Abstract
Introduction
The nectin family of transmembrane cell adhesion molecules contains four members, nectins 1-4
(in mice, Nectin-1, -2, etc., previously Pvrl-1 etc.; in humans,e.g. NECTIN1, previously PVRL1).
Each consists of an intracellular C-terminal tail with an afadin binding domain, a transmembrane
region, and an extracellular domain with three immunoglobulin-like (Ig-like) loops. Nectins form
calcium-independent cell-cell adhesions by homodimerizing in cis and preferentially forming
heterotypic dimer-dimer interactions in trans (Takai et al., 2008) (Figure 1). Cooperatively with
the adaptor protein afadin (Afdn, formerly Mllt4), nectins localize to adherens junctions (AJs),
where calcium-dependent, cadherin-based adhesions are also located. Additionally, nectins can
regulate tissue patterning via a cell sorting mechanism. Hair cells and supporting cells in the
mouse cochlea sort into a checkerboard pattern based on differential nectin expression (Togashi
et al., 2011), and a similar nectin-dependent cell sorting mechanism patterns the olfactory
epithelia (Katsunuma et al., 2016). Furthermore, the Drosophila Nectin homolog, Echinoid (Ed)
promotes similar cell sorting function, encouraging the formation of AJs between cells with
similar Ed expression patterns (Wei et al., 2005) as well as directing the assembly of an
actomyosin cable at the interface of cells that do and do not express Ed (Laplante and Nilson,
2006). During dorsal closure, sheets of Ed+ epithelial cells migrate over Ed- amnioserosa and
fuse at the dorsal midline. The boundary of Ed expression defines the leading edge of the
migrating epithelia and establishes a contractile actomyosin cable that ensures controlled
zippering of the epithelial sheets (Laplante and Nilson, 2006). Loss of the Ed-expression
boundary abolishes the actomyosin cable and results in defective dorsal closure, characterized by
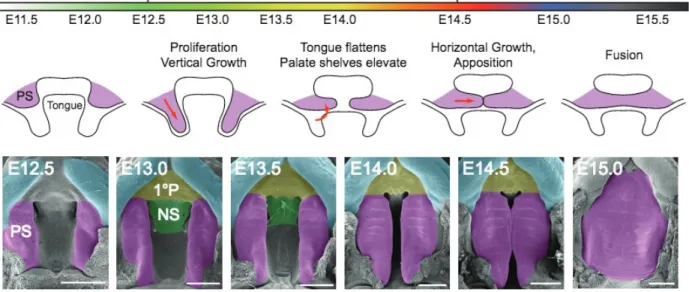
Much like Drosophila dorsal closure, secondary palate formation in mammals involves
the fusion of two distinct epithelial fronts to form a single continuous tissue (Jacinto et al., 2002).
The secondary palate begins as two protrusions, called palatal shelves, which grow downward
from the maxillary prominence toward the floor of the mouth, adjacent to the tongue. The
shelves then elevate above the tongue, approximate, and fuse. The epithelium at the site of fusion
(the medial edge epithelial, or MEE, before fusion; called the medial epithelial seam, or MES,
after fusion) is replaced by a continuous mesenchyme that later ossifies (Figure 2). The
mechanism by which MES is replaced is not fully understood, although apoptosis, migration,
extrusion, and epithelial-mesenchymal transitions have all been reported (Ahmed et al., 2007; Jin
and Ding, 2006; Kim et al., 2015; Nawshad et al., 2004; Shuler et al., 2004; Yu et al., 2009).
Cleft palate (CP) is a complete or partial failure of the palatal shelves to fuse, resulting in a
shared oral cavity and nasal sinus.
A growing body of clinical and experimental evidence suggests that nectins play an
important role in mammalian palatogenesis. Cleft lip/palate-ectodermal dysplasia syndrome
(CLPED1; OMIM 225060; also called both Zlotogora-Ogur syndrome and Margarita Island
ectodermal dysplasia) has been linked to nonsense and frameshift mutations in the NECTIN1 that
truncate the protein before the C-terminal afadin binding domain (Suzuki et al., 2000).
Additionally, Suzuki et al. (2000) observed that NECTIN2 is located close to a genomic locus
associated with non-syndromic CP. Homozygous mutations in NECTIN4 have been identified in
affected members of two families displaying ectodermal dysplasia-syndactyly syndrome-1
(EDDS1; OMIM 613573) (Brancati et al., 2010), although these individuals do not present with
It has been previously reported that nectin-1 and -2, but not -3, are expressed in the MEE
(Yoshida et al., 2012), although nectin-4 expression in the palate has not been previously
described. Here, I address this knowledge gap with preliminary work characterizing nectin-4
expression in the developing mouse palate, which suggests that it is expressed in the palatal
epithelium. Furthermore, isolated nectin-4+cells in the mesenchyme and cell clusters in the nasal
sinus, directly above the MES, were observed, and nectin-4 expression was in the apical contacts
between tongue and palatal shelf epithelial cells. These observations suggest that the
sensing/sorting functions of nectins, particularly nectin-4, might be important for palate fusion.
Our lab has shown that loss of Afdn in the palatal epithelia of mice in early development
causes highly penetrant CP (Lough et al., in review) (Figure 3). Since afadin links nectins to
other AJ components and anchors nectin-based adhesions to actin cytoskeleton, and given the
known clinical correlation between nectins and CP, we expect that nectin loss in mice should
functionally recapitulate Afdn loss and these mice should present with CP. Single knockouts of
Nectin-1, -2, and -3 (no knockout model of Nectin-4 currently exists)in mice fail to support this
hypothesis, although this may be the result of redundancy since multiple nectins are frequently
expressed in overlapping patterns. Additionally, analysis of compound nectin mutants is
technically challenging, as germline Nectin-1/3 knockouts are embryonic lethal (Yoshida et al.,
2010), and no floxed Nectin alleles exist.
I have designed and produced short hairpin RNA (shRNA) lentiviral vectors that target
Nectin-2 and -4. Lentiviruses are a class of retroviruses that stably integrate into the host genome
upon infection, and thus can be readily engineered for gene therapy purposes (Escors and
Breckpot, 2010). The lentiviruses we use harbor shRNA cassettes containing 21-mers
through the Drosha/Dicer RNAi pathway (Wilson and Doudna, 2013). Our lentiviral constructs
also contain a puromycin-resistance gene that allows the generation of stably-transduced
keratinocyte cell lines, when grown in low (1-2 µg/ml) concentrations of puromycin. Stable cell
lines were used to quantify knockdown efficiency by qPCR. A graduate student in the lab,
Kendall Lough, is also generating shRNAs to target Nectin-1 and -3.
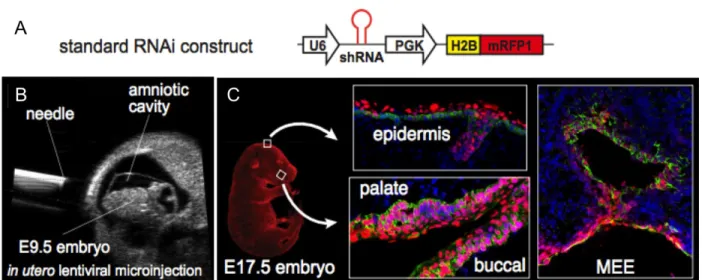
The long-term goal of this project is to adopt efficient shRNAs targeting Nectin-1-4 into
our in vivo pipeline utilizing our novel LUGGIGE (Lentiviral, Ultrasound-Guided
Gene-Inactivation and Gene Expression) technique. This technique utilizes ultrasound to deliver
high-titer lentivirus non-invasively into the amniotic fluid surrounding E9.5 mouse embryos, allowing
for efficient transduction of surface epithelial tissues including the skin and oral epithelia
(Beronja et al., 2010) (Figure 4). The LUGGIGE method is quicker and more efficient than
traditional homologous recombination-mediated methods of gene knockout or nuclease-based
gene-editing approaches, generating epithelial-specific conditional mouse models in a matter of
weeks rather than months or years. Importantly, LUGGIGE is more efficient than CRISPR-Cas9.
It allows us to transduce up to 95% of cells in exposed epithelia (Beronja et al., 2010; Byrd et al.,
2016)whereas the latter system is reported to have extremely low rates of recombination in vivo
(Nelson et al., 2016). Moreover, LUGGIGE has the potential to improve on existing Nectin
knockout models by selectively transducing the skin and oral epithelia, potentially circumventing
the lethality issues seen in compound Nectin knockout animals. With LUGGIGE,
epithelial-specific knockdowns can be generated earlier in development than would be possible through
genetic methods such as K14-Cre, which is especially important for studying palate fusion, a
process that occurs very shortly after K14 is expressed in the palatal epithelium (Figure 2). By
viruses harboring distinct fluorescent reporters (or drug resistance genes), it is possible to
simultaneously target multiple nectins both in vivo and in vitro. Therefore, this technique makes
our lab uniquely equipped to determine which complement of nectins, if any, can recapitulate the
CP phenotype observed in human disease and Afdn-deficient mice.
Results
Nectin-4 has distinct expression patterns in the palatal epithelium and mesenchyme
Nectin-4 expression in the developing palate has not previously been characterized. To address
this knowledge gap, nectin-4 expression in the palate was examined in E13.5 and E14.5 wild
type and conditional knockout (cKO) embryos by immunohistochemistry and confocal
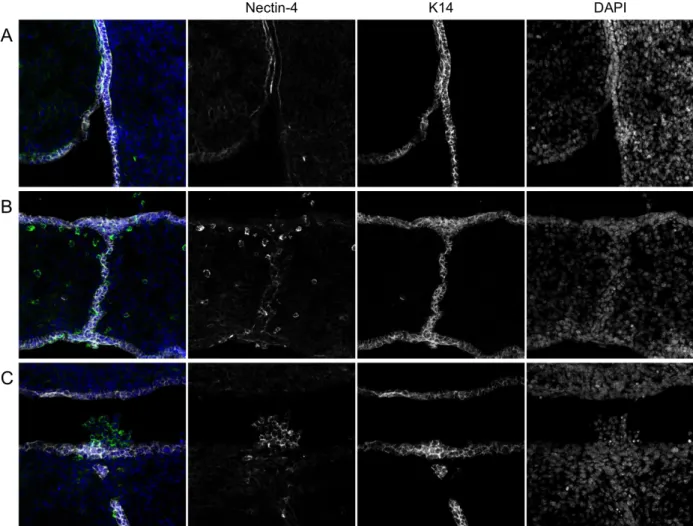
microscopy. At E13.5, prior to PS elevation, nectin-4 expression was observed in the basolateral
domains of palatal and lingual epithelium. Additionally, nectin-4 accumulated at points of
contact between the apical surfaces of these two tissues (Figure 5A). This demonstrates for the
first time that nectin-4 is expressed in medial edge epithelium (MEE) cells, and thus is positioned
appropriately to play a role in palate closure. A caveat of this finding is that the embryos
analyzed had shRNA-mediated Afdn knockdown, and thus it is possible that the expression
pattern may be different in wild-type embryos. Later, in E14.5 palates, nectin was weakly
expressed in the dissipating palatal epithelia, including the MES, but exhibited strong cortical
accumulation in some isolated nonepithelial (K14-) cells in the mesenchyme (Figure 5B). Strong
nectin-4 expression was also seen in clusters of cells in the sinus cavity directly above the MES
(Figure 5C). As MES cells have been shown to undergo a mesenchymal-epithelial transition
(losing K14 expression) following adhesion (Jin and Ding, 2006), these cells may represent
Cre-RFP or H2B-mCre-RFP1 by LUGGIGE are present in the palatal mesenchyme following palate
closure, and some of these are also Nectin-4+ (data not shown).
Design, cloning, and assembly of shRNA constructs
Delivery of lentiviral shRNA vectors into cells in culture or developing embryos in utero is a
promising technique for knocking down expression of target genes. No constructs targeting
Nectin-2/4 existed prior to this study; therefore, these had to be synthesized de novo. To prepare
for assembly of lentivirus targeting Nectin-2 or -4 for RNAi-mediated destruction, I generated
lentiviral expression constructs containing distinct Nectin-2 or -4 shRNAs (Table 1). Ten DNA
oligonucleotides (oligos) were selected from the RNAi Consortium based on predicted targeting
efficiency of the corresponding shRNA and evaluated by BLAST for complementarity to
off-target sequences (any candidate with >75% match to an off-off-target sequence was excluded).
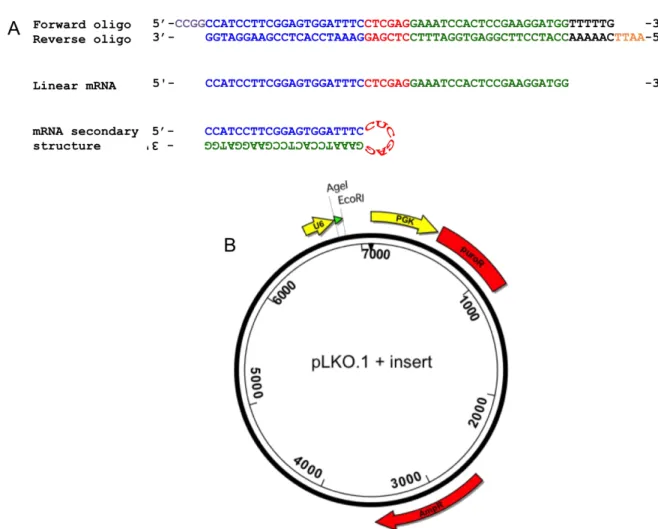
Forward and reverse complementary oligos were designed such that, once annealed, the
double-stranded DNA oligo would contain upstream AgeI and downstream EcoRI 5’ overhangs to
facilitate cloning into the pLKO.1 vector (Addgene plasmid 10878; Moffat et al., 2006). The
corresponding shRNA would form a hairpin secondary structure that targets a 21bp region in the
Nectin-2 or -4 transcripts (Figure 6A). I cloned each shRNA oligonucleotide into a pLKO.1
lentiviral backbone, which drives shRNA expression from the U6 (Pol III) promoter and contains
a puromycin-resistance gene to allow for drug selection and generation of stable cell lines
(Figure 6B). The resultant plasmids were cloned in the Stbl3 E. coli strain, which is recA
-deficient and thus suppresses potential recombination between lentiviral LTRs. Positive
were sequenced and compared to sequences of the desired products using Lasergene DNAStar
13 SeqMan Pro. Four pLKO.1 constructs per Nectin gene were successfully cloned (Table 1).
To assemble shRNA lentivirus, I co-transfected human embryonic kidney (HEK) 293FT
lentiviral packaging cells using a calcium phosphate method (Tiscornia et al., 2006) with the one
of the previously cloned pLKO.1 Nectin-shRNA constructs, a lentiviral packaging plasmid, and a
lentiviral VSV-G envelope plasmid. HEK 293FT cells are efficient at lentiviral assembly and
were used to package the desired lentiviral products. Replicates were done for each pLKO.1
Nectin-shRNA construct and one non-targeting (scramble) shRNA construct, which activates the
endogenous RNAi machinery but does not target any mouse or human transcripts. cEGFP, a
plasmid that induces Green Fluorescent Protein (GFP) expression in HEK 293FT cells but is not
used for viral assembly, was also included in the transfection cocktail to fluorescently label cells
that were successfully transfected. All replicates were GFP+, indicating successful transfection.
Viral supernatant was collected 46 hours post-transfection.
shRNA lentiviruses reduce Nectin-2/4 mRNA levels in keratinocytes
To assay the effectiveness of these viruses at reducing Nectin-2 or -4 expression in vitro, I
infected keratinocytes with virus at a low multiplicity of infection (MOI <1, or ~20%
transduction efficiency). This ensured that only one copy of the virus was integrated/expressed in
each cell, preventing overestimation of knockdown efficiency due to expression of multiple
copies of the shRNA. Two days after infection, keratinocytes were selected in media containing
puromycin; wild-type mock-infected keratinocytes cells were used as a negative selection control
to ensure that the puromycin regimen was effective at killing ~100% of uninfected cells. Once all
the pure infected keratinocytes, and generated cDNA using the Bio-Rad iScript cDNA synthesis
kit. Nectin-2 and -4 transcript levels were assessed by quantitative PCR using duplicate primer
sets for each nectin (Figure 7). Residual mRNA levels were determined by the 2-ΔΔCT method
(Livak and Schmittgen, 2001) using Ppib as a housekeeping gene, and relative to scramble
controls. Data were the obtained by calculating the geometric mean of triplicates using two
independent target primer pairs for each Nectin mRNA. All constructs reduced mRNA levels
with the exception of Nectin-2 1079, for which one of the primer sets indicated an unexpected
increase in transcript abundance. The constructs that yielded the lowest mRNA levels relative to
scramble for each nectin – 1028 and 515 for Nectin-2, 2631 and 2583 for Nectin-4;
corresponding to knockdown efficiencies of 64.1%, 75.4%, 82.9%, and 89.0%, respectively
(Supplemental Table 1) – were identified as the most promising candidates for use in future
applications.
Discussion
It has been long established that the nectin-afadin pathway is important for cell adhesion
(although the downstream signaling pathways remain poorly understood), and a growing body of
clinical and experimental evidence suggests that this pathway is also critical at the tissue and
organ scale for developmental processes such as palatogenesis. I examined the expression of
nectin-4 in the E14.5 palate immediately after fusion; it should be noted that I observed some
degree of nonspecific staining by the secondary antibody used to probe nectin-4 (data not
shown), and the observed staining patterns will need to be validated by repeat staining using a
Nectin-4 knockdown as a negative control. In addition, we have recently obtained a second
preliminary findings. The E14.5 embryos examined were mix of lentiviral Cre-RFP injected and
uninjected littermates with a floxed Afdn genetic background, meaning some were functionally
wild type whereas other others had conditional knockout of afadin in the surface epithelia. RFP
expression should distinguish injected (knockout) from uninjected (functionally wild type);
however, we did not assess the effect of Afdn loss on nectin-4 expression.
We observed weak nectin-4 expression in palatal epithelia, including the MES, of E14.5
mice (Figure 5B). Strong nectin-4 expression was observed in individual cells in the
mesenchyme and clusters of cells in the sinus cavity (Figure 5C). In both the mesenchyme and
sinus cavity these cells were K14-negative, however this does not exclude the possibility that
these cells are epithelial-derived and undergone an epithelial-mesenchymal transition. In support
of this, some cells from both populations of Nectin-4+ cells—clusters of “extruded” cells within
the sinus and isolated cells within the mesenchyme—were Cre-RFP+ (data not shown). Since
LUGGIGE only transduces epithelial cells and their progeny (such as the periderm layer that
coats MEE cells) and not underlying mesenchyme, it is highly likely that any Cre-RFP+ cells
observed in the mesenchyme following palate closure were initially epithelial cells. This could
be independently confirmed and pursued further by using genetic lineage tracing techniques.
In an E13.5 embryo, nectin-4 was seen in the palatal and lingual epithelium. The
presence of Nectin-4 on palatal epithelium prior to palate closure further supports our hypothesis
that it may play a role in fusion and resolution. Expression was largely excluded from the apical
domains of these cells except at points of contact between two tissues, where nectin-4 appears to
be strongly expressed (Figure 5A). This result is interesting since it demonstrates a possible
neighbor-sensing role of nectin-4. Since the E13.5 embryo had epithelial knockdown of afadin
type animals. However, there is no precedent in the literature for afadin loss leading to
upregulation of nectin expression, and in fact, other studies have shown that afadin loss leads to
decreased expression of nectins (Tanaka-Okamoto et al., 2011; Toyoshima et al., 2014;
Yamamoto et al., 2013).
The unique expression patterns of nectin-4 suggest possible cell-sorting activity. The
peculiar location of the nectin-4+, K14-negativecell clusters in the nasal sinus above the MES
(Figure 5C) suggests that these cells may be recently extruded. Kim et al. (2015) recently
reported that extrusion is a normal and necessary part of proper palate fusion. That this process
relies on the formation of an actomyosin cable at the boundary of mesenchymal and MES cells is
intriguing – differential expression of Ed in Drosophila also defines the leading edge of the
migrating epithelia, directs assembly of an actomyosin cable, and promotes proper
morphogenesis (Laplante and Nilson, 2006; 2011).
Studies on the effects of nectin loss could provide valuable information about the role of
nectins in development and disease. This prompted us to develop new molecular tools to study
the effects of single and compound nectin loss in vitro and in vivo while circumventing the
limitations of preexisting models of nectin loss. Here I report that eight shRNA lentiviruses
designed to target Nectin-2 or -4 have been generated. Assaying the effectiveness of these
viruses in vitro by measuring transcript abundance of infected keratinocytes by qPCR showed
that these constructs reduce transcript abundance, although with varying degrees of efficiency. A
notable exception to this trend is seen in the apparent two-fold increase of Nectin-2 transcript in
cells infected with construct 1079 when measured with one of the two primer sets. This result is
unexpected – an ineffective shRNA should, at worst, lead to no change in transcript abundance.
amplified using the second primer set, it is likely an error in the qPCR reaction rather than a true
increase in Nectin-2 transcript. Constructs 1028 and 515 appear to be most efficient at reducing
Nectin-2 transcripts in vitro; 2631 and 2583 at reducing Nectin-4 (Figure 7).
It is not yet known if these viruses will be equally effective at targeting the Nectin-2/4
transcripts in vivo, although strong correlations between in vivo and in vitro knockdown
efficiency has been observed before (Beronja et al., 2010; Williams et. al, 2011). This has
informed our decision to modify these four constructs for LUGGIGE by addition of a
fluorescently labeled histone marker (H2B-mRFP1) and high-titer concentration. This approach
will be used to conditionally knock down nectin expression in surface epithelia of developing
embryos. We hypothesize that knockdown of Nectin-4, or combination loss of nectins-1-3 early
in development will cause CP, although it is possible that knockdown will not be sufficient.
CLPED1 is observed in individuals heterozygous for W185X, a mutant allele coding for a
truncated NECTIN1 protein (Suzuki et al., 2000; Sözen et al., 2001). This suggests a possible
dominant-negative mechanism for the disease that would not be replicated by nectin knockdown
models. LUGGIGE could be used to deliver lentivirus to exogenously express the W185X
mutant allele, which could provide valuable information on the contribution of nectins to disease.
Methods
Immunohistochemistry and microscopy: Fresh frozen sections (8-10 µm) were prepared as described by Byrd et al. (2016). The following antibodies were used: Mouse anti-nectin-4
(Millipore, MABT64, 1:500), guinea pig anti-cytokeratin 14 (Acris, BP5009, 1:1000), chicken
anti-RFP (Millipore, AB3528, 1:500), donkey anti-guinea pig Cy5 (Invitrogen, 1:400), donkey
(Invitrogen, 1:1000). DAPI was used at 1:4000. Images were acquired using LAS AF software
on a Leica TCS SPE-II 4 laser confocal system on a DM5500 microscope with an ACS
Apochromat 20×/0.60 multi-immersion objective.
Cloning: DNA oligonucleotides were identified from The Broad Institute’s Mission TRC-1 mouse library. Oligos were thermally annealed in NEB Buffer 2.1.Addgene plasmid #10878 was
used as the pLKO.1 lentiviral backbone. Ligations were performed using a Roche Rapid DNA
Ligation kit. Stbl3 (Invitrogen C7373-03) E. coli cells were used for transformation. Plasmid
isolation was done using a Qiagen QIAprep Spin Miniprep Kit. Sequencing was done by
GeneWiz (Primer: CAAGGCTGTTAGAGAGATAATTGGA).
Transfection: Approximately 1 week prior to transfection, HEK 293FT cells were cultured in D10 medium (DMEM + 10% v/v FBS + 1% v/v Pen-strep/L-glut mix + 1% v/v 100 mM Sodium
Pyruvate + 1% v/v 7.5% sodium bicarbonate) supplemented with 500 µg/mL G418 (1% v/v of
100x frozen 50 mg/mL stock). 24 h prior to transfection, cells were plated in 6-well plates and
fed D10 medium (without G418). When ~90% confluent, calcium phosphate-mediated
transfection was used to deliver the component vectors (pLKO.1+insert; psPAX, Addgene
plasmid 12260; and pMD2.G, Addgene plasmid 12259) and a GFP expression plasmid (cEGFP).
16 h post-transfection, cells were switched to Viral Production Medium (VPM) (Ultraculture
[Lonza BioWhittaker 12-725F] supplemented with 1% v/v Pen-strep/L-glut mix + 1% v/v 100
mM Sodium Pyruvate + 1% v/v 7.5% sodium bicarbonate, and 5 mM sodium butyrate). At 46 h
post-transfection, cells were assessed for GFP expression to confirm transfection and viral
supernatant was collected, filtered, and stored at -80°C.
administered an infection cocktail consisting of 100µL viral supernatant and 2.6 mL low Ca2+
medium, supplemented with 300µL polybrene/serum (10% v/v polybrene [10 mg/mL stock in
PBS], 90% FBS(-) serum). Cells were centrifuged for 30 min at 300G to improve infection
efficiency, then transferred to low Ca2+ medium. After 48 h cells were treated with 1µg/mL
puromycin to select pure cultures of infected cells. Puromycin concentration was gradually
increased to 1.5 µg/mL, then 2.0 µg/mL until all uninfected negative control cells were killed.
RNA was collected from the pure keratinocyte cultures using a Qiagen RNeasy Micro Kit.
Whole RNA cDNA synthesis was performed using Bio-Rad iScript cDNA Synthesis Kit.
Abundance of target transcripts (Nectin-2 or Nectin-4) was determined for each sample in
triplicate by quantitative PCR using the following primers:
Primer name Forward sequence Reverse sequence
05 (Nectin 2) AGAGTCATAGCCCAGCCTGAGAA CCACGGGCACCAAGGAGTAT
06 (Nectin 2) ATACAGGCTGGCACCGTCACTAT GGGCTCTGGGTTGCTTCGTA
03 (Nectin 4) CAGCCCCCTCCCAAATACAA TATGATCACTGAGGCGGACACC
07 (Nectin 4) AGATGTGGGGCCCTGAAGC GCATTCGTACTCGCCCTCATC
pPIB (Housekeeping control) GTGAGCGCTTCCCAGATGAGA TGCCGGAGTCGACAATGATG
Residual mRNA levels were determined by the 2-ΔΔCT method (Livak and Schmittgen, 2001)
Author Contributions
Kendall Lough identified the shRNA targets. Experiments were performed by the author with the
assistance and guidance of Kendall Lough and Scott Williams, who both provided suggestions
and revisions that greatly improved this report.
Acknowledgments
Many thanks to my wonderful colleagues in the Williams lab: Scott Williams, for being an
exemplary leader, teacher, and role model; Kendall Lough, for exceptional mentorship, kindness,
patience, and support; and Kevin Byrd, Carlos Patiño Descovich, Jeet Patel, Abby Bergman, and
Jina Yom for constant encouragement, helpful conversations, and technical support. I am
grateful to Mark Peifer for sponsoring this project and providing valuable input and advice. I am
thankful to my BIOL 692H Honors Thesis classmates – especially my editing partner, Ben Lowe
– and our instructor, Amy Maddox, for providing critical feedback and kind support throughout
References
Ahmed, S., Liu, C. C., & Nawshad, A. (2007). Mechanisms of palatal epithelial seam
disintegration by transforming growth factor (TGF) β3. Developmental biology, 309(2), 193-207.
Beronja, S., Livshits, G., Williams, S., & Fuchs, E. (2010). Rapid functional dissection of genetic networks via tissue-specific transduction and RNAi in mouse embryos. Nature medicine, 16(7), 821-8 27.
Brancati, F., Fortugno, P., Bottillo, I., Lopez, M., Josselin, E., Boudghene-Stambouli, O., ... & Rossi, A. (2010). Mutations in PVRL4, encoding cell adhesion molecule nectin-4, cause ectodermal dysplasia-syndactyly syndrome. The American Journal of Human Genetics, 87(2), 265-273.
Byrd, K. M., Lough, K. J., Patel, J. H., Descovich, C. P., Curtis, T. A., & Williams, S. E. (2016). LGN plays distinct roles in oral epithelial stratification, filiform papilla morphogenesis and hair follicle development. Development, 143(15), 2803-2817.
Escors, D., & Breckpot, K. (2010). Lentiviral vectors in gene therapy: their current status and future potential. Archivum immunologiae et therapiae experimentalis, 58(2), 107-119.
Jacinto, A., Woolner, S., & Martin, P. (2002). Dynamic analysis of dorsal closure in Drosophila: from genetics to cell biology. Developmental cell, 3(1), 9-19.
Jin, J. Z., & Ding, J. (2006). Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development, 133(17), 3341-3347.
Katsunuma, S., Honda, H., Shinoda, T., Ishimoto, Y., Miyata, T., Kiyonari, H., ... & Togashi, H. (2016). Synergistic action of nectins and cadherins generates the mosaic cellular pattern of the olfactory epithelium. The Journal of cell biology, 212(5), 561-575.
Kim, S., Lewis, A. E., Singh, V., Ma, X., Adelstein, R., & Bush, J. O. (2015). Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol, 13(4), e1002122.
Laplante, C., & Nilson, L. A. (2006). Differential expression of the adhesion molecule Echinoid drives epithelial morphogenesis in Drosophila. Development, 133(16), 3255-3264.
Laplante, C., & Nilson, L. A. (2011). Asymmetric distribution of Echinoid defines the epidermal leading edge during Drosophila dorsal closure. The Journal of cell biology, 192(2), 335-348. Litvak, K. J., & Schmittgen, T. D. (2001). Analysis of relative gene expression data using
real-time quantitative PCR and the 2 (-Delta Delta C (T)) method. Methods, 25(4), 402-408 Lough, K. J., Byrd, K. M., Spitzer, D. C., Williams, S. E. (In review) Closing the gap: cell-cell
adhesion in mammalian secondary palatogenesis.
Moffat, J., Grueneberg, D. A., Yang, X., Kim, S. Y., Kloepfer, A. M., Hinkle, G., ... &
Carpenter, A. E. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell, 124(6), 1283-1298.
Nawshad, A., LaGamba, D., & Hay, E. D. (2004). Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Archives of oral biology, 49(9), 675-689.
Nelson, C. E., Hakim, C. H., Ousterout, D. G., Thakore, P. I., Moreb, E. A., Rivera, R. M. C., ... & Asokan, A. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science, 351(6271), 403-407.
Shuler, C. F., Guo, Y., Majumder, A. S. I. M. A., & Luo, R. Y. (2004). Molecular and morphologic changes during the epithelial-mesenchymal transformation of palatal shelf medial edge epithelium in vitro. International Journal of Developmental Biology, 35(4), 463-472.
Sözen, M. A., Suzuki, K., Tolarova, M. M., Bustos, T., Iglesias, J. E. F., & Spritz, R. A. (2001). Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nature genetics, 29(2), 141.
Suzuki, K., Hu, D., Bustos, T., Zlotogora, J., Richieri-Costa, A., Helms, J. A., & Spritz, R. A. (2000). Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nature genetics, 25(4), 427-430.
Takai, Y., Ikeda, W., Ogita, H., & Rikitake, Y. (2008). The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annual Review of Cell and Developmental Biology, 24: 309-342.
Tanaka, Y., Nakanishi, H., Kakunaga, S., Okabe, N., Kawakatsu, T., Shimizu, K., & Takai, Y. (2003). Role of nectin in formation of E-cadherin–based adherens junctions in keratinocytes: analysis with the N-cadherin dominant negative mutant. Molecular biology of the cell, 14(4), 1597-1609.
Tanaka-Okamoto, M., Hori, K., Ishizaki, H., Itoh, Y., Onishi, S., Yonemura, S., Takai, Y. and Miyoshi, J. (2011). Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia. J Cell Sci, 124(13), 2231-2240.
Tiscornia, G., Singer, O., & Verma, I. M. (2006). Production and purification of lentiviral vectors. Nature protocols, 1(1), 241.
Togashi, H., Kominami, K., Waseda, M., Komura, H., Miyoshi, J., Takeichi, M., & Takai, Y. (2011). Nectins establish a checkerboard-like cellular pattern in the auditory epithelium. Science, 333(6046), 1144-1147.
Toyoshima, D., Mandai, K., Maruo, T., Supriyanto, I., Togashi, H., Inoue, T., Mori, M. and Takai, Y. (2014). Afadin regulates puncta adherentia junction formation and presynaptic differentiation in hippocampal neurons. PloS one, 9(2), e89763.
Wei, S. Y., Escudero, L. M., Yu, F., Chang, L. H., Chen, L. Y., Ho, Y. H., ... & Hsu, J. C. (2005). Echinoid is a component of adherens junctions that cooperates with DE-Cadherin to mediate cell adhesion. Developmental cell, 8(4), 493-504.
Williams, S. E., Beronja, S., Pasolli, H. A., & Fuchs, E. (2011). Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature, 470(7334), 353-358.
Yamamoto, H., Maruo, T., Majima, T., Ishizaki, H., Tanaka-Okamoto, M., Miyoshi, J., ... & Takai, Y. (2013). Genetic deletion of afadin causes hydrocephalus by destruction of adherens junctions in radial glial and ependymal cells in the midbrain. PLoS One, 8(11), e80356. Yoshida, M., Shimono, Y., Togashi, H., Matsuzaki, K., Miyoshi, J., Mizoguchi, A., Komori, T.
and Takai, Y. (2012). Periderm cells covering palatal shelves have tight junctions and their desquamation reduces the polarity of palatal shelf epithelial cells in palatogenesis. Genes to Cells, 17(6), 455-472.
Yoshida, T., Miyoshi, J., Takai, Y., & Thesleff, I. (2010). Cooperation of nectin-1 and nectin-3 is required for normal ameloblast function and crown shape development in mouse teeth.
Developmental Dynamics, 239(10), 2558-2569.
Tables and figures
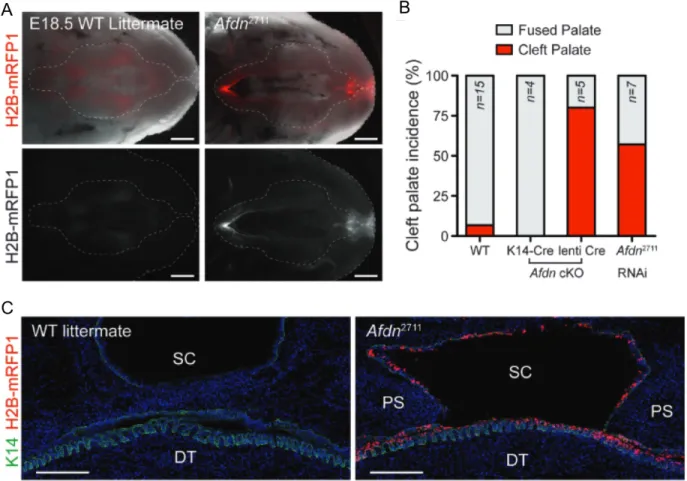
Figure 3. Afadin loss early in development leads to highly penetrant cleft palate in mice. A) Cleft palate was observed in mice whose oral epithelia had been transduced by with Afdn2711, an shRNA
targeting Afdn, but not in wild type littermates. Afdn2711 was introduced by a technique termed
LUGGIGE, which is detailed in Figure 4. B) Cleft palate is highly penetrant in mice with Afdn knockout induced via LUGGIGE (lenti Cre-mediated conditional knockout [cKO] and Afdn2711 RNAi),
Figure 5. Nectin-4 expression in the developing palate. A) Coronal section of an E13.5 embryo at the boundary of a palatal shelf (left) and the tongue (right). Nectin-4 is expressed in the basolateral domains of palatal and lingual epithelia. It is also strongly expressed at apical contacts between the palatal and lingual epithelia. B) Coronal section of an E14.5 palate. Nectin-4 is weakly visible in the epithelia, including the MES. Individual cells strongly expressing nectin-4 are visible in the mesenchyme. C) Coronal section of an E14.5 palate. A cluster of K14- cells strongly expressing nectin-4 is visible in the
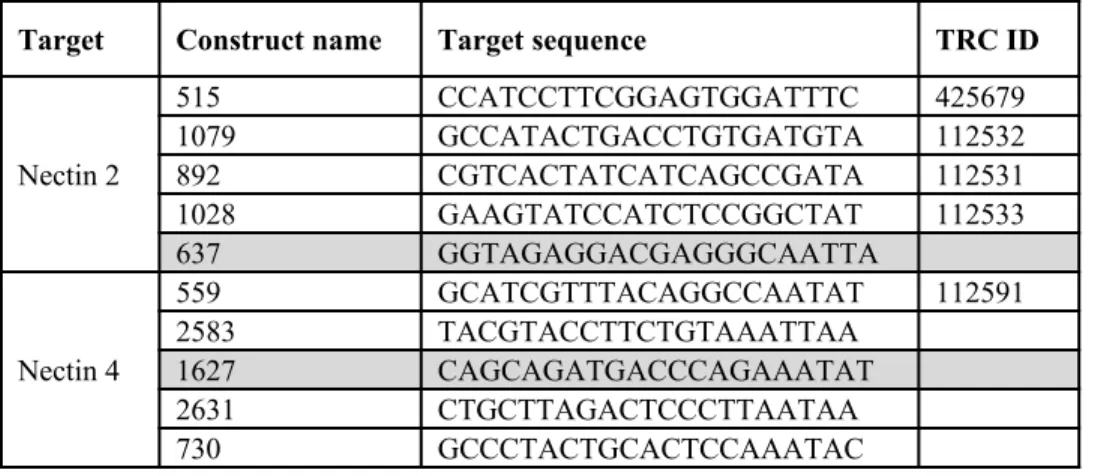
Table 1. Overview of shRNA lentivirus targeting Nectin-2 and -4. Oligonucleotides containing 21bp sequences complementary to regions in Nectin-2 or -4 transcripts were selected from The Broad Institute’s Mission TRC-1 mouse library. Preexisting TRC constructs (ID shown) and novel constructs were used. Five constructs per gene were selected; all but constructs 637 and 1627 (shaded) were successfully cloned and used for viral assembly.
Target Construct name Target sequence TRC ID
Nectin 2
515 CCATCCTTCGGAGTGGATTTC 425679
1079 GCCATACTGACCTGTGATGTA 112532
892 CGTCACTATCATCAGCCGATA 112531
1028 GAAGTATCCATCTCCGGCTAT 112533
637 GGTAGAGGACGAGGGCAATTA
Nectin 4
559 GCATCGTTTACAGGCCAATAT 112591
2583 TACGTACCTTCTGTAAATTAA
1627 CAGCAGATGACCCAGAAATAT
2631 CTGCTTAGACTCCCTTAATAA
Supplementary Information
Supplemental Table 1. Percent knockdown of each lentivirus was assessed by 2-ΔΔCT analysis following
quantitative PCR of whole RNA from infected keratinocytes using duplicate primer sets. The reported percent knockdown for all constructs except 2583 was the mean of that found using each of the two experimental primer sets (05 and 06 for Nectin-2, 03 and 07 for Nectin-4). The reported percent
knockdown of construct 2583 was determined using only primer set 07 as the cycle threshold from primer set 03 for this construct was undetermined.
Target Construct name Percent knockdown
Nectin 2
515 75.44550148
1079 -21.87397278
892 58.21065753
1028 64.05271123
Nectin 4
559 60.15248896
2583 88.96961081
2631 82.89137003