Medical Sciences
Cellular latency in human immunodeficiency virus-infected
individuals
with high CD4 levels can be detected by the
presence of
promoter-proximal transcripts
(AIDS/Tat/transcriptIonelongaton/Ul cells)
MELANIE
ADAMS*t,
LAMIASHARMEEN*,
JACULYNKIMPTONt, JOSEPH
M.ROMEO*, J. VICTOR GARCIA§,
B. MATUA
PETERLIN*, MARK GROUDINE*,
ANDMICHAEL
EMERMAN4¶*HowardHughesMedicalInstitute,DepartmentsofMedicine, Microbiology, andImmunology, andtLaboratoryMedicine, University ofCalifornia,San
Francisco, CA94143;tPrograminMolecularMedicine and Division of Basic Sciences,Fred Hutchinson Cancer Research Center, Seattle, WA 98104; andIDepartmentofVirologyand MolecularBiology,St. Jude Children's ResearchHospital, Memphis,TN 38105
CommunicatedbySeymourKlebanoff,January 4, 1994(receivedforreviewOctober 22, 1993) ABSTRACT We haveinvestigatedthemolecular basis of
human
immunodeficlency
virustype
1 (HIV-1) latency In atissueculturemodelandIn
HIV-infected
people.We show thatincreasedlevelsofTat, but not Rev, can release theproviruses from latencyinU1 cells. The absence of Tat in thesecells is manifested by the accumulation of promoter-proximal viral
transcripts, whereas thepresence of Tatcorrelates with in-creased expression of viral proteins and an increasein
pro-moter-distal tnscripts. The presence ofpromoter-proximal transcriptsalsoserves as amarker forlatencyin humans. We observed the exclusive presence of promoter-proximal viral transcripts in peripheral mononuclear cellsfromthe
majiority
(10/11)of asymptomaticIlV-infected individualsexamined. Activation of these cells in vigro, andviremiainvivo, correlated
with a switch frompromoter-proimal anscription to pro-moter-distal ascription.These resultssuggest that the con-trol betweenlatencyandreplicationof HIV in vivo isatthe level oftranscription elongation.
Although the progression from sero-conversion to the
ac-quiredimmunodeficiency syndrome(AIDS)frequentlytakes years, viralreplication occurs atall stagesof the infection
(1-4). Inparticular, high levels ofvirus canbedetected in lymph nodes during the asymptomatic stages of disease progression (4, 5).Nonetheless, the factthatthere is
persis-tent replication of human immunodeficiency virus (HIV)
eveninstagesof clinicallatency doesnot meanthat
individ-ual cells do not harbor latent proviruses (called "cellular latency").Indeed, individual infected cells harborproviruses
thatare notexpressed until further cellular activation
(6-8).
Moreover, large numbers oflatently infected cells can be detected both inlymphnodes(5)and in the bloodbeforethe actual onsetof AIDS (4).U1 cells(9)havebeen usedasaconvenienttissueculture
modelofHIV-1latency becausetheir
proviruses
arepoorlyexpressed until cellularactivationbyanumber of
cytokines/
lymphokines orphorbolesters thatactthrough the cellulartranscriptionfactor NF-#cB(reviewedinref.10). U1 cellsare derivedfrom U937cells, which representimmature human
CD4-positive monocytes (11), and contain two integrated
HIV-1proviruses.
Here, we find that constitutive expression of the viral
protein Tat induces the expression of all major viral
tran-scriptsandproteinsinU1cells,whereas the viralproteinRev hasnoeffect. This suggests that U1 cells are held inlatency because ofalack ofTat protein. We developed a reverse
transcription-polymerase chain reaction (RT-PCR) method
todetect the short, promoter-proximal transcripts that are
made fromthevirallong terminalrepeat(LTR)in the absence ofTatand showed thatU1cellssynthesize largeamountsof promoter-proximal transcripts relative to promoter-distal
transcripts.Cellularactivation with phorbolesters, or
intro-duction ofTat alone, increased the relative abundance of promoter-distaltranscripts.
We used this RT-PCR assay to determine the
transcrip-tionalstateofprovirusesof HIV-infectedindividuals in vivo. In 10 of11 HIV-infected individualswith high CD4 levels (>400), promoter-proximal transcripts could be readily de-tected in the absence ofpromoter-distal transcripts in
pe-ripheral blood mononuclear cells (PBMCs). Activation in
culture oflatently infectedcellsfromanasymptomatic
HIV-infectedindividual correlated with virusproduction andthe
induction ofpromoter-distal transcription. Theseresults in-dicatethat cellularlatencybyHIV in vivocanbedetectedby
the presenceoftranscriptionally active provirusesthat tran-scribeonlypromoter-proximalviral RNA.
MATERIALS AND
METHODS
Cells.U1cellsweregrowninRPMI medium with
10%o
calfserumandantibiotics. PBMCswereseparatedfrom antico-agulated wholeblood with Sepracell-MN (Sepratech,
Okla-homaCity, OK).Whencultured,4x
106
cellswereadded to 4 ml ofRPMI medium with 20% fetal bovine serum, 5% interleukin2, and0.12% Polybrene.Onemicrogram ofphy-tohemagglutinin (PHA) per ml and 50 ng of phorbol
12-myristate13-acetate(PMA)per ml wereaddedtothe medium
tostimulate PBMCs or U1 cells.
Reverse Transcription and PCR. RNA and DNA were isolated from 2 x
106
culturedcells or freshly isolated PBMCs as described (12). RNA and DNA were serially diluted separately inwaterwith 1pg
oftRNAper ml. Each serial dilution was separatelyamplified.cDNAsynthesis wasper-formedwith randomprimers ina
20-t4
reaction volume. Acommercially availablePCR carry-overprevention kit,and
the
"hot-start" system Ampli-wax (Perkin-Elmer/Cetus),wasalso added to each sample andcDNAs wereamplifiedin afinalvolumeof 100
td
inanamplification mixturecontaining1 unit of Taq polymerase, 1 unit of uracil N-glycosylase, Abbreviations: AIDS, acquired immunodeficiency disease syn-drome; HIV, humanimmunodeficiency virus; LTR, long terminal repeat;PBMC, peripheralblood mononuclear cell; RT-PCR, reverse transcription-polymerase chain reaction;MuLV, murine leukemia virus; PHA, phytohemagglutinin; PMA, phorbol 12-myristate 13-acetate.
ITowhom reprint requests should beaddressed at: Fred Hutchinson Cancer Research Center,Program in Molecular Medicine, Room C2-023, 1124ColumbiaStreet, Seattle, WA98104.
Proc. Natl.Acad. Sci. USA 91 (1994) 3863
NEO
LXSN I I
-E
tat NEO
LtatSN I =r
LrevSNv
rev NEO
FIG. 1. Structure of retroviral vectors. Vectors were based on the MuLV genome asdescribed (15).Open boxes representthe LTRs; black boxes represent the simian virus 40early promoter, striped boxes are either the tat cDNA or the revcDNA, and NEO is the G418-resistancegene.There was a50-foldincrease in chloramphen-icol acetyltransferase activity in U937 cells infected with LTatSN compared with U937 cells infected with LXSN when both were transiently transfected with a plasmid containing an HIV-1 LTR 5' to the cat gene. There was a>200-foldincrease inp24P8in U937 cells infected with LrevSN compared with U937 cells infected with LneoSN when both were transiently transfected with a HIV-1 proviruscontaining a frame-shift mutation in the second exon of Rev. 0.8-1.0pmol of eachprimer, 200 pM(each) dATP, dGTP, anddCTP,100
AM
dUTP,10mM Tris (pH 8.3), 3mMMgCl2,50mM
KCl,
and 200 ,ugofgelatinperml,for30cyclesusingathermalprofilefor 20 sec at 95°C, for 20 sec at 56°C, and for 40 sec at72°C.Sequences ofprimersandprobes were derived from the
HIV-1..j
LTR. They were as follows: primer 1,GGGTCTCTCTGGTTAGA (positions 1-16); primer 2,
GGGTTCCCTAGTTAGCC(positions 58-42); primer 3,
CT-GCTAGAGATTTTCCACACTGAC (positions 181-158), where +1is the first base of R.
Theamplified product of primer pairs 1 and 2 was 59 bp in length and the product of primer pairs 1 and 3 was 182 bp in length. Amplified products were detected by liquid hybrid-ization as described (13). Each sample was separately am-plified but electrophoresed in
10%o
nondenaturing polyacryl-amide gels and hybridized together in the same lanes for ease ofcomparison. The radiolabeled probe was complementary to the TARloop region of the HIV-1 LTR: TAR loop probe,GCCTGGGAGCTCTCTGG (positions 27-43).
Theprobe was end-labeled with
[y-<32P]ATP
andadded to afinal concentration of 30 pmol to samples that were dena-turedfor 1 minat950C
andhybridized for 3 min at 56°C.Some sampleswere done by an alternative protocol that has equal sensitivity and specificity but is more rapid. Briefly, simultaneous RNA and DNA isolation was done
usingaSnap-o-sol RNA/DNAisolation kit (Biotecx Labo-A
1(1
e" I
{>.1
ratories, Houston), and RNA samples were treated with DNase I. Primer 1(above) was end-labeled with[y-32P]ATP
andgelpurified. The PCR Gem-mediated hot-starttechnique
(Perkin-Elmer) was used to increase the specificity of the amplification. PCR reactions were carried out for 25cycles,
andone-fifth of theproducts were loaded on a 8%denaturing polyacrylamide gelthat was run at 25 mA for 2hr, dried,and exposed to film. Negative controls without RT were per-formed for all samples in addition to an RNA standard.
RESULTS
Tat Expression Induces the HIV-1 Proviruses in U1 Cells in theAbsence ofCellular Activation. Togain insight into the molecularbasis of latency in HIV-infectedpeople, wefirst investigated atissueculture model of cellular latency. The activation of U1 cells by phorbol esters has been shownto increase levels of total viral RNA and especially oflarger singly splicedandunspliced viraltranscripts(14). To deter-mine ifthe maintenance ofproviral latencyin U1 cells is due toinsufficient levels of a viralgene product, specifically Tat orRev, wetested whether or not theincreased expressionof Tator Revfromheterologous promoters could induce these proviruses in U1 cells. To this end, retroviral vectors that expressed either Tat or Rev of HIV-1 were constructed using the genomeof the murineleukemiavirus (MuLV)(Fig. 1).
Hightiter viral stocks wereobtainedthat couldinfecthuman epithelial andlymphoidcell linesand transfer functional Tat or Revinto these cells(Fig. 1 and ref. 15).
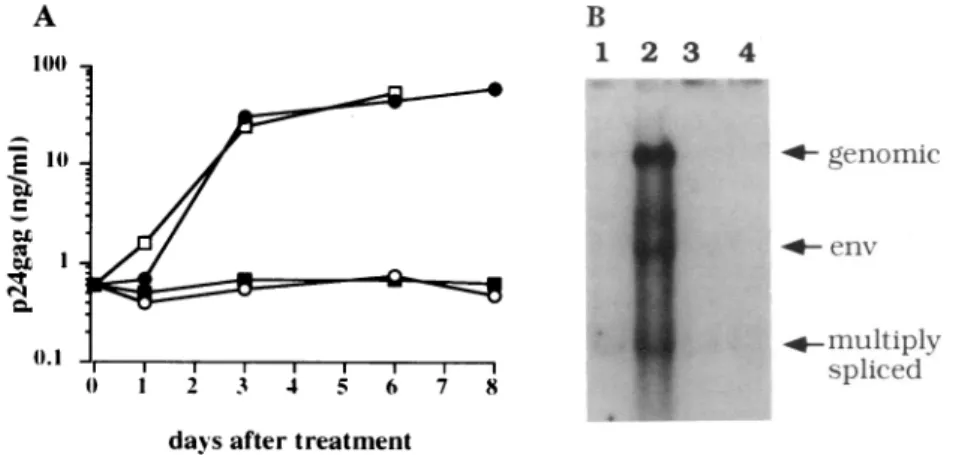
Afterinfection withretroviral vectors thatcontainedTator Rev, viralreplication was assessed by measuring levels of secreted p24w in culture supernatants of U1 cells(Fig.2A).
Some cultures were also infected withwild-type amphotropic MuLVtocontrol for possible effects of MuLV proteins (Fig. 2). Infections of U1 cells with thewild-type MuLV and the retroviral vector encoding Rev did not increase levels of
p24Pm (Fig. 2A). On the other hand, infections with the retroviral vector encoding Tat led to rapid and sustained increases in levels ofp24w. By day 8 after infection, these levels were 100-fold higher than in the control U1 cells and werenearly equivalent to that observed with PMA (Fig. 2A). Uninfected U1 cells express small amounts of doubly splicedviral mRNAs (Fig. 2B, lane 1). We found that Tat aloneincreased the total amounts of viral RNA and increased levelsofsingly spliced and genomic viraltranscriptsrelative tothoseofdoubly spliced mRNAs (Fig. 2B, lane 2). On the
B
1 2 3 4
4- genornic
4. enr
+. nultiplv
spliced daysafter treatment
FIG. 2. Effects ofTatontheexpression of viralproteins in U1 cells. (A)Tatbrings U1 cells out of latency. U1 cells (1X 106)wereinfected with 2 X 105 G418 transforming units of virus (for LtatSN and Lrev SN) or with 1 x 106TCID50 units ofwild-type MuLV. An aliquot of cells wasalso treated withPMA/PHA. The y axis is a logarithmic scale. e, LtatSN-infected cells; o, LrevSN-infected cells; *, wild-type amphotropic MuLV-infectedcells;o, PHA/PMA-treated cells.(B) Sevendaysafterinfection,RNAwascollectedfrom cultures infected with the same retroviralvectors asinA.Tenmicrogramsof total RNAwasloaded per lane. Lane1, unstimulated U1cells;lane2,U1 cells infected withLtatSN; lane 3, U1 cells infected withLrevSN;lane 4, U1 cells infected withwild-type MuLV. Sizes of themajorRNAspecies,which representgenomic, env, andmultiply splicedviraltranscripts are marked.
other hand, the quantity and splicing patterns of these mRNAs did not change when rev or neo genes were intro-duced into these cells (Fig. 2A, lanes 3 and 4). These data
indicate that constitutive expression of Tat induces the
expression of integrated proviruses and can substitute for cellular activation of U1 cells. Thus, the previously observed phenotype of increased expression of singly spliced and
genomicviral transcripts that followed cellular activation of U1 cells (14) probably reflected the increased synthesis of Rev that occurred after sufficiently high levels of Tat were
achieved.
The presence of functional Tat in activated and
nonacti-vatedU1 cells was indirectly assayed by fusing U1 cells with
polyethylene glycolto anindicator cell line that contained a
single integrated copy of the HIV-1 LTR linked to the
P-galactosidase
reporter gene thatis sensitive tolevels of Tat(16). The results of these experiments (not shown)
demon-strated thatnonactivated U1 cells express little Tat but that
cellularactivation increases the synthesis of Tat. This sug-gests that activation of U1 cells by phorbol esters acts, at
least inpart, through increasingTat levels.
Tat Affects the Ratio ofPromoter-Proximal to
Promoter-Distal Viral Transcripts in Ul Cells. Nuclear run-on experi-mentsdemonstrated a steep polarity of HIV-1 transcription
from the LTR that was reversed by Tat (17-20). These
transcriptional states were reflected in the accumulation of short, prematurely terminated TAR transcripts in the ab-senceofTat and of longpolyadenylylatedviral transcripts in
thepresenceofTat(18,21). Given our results that insufficient
levels ofTat were indicative of proviral latency in U1 cells (Fig. 2),wetestedwhether these different viral RNAs could
be usedas markers forproviral latency.
Todeterminelevelsof short and long viral transcripts, we
used quantitativeRT-PCR (12, 13). First, pairs of primers that
correspondedto 5'and 3' ends of TAR (primers 1 and 2)and
tothe 3' end ofthe U5 region (primer 3) were synthesized
(Fig. 3A). Primers1and 2 would amplify both short and long
transcripts, while primers 1and 3 would amplify only RNA
that was longer than the TAR region (Fig. 3A). Given the steeptranscriptional polarityin the absence of Tat, and the
fact that only prematurelyterminated transcripts that contain
theTARRNAstem-loop are stable in cells (17, 19, 22), the
secondsetofprimers(1and 3) should detect very few, if any,
prematurely terminated transcripts. Both sets of primers
amplified with equal efficiencies in vitro transcribed RNA
and plasmid DNA and were sensitive to <100 copies of
nucleic acid (Fig. 3B and data not shown).
Ratios ofshort to longtranscripts were assessed by
com-paring autoradiographsofamplified DNA obtained with both sets ofprimers (Fig. 3B, left). Nonactivated U1 cells
tran-scribed predominantly short transcripts (Fig. 3B, righthand
panel).Thiscorrelatedwith the low levels of doubly spliced
transcripts in U1 cells (Fig. 2B, lane 1). However, 4 and 8
days after infectionwithamphotropic retroviruses coding for Tat(Fig. 1), ratios ofshort to long transcripts decreased by
10-to 100-fold (Fig. 3B, central panels). Moreover, 4 days after the administration ofPHA/PMA,theratios of short to
longtranscripts decreased similarly (Fig. 3B, righthand
pan-el). Activation ofU1 cells with PMA led to a more rapid
qualitative change in HIV-1 transcription, which suggests that activated U1 cells expressed Tat earlier than those
infectedwith amphotropic retroviral vectors. These experi-mentssuggest that escape from virallatency is accompanied
by Tat-mediated increase in elongation efficiency of RNA
polymerase II.
DetectionofPromoter-ProximalTranscriptsin PBMCs from
H1-Infocltd
Individualswith HighCD4Levels. Because of theconcordance between assaysofTatfunctionandRT-PCR inU1cells,
wenextasked whether RT-PCR could be used to detect thisform ofproviral
latencyin PBMCs fromHIV-1-U\tiltI32vlt2lnrncis
--- - .1.i1 5g
TAAR
Primers I and 2 9-;9bp
PTiniers
and3B
DNA(cops #)
1d
I-iIlSeTfl iS
182 ant
U
-TA4F
d(I io 18
PHA PMA
(14
up
ft~~ :W 0
ST-- _
LT-SI' (X)
FIG. 3. Detection of transcripts initiated from the HIV-1 LTR in U1 cells by RT-PCR. (A) Schematic representation of the HIV-1 LTR,oligonucleotideprimers used to amplify viral transcripts, and expectedsizesofPCRproducts. The three primers are shown above the boxes representing U3,
R,
and U5 regions of the HIV-1 LTR (TAR iscontained withinR). Short, prematurely terminated, non-polyadenylylated transcripts (ST) of59 nucleotides and long tran-scripts(LT)of 182 nucleotides, which are amplified by primer pairs 1 and2and1 and3, respectively, are diagramed below the HIV-1 LTR.Thefilled boxrepresentsthe TAR region. (B) Ratios of short tolongtranscriptsin U1cells infected withretroviruses encoding Tat and treated withPHA/PMA. Ten-fold dilutions ofplasmid DNA amplifiedwithbothprimerpairsare shown on the left. On the right, RNAfrom U1cellswasamplified with the primer pairs shown in A. dO, Nonactivated U1 cells;TATd4,U1 cells 4 days after infection with LtatSN (Fig. 1); Tatd8,U1 cells 8days after infection with LtatSN;PHA/PMAd4, U1cells 4 days after stimulation with PHA andPMA.Ratiosofthelongtranscripts (LT) to short transcripts (ST) aregivenbeloweachlane.infected individuals. To this end, RNA and DNA were extracted from PBMCs of 9 HIV-1-infected individuals with high CD4 levels (CD4 cells per mm3 ranged from 1051 to 502, with a median level of 620). None of these individuals was viremic, and all were asymptomatic except for oral candida in two individuals.
In all nine cases, viral DNA could be amplified from cells of these individuals with both primer pair 1 and 3 (Fig. 4C) and primer pair 1 and 2 (Fig. 4D). This indicates that the PBMCs of each individual harbored HIV-infected cells. Moreover, in all nine cases the presence of promoter-proximal transcripts (short transcripts) could be readily de-tected (Fig. 4B) in the absence of promoter-distal transcrip-tion (Fig. 4A). Controls of the reactranscrip-tions without reverse transcriptase verified that the promoter-proximal signal was due to RNA rather than DNA (Fig. 4B, lanes marked with a minus sign). This result indicates all (9/9) of these non-AIDS individuals with high CD4 counts harbored latent proviruses that were transcriptionally active, but deficient in transcrip-tion elongatranscrip-tion.
Proc.Natl. Acad. Sci. USA 91 (1994) 3865
Ul 1 2 3 4 5 6 7 8 9
A a
Ul 1 2 3 4 5 6 7 8 9
B
El
*4W
W
-+ -+ -+ -+ -+ -+ -*1- -+ .+ -+
3 4 5 6 7 8 9
______*TP _*
0 to
-j
z Ul 1 2 3 4 5 6 7 8
D
FIG. 4. Analysis of HIV-1 transcripts in PBMCs of infected individuals with high CD4counts.RNA and DNAwereisolated frombuffycoats ofindividuals labeled1-9 and subjectedtoPCR in thepresence orabsenceofreversetranscriptase (see text). (A) RT-PCR with primer pair 1
and 3(longtranscripts, Fig. 3A) usinganRNAtemplate from the PBMCs of individuals 1-9. The lane marked U1 is RNA from activated U1
cells.(B) RT-PCR with primer pair 1 and 2 (short transcripts, Fig. 3A) usinganRNAtemplate from the PBMCs of individuals 1-9. The lanes
marked U1areRNAfrom activated U1 cells. Theminusorplus sign indicates whetherornotreversetranscriptasewasaddedtothe reaction priortothe PCR. (C) PCR with primer pair 1 and 3 usingaDNAtemplate from the PBMCs of individuals 1-9. HL60 is DNA from HL60 cells
as anegative control. (D) PCR with primer pair 1 and 2 usingaDNAtemplate from the PBMCs of individuals 1 through 9. CD4countsof individuals 1-9were1051, 636, 545, 771, 595, 620,840, 546, and 505, respectively. All patients had received AZTexceptno.9.
promoter-distal transcription, the PBMC with aCD4count of 420wereactivated
autologous cellsinthepresenceof PHA
promoter-proximal transcripts could be unculturedcells (Fig. 5, day 0). Howevc ratios ofshorttolong transcripts decline 5, day 3 andday 14). Increased levels of lo also correlated with the appearance oi
supernatants(undetectableatday 0; 9 pg/
CD4 COUNT 420
Activated I
do d3 d14
Isofoneindividual ondays 3 and 14 after cellular activation,respectively). RNA
Iby coculturewith wasalso extracted from PBMCs oftwoAIDSpatientswith As inFig. 4, only CD4 countsof 10 and 36(Fig. 5).As expected,because of detected in these increased levels of viral replication observed late in the or, after activation, disease(23, 24),the ratio of short tolong transcriptsinthese Ato<10-fold(Fig. patients approachedone(Fig. 5).Promoter-distaltranscripts
)ngtranscriptswere could alsobe detected in the PBMCs ofanasymptomaticand f p24w* in culture p24w negative individual with aCD4 countof 412(Fig. 5).
(mland>100pg/ml These results indicate that cellular latency in vivo can be detected by thepresence ofpromoter-proximal transcripts
412 10 36 and that activation ofproviruses,either in cultureor invivo
during disease progression,marksatransitionto promoter-dO do do distaltranscription.
LT _
ST o
LT:ST <1:1000
FIG.5. Activation of lat distaltranscription. RT-PCJ asymptomatic patients (CE patients (CD4countsof 10 patientswereWalterReed therapyatthe time theblot Walter Reedstage Vandsl
deoxythymidine (AZT)atthe
one asymptomatic patient
PHA/PMA for 3 days and/o cells for 14days. Ratios of estimatedasinFig.3 anda
DISCUSSION
Weshowthat the HIV-1provirusinU1 cellscanbereleased
fromlatency by anincrease in theintracellularlevel of the
viral transactivator, Tat. The absence of Tat is correlated withapredominance of promoter-proximaltranscriptsover
;^.
promoter-distal transcripts.
The addition of Tatalone,
or*w"t | stimulation of the cells with
phorbol
esters,changes
the ratio ofpromoter-proximal topromoter-distal transcripts. These results allowedustodevelop anassayfor the detection ofHIVcellularlatencyininfectedpeople.
1:10 1:10 1:100 1:10 1:1 The molecular basis of HIV-1 latency in cells can be
explained by blocks atseveralstagesof the viral lifecycle. tentlyinfected cells results inpromoter- There is evidence to support incompletely reverse tran-R productsfrom PBMCs ofinfected but scribed RNA(25), unintegrated proviralDNA(7),and
inte-)4 counts of 420 and 412) and AIDS grated proviruses that are either transcriptionally silent or
and36) areshown. Theasymptomatic express only doubly spliced viral mRNAs (26). Our study
stageIandwerenotreceivingantiviral suggeststhe existence of anotherstateinwhich
promoter-odwastaken. TheAIDSpatients were proximal transcription from the HIV-1 LTRpredominates.
tageVI andwerereceiving 3'-azido-3'- Thesestatesof virallatencyandreplicationaredistinguished
etime the bloodwastaken. PBMCs from elongatioficient andelnaicompete
transcrd
(CD4 count 420) were activated with by elongation-deficient andelongation-competent
transcrip-orbycocultivation with stimulated feeder tioncomplexes that produce short transcripts and long tran-longtoshorttranscripts(LT:ST)were scripts, respectively. In all individuals with a CD4 count Ireshown under each lane. above 500 studied here,thepresenceofpromoter-proximal
0 -j
x Ul 1 2
C
9
'Adwwuiwbw&-, , .
.1
;"Mw,-p -f"
transcripts could be detected in the absence of promoter-distal transcripts.
In Ul cells, and in the PBMCs of an asymptomatic HIV-1-infected individual, cellularactivation correlated with
de-creased ratios of short tolong transcripts and new synthesis of viralproteins. We also observe this phenotype in AIDS
patients andinoneHIV-infected individual withan interme-diate CD4 level (Fig. 5). It is possible that detection of
promoter-distal transcriptsmight be a reflection ofthe escape of the activated infected cells from the lymph nodes and, therefore,detection ofpromoter-distal transcripts might bea sensitive markerforthedestruction ofthefollicular dendritic
cell networksthat occurs earlyin disease progression (re-viewed in ref. 27).
Various mechanisms have been proposed to explain the
regulation of assembly ofelongation-competent polymerase complexes (28). Presumably, in latently infected cells that transcribe only promoter-proximal viral RNA, the basal levels of NF-KB (or othertranscription factorsthatact onthe
LTR)aresolow thatlevels of Tat arenotreachedthat would
affect achange frompromoter-proximal topromoter-distal transcription. This might occur in T cells that were once
activated suchthatviralintegration occurred (25, 29) but then becamequiescent and part of the poolof infected"memory"
T cells. Indeed, these T cells as defined by the CD45RO
markercontain abundantproviralDNA(30, 31). Activation
of these cells by antigen would increase the basal level of transcription from the LTR,which,inturn, wouldincrease
the level ofTatand activateproviral expression. Given that
increased levelsofviremiaare observed latein the disease
(23, 24),it ispossible that transcriptional activation ofthese
latent proviruses in the periphery plays a role in T-cell
depletion and in the
progression
toAIDS.M.A. and L.S. contributed equallytothis work. We thank H. Eisen, P. Neiman, K. Peden, members of our laboratories, for discussionsandcomments onthemanuscript,A.Collier for clinical samples,andM. Busch for supportanddiscussion. U1 cellswere obtainedfrom the AIDS Research and ReferenceReagent Program contributed by T. Folks. This work was supported by National Institutes of Health GrantsA130927 (to M.E.), A127291 (toM.G.), and CA59175andALSAC(toJ.V.G.).M.A. wassupported by Grant T32-HL-07100. M.E.is aScholar ofthe American Foundation for AIDS Research.
1. Michael, N.L., Vahey, M.,Burke, D.S. & Redfield, R. R. (1992)J. Virol.66, 310-316.
2. Bagnarelli, P., Menzo, S., Valenza, A., Manzin, A., Giacca, M., Ancarani, F., Scalise, G., Varaldo, P. & Clementi, M. (1992)J. Virol. 66, 7328-7335.
3. Piatak, M., Saag,M.S., Yang, L.C., Clark, S.J., Kappes, J.C., Luk, K.-C., Hahn,B.H.,Shaw,G. M.&Lifson,J. D. (1993)Science259, 1749-1754.
4. Pantaleo, G., Graziosi, C., Demarest, J. F., Butini, L., Mon-troni, M., Fox, C. H., Ornestein, J. M., Kotler, D. P. & Fauci, A.S.(1993) Nature (London) 362, 355-358.
5. Embretson,J.,Zupancic,M.,Ribase,J. L., Burke, A., Racz, P., Tenner-Racz, K.&Haase, A. T. (1993) Nature (London) 362,359-362.
6. Bednarik, D. P. &Folks,T. M. (1992) AIDS6, 3-16. 7. Bukrinsky,M.I.,Stanwick,T.L., Dempsey, M. P. &
Steven-son, M. (1991)Science 254, 423-427.
8. Saksela,K.,Muchmore, E.,Girard,M., Fultz, P.& Baltimore, D.(1993)J. Virol.67,7423-7427.
9. Folks,T.M.,Justement, J.,Kinter, A.,Schnittman, S., Oren-stein, J., Poli, G. & Fauci, A. S. (1988) J. Immunol. 140, 1117-1122.
10. Poli, G. & Fauci,A. S. (1992) AIDS Res. Hum. Retroviruses 8, 191-197.
11. Sundstrom,C. & Nilsson, K.(1976) Int.J. Cancer 17, 565-577. 12. Romeo, J. M., Ulrich, P. P., Busch, M. P. & Vyas, G. N.
(1993)Hepatology17,188-195.
13. Lee, T.-H.,Sunzeri,F.J.,Tobler,L. H., Williams, G. G. & Busch,M. P.(1991)AIDS 5,683-691.
14. Pomerantz, R.J.,Trono,D., Feinberg,M. B.& Baltimore,D.
(1990)Cell61,1271-1276.
15. Garcia, J. V.& Miller,A. D.(1994)AIDSRes. Human Ret-roviruses 10, 47-52.
16. Kimpton,J.& Emerman,M.(1992)J. Virol. 66, 2232-2239. 17. Feinberg,M.B.,Baltimore,D. &Frankel,A. D.(1991)Proc.
Nati.Acad. Sci. USA88, 4045-4049.
18. Kao, S.Y., Calman, A. F., Luciw, P. A. & Peterlin, B. M. (1987)Nature(London) 330,489-493.
19. Laspia,M. F.,Rice, A. P.& Mathews,M. B.(1989) Cell 59, 283-292.
20. Marciniak, R. A. &Sharp, P. A. (1991)EMBOJ. 10, 4189-41%.
21. Lu, X.,Welsh,T. M. &Peterlin, B. M. (1993)J. Virol. 67, 1752-1760.
22. Selby, M.J., Bain, E.S., Luciw, P. A. & Peterlin, B. M. (1989)Genes Dev.3, 547-558.
23. Coombs, R.W., Collier, A.C., Allain, J. P., Nikora, B., Leuther, M.,
GQerset,
G. F. & Corey, L. (1989)N.Engl. J. Med.321,1626-1631.24. Ho,D. D.,Moudgil,T. &Alam,M. (1989) N.Engl.J.Med. 321, 1621-1625.
25. Zack,J. A.,Arrigo,S. J.,Weitsman, S.R.,Go,A.S., Haislip, A. &Chen, I. S.Y. (1990)Cell 61,213-222.
26. Seshamma, T., Bagasra, O., Trono, D., Baltimore, D. & Pomerantz,R. J.(1992)Proc.Natl. Acad.Sci. USA 89,10663-10667.
27. Fauci,A. S.(1993)Science262, 1011-1018.
28. Krum,A.,Meulia,T.&Groudine,M. (1993)BioEssays 15, 1-7. 29. Stevenson, M., Stanwick, T.,Dempsey, M.& Lamonica, C.
(1990)EMBO J.9, 1551-1560.
30. Willerford, D. M.,Gale, M. J.J., Benveniste, R.E., Clark, E. A.&Gallatin,W. M. (1990)J.Immunol. 144, 3779-3783. 31. Schnittman, S.M., Lane, H.C., Greenhouse,J., Justement,