amines; and fu rther consideration must be given to chemical structural constraints, chemical environment, or experimental data in the assessment of potential genotoxicity. Due to the uncertain rel evan ce o f structural alerts, regulatory action should not be based solely on the presen ce of a p articular fun ctional group; rather the accuracy fo r predicted genotoxicity should be evaluated case-by-case b ased on the av ailable scientifi c literature, additional unpublished (proprietary) dat a on the chemical class and further av ailable (genotoxicity) test results on closely related structures.
This group includes impurities that contain an alerting functional moiety that is shared with the p arent stru cture. The genotoxicity o f the isolated impurity is unknown, but the genotoxicity of the active p rinciple has
been ch aracterized through conventional g enotoxicity testing. Similar chemical constraints and ch emical environment exist fo r the alerting substructure in the impurity and the API.
Alkyl, Aryl, or H, Halogen = F, Cl, Br, I
EWG = Electron withdrawing group (CN, C=O, ester, etc).
Some ex amples of structurally alerting functional groups that are known to be involved in reactions with DNA (this list is not ex haustive) (Muller et al., 2006).
Class 5—No alerting structure or sufficient evid ence for absence of genotoxicity:
This group would be adequately covered by existing ICH Q3A(R), Q3B(R), and Q3C guidelines. It has to be emphasized that this classi fication system would be used sol ely for the pu rpose to decide wh ether an impurity possesses a high level o f risk and is therefo re to be controlled at very low l evels o f daily intak e. Hen ce, this classi fication is not a gen eral classi fication o f genotoxicity.
USFDA Center for Drug Evaluation and Research Guidance on Genotoxic Impurities:
ICH guidance‟s do not provide clear recommendations for handling these types o f impurities. The Center for Drug Ev aluation and Research (CDER) o f th e USFDA is developing guidance to address issues with genotoxic impurities in ph armaceutical produ cts. The CDER is considering the proposals o f the EMEA and PhRMA in developing its guidance. The presence o f genotoxic impurities should be avoided if possible. However, it is recognized that complete remov al is o ften not possible. In these cas es, the amounts of genotoxic impurity present should be limited to a level that represents an insignifi cant increase in risk to clinical t rial subjects or patients.
This level may be based on adequ ate compound-speci fic d ata to calculat e an acceptable risk-sp eci fi c dose or may b e bas ed on a toxicological threshold derived from a robust carcinogeni city databas e. A staged implementation o f the th reshold approach is considered acceptable for products that are und er d evelopment. In applying qu ali fication thresholds, consideration should be giv en to the produ ct's stage o f clinical dev elopment, the maximum duration of d rug administration at that stage, and the p roposed indication. In some cases, increases in the recommended thresholds may be supported in the presence o f a potential pharmacological benefit to patients.
Analysis of Genotoxic Impurities:
In general, impurities should be quantitated at levels ≥0.03 or 0.05% by weight according to ICH guidelines. Genotoxic impurities or potential genotoxic impurities must be controlled at levels significantly lower than the 0.03–0.05% levels that are typically report ed by an HPLC impurity assay. Typically, developing limit tests (e.g. <50 ppm) fo r highly toxic impurities is readily achievable, however it can be di fficult to dev elop a test to control a particular genotoxic impurity at 1 ppm (0.0001% w/w).
“For genotoxic impurities we need very sensitive and sel ective methods. One needs higher sel ectivity to determine ppm – level impurities and selective methods to sep arate low lev els o f g enotoxic impurities from base line noise and other organi c impurities. The typical HPLC methods with a nonspeci fic d etector (e.g. UV) that are used to measure o rganic impurities may not be appropriate to quantitated low ppm levels of genotoxic impurities. The quantitation of low levels (in the range of ppms) o f impurities is the challenging part, and using specific detectors such as MS or MS–MS with LC will significantly improve the method selectivity and the quantitation limit. The goal for scientists is to identify potential genotoxic impurities early in dev elopment, develop an alytical methods to test for these impurities in the intermediates, and if possible, to demonstrate that the manu factu ring process cont rols them before reaching the final drug substance. “ If you eliminate them early enough, then your actual active drug substance is pure, free o f genotoxic impurities”.
Genotoxic Impurities in Food Products:
that may b e g enotoxic carcinogens in a written opinion. The SCF has evaluated genotoxic carcinogens in the diet (such as contaminants and natural toxicants) on a case by-case b asis using a „„weight of evidence‟‟ approach, whereas in the USA (e.g. US EPA), as well as in some European countries (e.g. Norway), a quantitative risk charact erization is commonly performed by mathematical low dose extrapolation o f animal data. It is recognized that current risk analysis approaches to compounds in food that are genotoxic and carcinogenic in exp erimental animals may som etimes incur disproportionate or even unnecessary measu res on the part of regulato rs and industry.
The risk assessment of genotoxic carcinogens h as been considered recently by the European Food Safety Authority (EFSA) and by the WHO/FAO Joint Expert Committee of Food Additives (JECFA). In 2003, EFSA established a Working Group to consider how to improve advice given on the h ealth risks arising from the pres ence in food o f compounds th at are both genotoxic and carcinogenic.
DNA-reactive carcinogens have long been known to be present in the human diet. Most problems with DNA-reactive carcinogens in food arise from chemicals that are either natural food constituents (such as ethyl carb amate) o r cont aminants (such as acrylamide, hetero cyclic amines, polycyclic aromatic hydrocarbons and heterocyclic amines such as PhIP (2- amino-1-methyl-6-ph enylimidazo [4,5-b] pyridin e), which are fo rmed during cooking processes or by fungal toxins (su ch as aflatoxins) b ecaus e they cannot be completely eliminated from the human diet unless the food itsel f is banned. Compounds that are incorporated into food intentionally, either directly (e.g. additives) or indirectly (e.g. residues o f p rocessing aids, pesticides, veterinary drugs or migrants from food cont act materials) are assessed for their g enotoxic and carcinog enic potentials prior to marketing, and compounds that might be DNA-reactive carcinog ens would not be permitted.
Risk characterization:
Risk characterization has been defin ed as „„the quantitative or semi-quantitative estimate, including attendant uncertainties, of the probability of occurrence and sev erity o f adverse effect(s)/ev ent(s) in a given population under defin ed conditions bas ed on hazard identi fication, hazard characterization and exposure. At its best, a risk charact erization „„synthesizes an overall conclusion about risk that is complete, informative and useful for decision makers‟‟. The usual hazard characteri zation approach fo r compounds that cause cancer by non-DNA-reactive mechanisms is to calculat e a health-based guidan ce valu e, such as a tolerabl e daily intake, using the no-observed adverse effect level and uncertainty factors. Such an approach is not used for compounds that cause can cer by DNA-reactive mechanisms. The decision-making environment also may dictate the nature and focus o f the advi ce. Dep ending upon the circumstances, risk managers could be present ed a risk characterization containing only a qualitative judgment of risk. Alternatively, the risk ch aracteri zation may in clude on e or more quantitative estimates o f risk in addition to the qualitative judgment. Whichever method of risk ch aracteri zation is adopted, the output is only as reliable as the quality o f the data used. This applies to all data used, i.e. hazard identi fication, hazard characterization and dose–response an alysis and exposure estimation.
The decision-making environment also may dictate the n ature and focus o f the advi ce. Dep ending upon the circumstances, risk managers could be present ed a risk characterization containing only a qualitative judgment of risk. Alternatively, the risk ch aracteri zation may in clude on e or more quantitative estimates o f risk in addition to the qualitative judgment. Whichever method of risk ch aracteri zation is adopted, the output is only as reliable as the quality o f the data used. This applies to all data used, i.e. hazard identi fication, hazard characterization and dose–response an alysis and exposure estimation.
The only data essential for qu alitative ap proaches, such as ALARA (as low as reasonably achievabl e), is identification o f the compound as a genotoxic carcinog en. An important decision is that the genotoxicity arises via direct cov alent binding to DNA, rath er th an via a mechanism that would show a threshold in the dose–response relationship. Often data are available from a v ariety o f g enotoxicity tests and the d ecision is b ased on a weight of eviden ce appro ach, which takes into account the quality of the available d ata. Although the ALARA p rinciple is an easy to und erstand
concept, it poses some majo r di ffi culties for the risk man ager as it does not discriminate between very potent and very weak carcinog ens and does not take human exposure into account. It does not give any guidance on the magnitude of any risk that might be associated with a given „„reasonably achievable‟‟ low exposure level. Although these diffi culties would point to the alternative appro ach o f calculating the intake levels associated with „„acceptably small risks‟‟, the mathematical models necessary may give widely divergent answers and do not provid e a reliable basis fo r the fo rmulation of realistic risk management advice.
Conclusion: The control of impurities bearing a genotoxic potential in pharmaceutical p roducts and food produ cts has received more and more attention over the past years. The inherent difficulties o f true o r hypothesized linear dose effect rel ationships have led to div erse strategies and risk calculations to achieve a rational lev el o f control. Hence, a uni fied appro ach fo r product dev elopment and mark eting would be useful. The ultimate risk concern fo r genotoxicants is carcinogenicity but carcinogenicity d ata are not available in most cases. Hence, a risk assessment bas ed on surrogate d ata such as structure–activity relationships and limited genotoxicity testing in bacteri al rev ers e mutation tests, knowledge about the relationship between genotoxicity and carcinog enicity, and a generic det ermination o f virtually safe exposu re lev els for the wo rld o f genotoxic carcinogens is proposed. The risk assessment is based on Threshold o f Toxicological Concern (TTC) approach fo r the intake o f g enotoxic impurities over v arious periods o f exposure. This staged TTC is based on knowledge about tumorigenic potency o f a wid e range o f genotoxic carcinogens and can b e used for genotoxic compounds, for which cancer data are limited or not available. The delineated accept able daily intake values of between ~1.5 μg/day for ~ lifetime intake and ~120μg/day for < 1 month are virtually safe doses. Based on sound scienti fi c reasoning, thes e virtually s afe intake valu es do not pose an unacceptable risk to either human volunteers or p atients at any stage o f clinical dev elopment and marketing o f a ph armaceutical product. The intake levels are estimated to give an excess cancer risk of 1 in 100,000 to 1 in a million over a lifetime, and are extremely conservative given the cu rrent lifetime can cer risk in the population o f ov er 1 in 4. This approach, together with a proactive pro cess an alysis of the ch emistry behind the synthesis of the pharmaceutical product and matching analytical capabilities, ensures patient and volunteer safety and m ay not hinder inapp ropriately the fastest possible development o f new medicines to improve patient health.
Apart from pharm aceuticals various di fficulties are pres ented due to presence o f low levels o f food-born e DNA-reactive genotoxic carcinogens, some of which may b e di fficult to eliminate completely from the di et, thus a structured appro ach has been proposes fo r the evaluation o f such compounds. While the ALARA approach is widely applicable to all substances in food that are both carcinogeni c and genotoxic, it does not take carcinogenic potency into account and, therefore, does not permit prioritization based on potential risk or concern.
In the absence o f carcinogenicity dose–response data, an assessment based on comparison with an approp riate threshold o f toxicological concern may be possible. The above approaches were applied to selected food-born e genotoxic carcinog ens. The proposed appro ach is applicable to all substan ces in food th at are DNA reactive genotoxic carcinogens and en ables the formulation o f app ropriate semi-quantitative advice to risk managers.
APPLICATIONS:
Numerous applications have been sought in the areas o f drug designing and in monitoring Quality, stability, and safety o f pharmaceutical compounds, whether produced synthetically, Extract ed from natu ral products or produ ced by recombinant m ethods. The applications include Alkaloids,
amines, amino acids, analgesics, antibacterials, anticonvulsants,
antidepressant, Tranquilizers, antineoplastic agents, local an esthetics, macromolecul es, steroids, miscellaneous [82].
Current Marketed Formulation Which Contain Impurity:
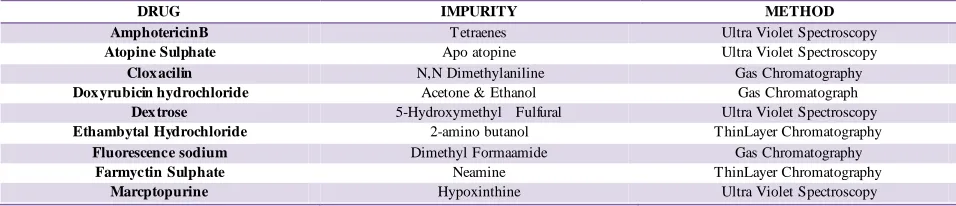
Indian pharmacopoei a speci fi es qualitative, quantitative or semi quantitative tests for limiting known impurities in certain drugs. The list of few such drugs and corresponding impurities is as follows:
Table No. 1: Current Marketed Formulation which contain Impurity
DRUG IMPURITY METHOD
AmphotericinB Tetraenes Ultra Violet Spectroscopy
Atopine Sulphate Apo atopine Ultra Violet Spectroscopy
Clox acilin N,N Dimethylaniline Gas Chromatography
Dox yrubicin hydrochloride Acetone & Ethanol Gas Chromatograph
Dex trose 5-Hydroxymethyl Fulfural Ultra Violet Spectroscopy
Ethambytal Hydrochloride 2-amino butanol ThinLayer Chromatography
Fluorescence sodium Dimethyl Formaamide Gas Chromatography
Farmyctin Sulphate Neamine ThinLayer Chromatography