in cystic fibrosis ferrets
Alicia K. Olivier, … , Andrew W. Norris, John F. Engelhardt
J Clin Invest.
2012;
122(10)
:3755-3768.
https://doi.org/10.1172/JCI60610
.
Diabetes is a common comorbidity in cystic fibrosis (CF) that worsens prognosis. The lack
of an animal model for CF-related diabetes (CFRD) has made it difficult to dissect how the
onset of pancreatic pathology influences the emergence of CFRD. We evaluated the
structure and function of the neonatal CF endocrine pancreas using a new
CFTR
-knockout
ferret model. Although CF kits are born with only mild exocrine pancreas disease,
progressive exocrine and endocrine pancreatic loss during the first months of life was
associated with pancreatic inflammation, spontaneous hyperglycemia, and glucose
intolerance. Interestingly, prior to major exocrine pancreas disease, CF kits demonstrated
significant abnormalities in blood glucose and insulin regulation, including diminished
first-phase and accentuated peak insulin secretion in response to glucose, elevated peak
glucose levels following glucose challenge, and variably elevated insulin and C-peptide
levels in the nonfasted state. Although there was no difference in lobular insulin and
glucagon expression between genotypes at birth, significant alterations in the frequencies of
small and large islets were observed. Newborn cultured CF islets demonstrated
dysregulated glucose-dependent insulin secretion in comparison to controls, suggesting
intrinsic abnormalities in CF islets. These findings demonstrate that early abnormalities
exist in the regulation of insulin secretion by the CF endocrine pancreas.
Technical Advance
Metabolism
Find the latest version:
Abnormal endocrine pancreas function
at birth in cystic fibrosis ferrets
Alicia K. Olivier,1 Yaling Yi,2 Xingshen Sun,2 Hongshu Sui,2 Bo Liang,2 Shanming Hu,3 Weiliang Xie,2
John T. Fisher,2 Nicholas W. Keiser,2 Diana Lei,2 Weihong Zhou,2 Ziying Yan,2 Guiying Li,4
Turan I.A. Evans,2 David K. Meyerholz,1 Kai Wang,5 Zoe A. Stewart,4
Andrew W. Norris,3,6 and John F. Engelhardt2,6
1Department of Pathology, 2Department of Anatomy and Cell Biology, 3Department of Pediatrics, 4Department of Surgery, 5Department of Biostatistics,
College of Public Health, and 6Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, Iowa, USA.
Diabetes is a common comorbidity in cystic fibrosis (CF) that worsens prognosis. The lack of an animal model
for CF-related diabetes (CFRD) has made it difficult to dissect how the onset of pancreatic pathology
influ-ences the emergence of CFRD. We evaluated the structure and function of the neonatal CF endocrine pancreas
using a new CFTR-knockout ferret model. Although CF kits are born with only mild exocrine pancreas disease,
progressive exocrine and endocrine pancreatic loss during the first months of life was associated with
pancre-atic inflammation, spontaneous hyperglycemia, and glucose intolerance. Interestingly, prior to major exocrine
pancreas disease, CF kits demonstrated significant abnormalities in blood glucose and insulin regulation,
including diminished first-phase and accentuated peak insulin secretion in response to glucose, elevated peak
glucose levels following glucose challenge, and variably elevated insulin and C-peptide levels in the nonfasted
state. Although there was no difference in lobular insulin and glucagon expression between genotypes at birth,
significant alterations in the frequencies of small and large islets were observed. Newborn cultured CF islets
demonstrated dysregulated glucose-dependent insulin secretion in comparison to controls, suggesting
intrin-sic abnormalities in CF islets. These findings demonstrate that early abnormalities exist in the regulation of
insulin secretion by the CF endocrine pancreas.
Introduction
Cystic fibrosis (CF) is caused by defects in the CF transmembrane conductance regulator (CFTR) chloride channel. Cystic fibrosis– related diabetes (CFRD) is a common complication of CF and affects 20%–25% of adolescents and 40%–50% of individuals over 30 years of age (1, 2). CFRD is associated with worsening clinical sta-tus, including reduced pulmonary function, increased frequency of pulmonary exacerbations, and a decline in nutritional status (3–7). Furthermore, CFRD leads to increased mortality compared with CF patients without diabetes (4, 8). Thus, early diagnosis and treat-ment are vital to improving clinical outcome of CFRD patients.
While the pathophysiology of CFRD is multifactorial, delayed insulin secretion appears to be a key hallmark of disease progres-sion (9–12), and the health of CF patients is improved by insulin therapy prior to and following diagnosis of overt diabetes (13, 14). Partial insulin deficiency occurs in part due to islet loss associated with exocrine pancreas disease (15–18). However, CF pancreata at CFRD autopsy demonstrate that remaining islets contain roughly half the number of insulin-positive cells found in non-CF controls (17, 18), and this degree of β cell loss is thought to be insufficient to explain diabetes (19). Thus, insulin deficiency in CFRD is rel-ative and not absolute. All stages of CFRD are characterized by abnormalities in circulating insulin levels (10, 12, 20, 21). Impaired first-phase insulin (IFPI) responses are common in CFRD patients, but also occur in approximately 50% of CF children with normal glucose tolerance (9). Current guidelines for CFRD screening
emphasize 2-hour blood glucose measurements following oral glucose tolerance testing (22).
Multiple non-pancreatic organ abnormalities, including those in the lung, liver, and intestine, may modify glucose disturbances in CF patients. For example, hepatic insulin resistance (15, 23, 24) and/or enhanced peripheral insulin sensitivity have been observed in CF patients (15, 25, 26). However, normal insulin sensitivity has also been reported in CF patients with impaired glucose tolerance or overt diabetes (12, 27–29). Altered entero-insular axis hormones such as cholecystokinin (CCK), glucagon-like peptide–1 (GLP-1), and glucose-dependent insulinotropic peptide (GIP) have been observed in CF patients (30–32) and may also play a role in CFRD, since each of these hormones participate in insulin regulation (33). Furthermore, chronic lung disease in CF is thought to contribute to abnormal glucose regulation and insulin resistance in concert with increasing metabolic demand (34).
Characterizing early structural and functional endocrine pan-creas abnormalities that precede glucose intolerance in CF has been difficult due to the lack of an animal model in which CFRD spontaneously develops. Furthermore, to our knowledge, no clini-cal studies have evaluated CF glucose abnormalities in the first years of life. Cftr-knockout mice demonstrate enhanced sensitivity to low-dose streptozotocin-induced hyperglycemia, but are eugly-cemic in the absence of streptozotocin (35, 36). The recent devel-opment of CFTR-knockout models in the pig and ferret provides new avenues for investigating the pathogenesis of CFRD. Both CF pigs and ferrets develop intestinal, hepatic, pancreatic, and lung disease that is pathologically similar to various stages of disease in CF patients (37, 38); however, the progression of pancreatic disease in the two models is very different. CF pigs are born with signifi-cant exocrine pancreatic destruction, inflammation, and fibrosis Authorship note: Alicia K. Olivier, Yaling Yi, and Xingshen Sun contributed equally
to this work.
Conflict of interest: The authors have declared that no conflict of interest exists.
of the exocrine and endocrine pancreas was associated with spon-taneous hyperglycemia, glycosuria, and glucose intolerance. These findings in the CF ferret model demonstrate that alteration of endocrine pancreas function occurs very early in CF and is charac-terized, at least in part, by abnormal glucose responsiveness of the islet to secret insulin. Such alterations in the regulation of insulin secretion may be involved in the pathogenesis of CFRD.
Results
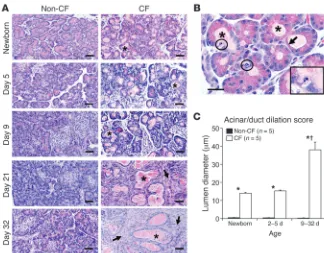
Progressive pancreatic inflammation and exocrine pancreas destruction are associated with impaired glycemic stability in CF ferrets. We previously reported that CF newborn ferrets have mild pancreatic pathol-ogy characterized by multifocal dilation of exocrine acini and lesser ducts by eosinophilic zymogen material (40). In the present study, we sought to better characterize age-dependent morphological changes in the pancreas in animals ranging from newborn to 1 month of age. Lumen dilation of acini and ducts resulted in epithelial changes including compression, attenuation, loss of zymogen granules, and nuclear morphologic changes consistent with apoptosis (Figure 1, A and B). Immunohistochemical staining for cleaved caspase-3 con-firmed enhanced apoptosis in CF exocrine ducts and acini at birth (Supplemental Figure 1, A and B), suggesting that apoptotic dam-age in these regions occurs very early after birth coincident with duct dilatation. However, there was no significant difference in apoptosis of CF and non-CF newborn islets as measured by cleaved caspase-3/ insulin co-staining (Supplemental Figure 1, C and D).
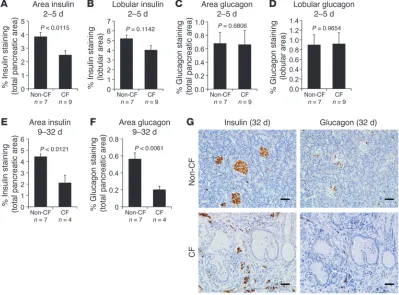
By 21 days, dilated lumens were lined by cuboidal epithelial cells lacking zymogen granules, making discernment of acini versus ducts difficult, and were therefore termed acinoductular units. The lumen diameter of the acinoductular units was significantly larger in CF compared with non-CF pancreata in all age groups, with a marked increase between P9 and P32 compared with pan-creata from younger CF kits (Figure 1C). By 21 days, there was loss of lobular parenchyma and replacement by bands of fibrous tissue characteristic of late-phase damage in CF patients, whereas CF
fer-rets have mild pancreatic histopathology at birth (38). For these reasons, the CF ferret model (39) may provide unique insights into the early pathogenesis of CFRD. CF newborn ferrets (kits) must be reared on antibiotics to prevent lung infections and on a proton pump blocker to raise gastrointestinal pH necessary for weight gain (40). As CF ferrets age, they require pancreatic enzyme sup-plementation to retain growth, similar to CF infants. CF kits are born with elevated markers of liver pathology, as occurs in human CF infants (41), and oral ursodeoxycholic acid (UDCA) therapy normalizes these markers in both species (40, 42, 43). Thus, the CF ferret model has several characteristics similar to the human CF phenotype. While rearing of adult CF ferrets is challenging (40), the model also represents an opportunity to evaluate the early CF phenotype less easily approachable in human infants.
[image:3.585.41.365.81.334.2]In the current study, we sought to determine whether altera-tions in insulin secretion and glucose homeostasis are present in newborn CF ferrets prior to structural loss of the endocrine and exocrine pancreas. Our studies demonstrate that fasted newborn CF ferrets have reduced first-phase insulin secretion and abnormal glucose tolerance. Structurally, newborn CF and non-CF endo-crine pancreata were indistinguishable in terms of size and percent insulin and glucagon-expressing cells in islets. However, there was a difference in the distribution of large and small islets between genotypes and enhanced apoptosis in acinar and duct cells of CF animals at birth. Random blood insulin and C-peptide levels in the nonfasted state were widely variant and higher in newborn CF animals, whereas they were tightly regulated and lower in non-CF controls, suggesting that β cell secretion of insulin is poorly regu-lated. Studies with isolated cultured islets from newborn animals confirmed dysregulated insulin secretion by CF β cells, demon-strating elevated insulin secretion at low glucose and impaired induced insulin secretion to high glucose challenge, as compared with non-CF controls. In juvenile ferrets, progressive destruction
Figure 1
Progressive exocrine histopathology in the neonatal CF ferret pancreas. (A) Representa-tive pancreatic histology from neonatal non-CF and CF ferrets. Multifocally the lumens of acini and lesser ducts of CF ferrets were dilated by pale eosinophilic material (*). Older CF animals exhibited loss of exocrine acini and replacement by fibrosis (arrows). Scale bars: 50 μm. (B) High magnification of acinar luminal dilation in a newborn CF pancreas exhibiting intraluminal material (*), loss of epithelial cytoplasmic zymo-gen granules (arrows), and apoptotic bodies (circled areas and higher-magnification inset) (scale bar: 50 μm; inset magnification: ×600). (C) Acinar/duct dilation scores for specific age groups. Values are mean ± SEM for n indepen-dent animals. *P < 0.001, age-matched CF ver-sus non-CF controls for the various groupings;
†P < 0.001, 9–32 day versus newborn or 2–5
underlies the prevailing hypothesis that CFRD arises from reduced islet mass and/or β cell injury caused by nonspecific “bystander damage” to islets during exocrine pancreatic destruction and inflammation (17–19). Alternatively, a more direct role for CFTR in the regulation of the endocrine pancreas has been proposed (35, 36). Our above results demonstrating a correlation between hyperglycemia onset and histopathologic damage to the CF pan-creas supports the bystander damage hypothesis (Figures 1–3), but does not disprove potential involvement of CFTR in regulating the endocrine pancreas. To explore this possibility, we characterized the early endocrine pancreas in newborn CF ferrets prior to overt exocrine pancreas pathology.
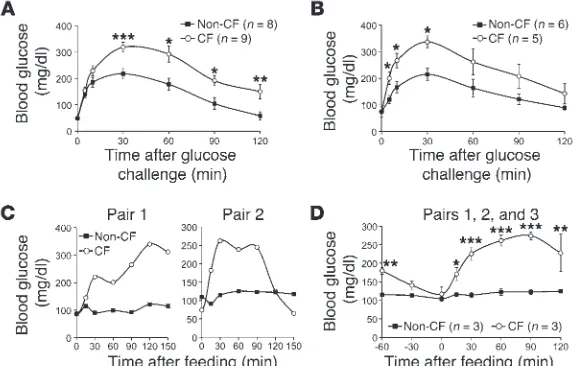
Fasted glucose tolerance tests (GTTs) on newborn animals 6–12 hours after birth demonstrated impaired glucose homeostasis in CF kits consisting of significantly elevated blood glucose at 30–120 minutes (Figure 4A) and significantly elevated glucose area under curve (AUC0–120min) (see Figure 4A legend). CF kits at
5–11 days of age also demonstrated significantly altered glucose tolerance as compared with controls (Figure 4B), including sig-nificantly elevated GTTGluc5–30min glucose and AUC0–60min values in
CF animals. Peak GTT values at 30 minutes were similar within a given genotype for newborn (CF 325 ± 23 mg/dl vs. non-CF 218 ± 20 mg/dl) and 5- to 11-day-old (CF 326 ± 25 mg/dl vs. non-CF 215 ± 23 mg/dl) kits. This was also true for AUCGluc0–120min
val-ues in both genotypes. Thus, there appears to be little change in glucose tolerance in CF kits during the first 2 weeks of life. These findings strongly support impaired glucose handling in neonatal CF kits prior to the emergence of overt pathology in the pancreas. Progression of abnormalities in glucose handling with age was evaluated by mixed meal tolerance test (MMTT) in CF and non-CF animals ranging from 45 to 90 days of age. Results from these that separated remnant dilated acinoductular units (Figure 2A).
Pancreas sections were scored for inflammation on a scale of 0 to 3 (Figure 2B). No significant differences in inflammation were noted between genotypes in newborn or early neonatal pancreata (Figure 2C). However, inflammatory cell infiltration into the interstitium and peripancreatic adipose tissue was significantly higher in CF as compared with non-CF pancreata by 9–32 days and was characterized by the infiltration of neutrophils, macro-phages, and lymphocytes (Figure 2C).
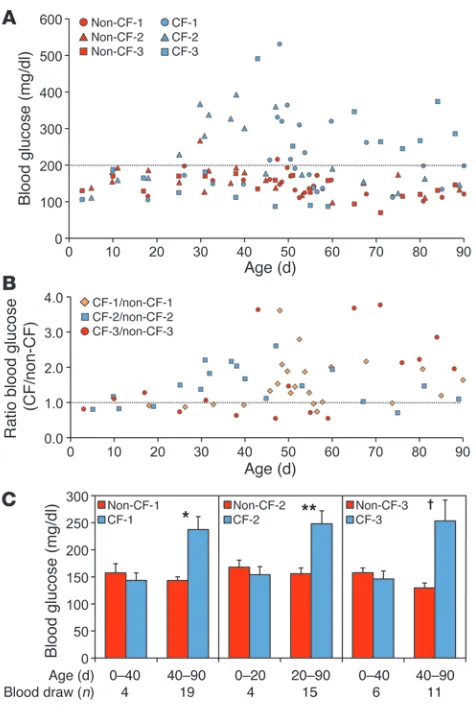
Major histopathologic changes in the exocrine pancreas pre-ceded episodes of hyperglycemia and glycosuria, which emerged at 1–2 months of age (Figure 3). To control for the tailored (and thus varied) health care provided each CF animal, we evaluated glucose and glucose ratios among 3 sibling pairs of CF and non-CF animals matched for provided care and timing of blood collec-tion. Roughly 44% of random nonfasted glucose measurements within the first 3 months of life were greater than 200 mg/dl in CF animals as compared with approximately 3% for sibling control lit-termates (Figure 3A). The CF/non-CF blood glucose ratio averaged 1.6 ± 0.1 across all time points and demonstrated a significant age-dependent rise for the combined 3 sibling pairs (P < 0.0152) (Fig-ure 3B). The onset of hyperglycemia (>200 mg/dl) ranged from 25 to 43 days and was preceded by a period of euglycemia (Figure 3C). As expected, hyperglycemia was associated with glycosuria in CF animals (ranging from 100 to 1,500 mg/dl), and glucose was never observed in the urine of non-CF controls. These findings demon-strate that the progressive stages of histologic pancreatic damage in the CF ferret model correlates with impaired glycemic regula-tion and suggest this model may be useful for studying CFRD.
[image:4.585.40.359.82.372.2]Glucose regulation by newborn and juvenile CF ferret. Exocrine pan-creas insufficiency is a CFRD risk factor (15, 44). This observation
Figure 2
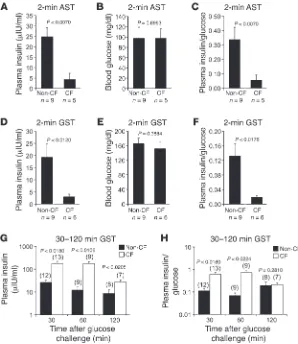
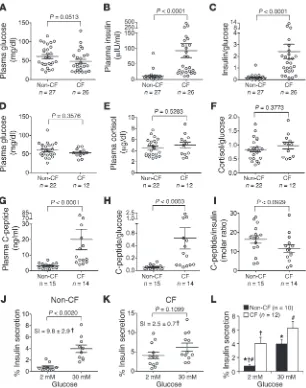
CF kits (3-hour-fasted) resulted in a significantly reduced 2-min-ute plasma insulin levels (5.7-fold) and insulin/glucose ratios (6.1-fold) in comparison to non-CF controls, despite similar blood glucose values (Figure 5, A–C). Similarly, reduced first-phase insu-lin response (6.5-fold) and insuinsu-lin/glucose ratio (7.0-fold) were also observed in 3-hour-fasted newborn CF kits following an i.p. injection of glucose (Figure 5, D–F). By contrast, the level of plas-ma insulin at 30, 60, and 120 minutes following an i.p. injection of glucose to 3-hour-fasted kits was significantly greater in CF kits (6.8-, 14.8-, and 3.2-fold, respectively) as compared with non-CF kits (Figure 5G). Insulin/glucose ratios during the 30- and 60-min-ute measurements were also significantly elevated in CF kits as compared with non-CF kits (5.5- and 10.5-fold, respectively) (Fig-ure 5H); at these time points, CF kits were hyperglycemic relative to controls (Figure 4A). By contrast, at 120 minutes the insulin/ glucose ratio between genotypes was similar (Figure 5H) despite persistent hyperglycemia in the CF group (Figure 4A), suggest-ing that elevated late-phase insulin secretion in CF animals is in response to hyperglycemia caused by an IFPI secretion.
Newborn CF ferrets have altered islet morphology but similar parenchy-mal insulin and glucagon expression. The observed insulin secretion abnormalities of newborn CF kits suggested that the endocrine pancreas was intact but functionally altered. To evaluate whether structural changes exist in the newborn CF ferret endocrine pancre-as, we performed quantitative morphometry assessing insulin and glucagon. Interestingly, per insulin staining, newborn CF pancreata studies demonstrated a variety of abnormal glucose tolerance
phe-notypes in CF animals (Figure 4C). Although all CF animals dem-onstrated postprandial peak glucoses greater than 200 mg/dl, the kinetics of the rise and fall in blood glucose was variable between animals. For example, 2 CF animals retained significantly elevated Gluc120min values (270 ± 44 mg/dl and 287 ± 106 mg/dl) as compared
with non-CF controls (129 ± 10 mg/dl and 116 ± 3 mg/dl), with a profile similar to that shown in Figure 4C (left panel). By contrast, the third CF animal maintained normal average Gluc120min values
(128 ± 36 mg/dl) as compared with non-CF control (130 ± 5 mg/dl), with a profile similar to that shown in Figure 4C (right panel). Despite these observed differences in MMTT profiles of CF animals, AUC0–120min values for each of the 3 CF animals were significantly
greater than in non-CF controls (see Figure 4 legend), and the aver-age glucose values at 30, 60, and 90 minutes after feeding were most significantly greater (P < 0.0001) in the CF as compared with the control group (Figure 4D). These studies demonstrate a progression in glucose abnormalities in CF ferrets with age.
Newborn CF ferrets have IFPI secretion. To evaluate whether impaired glucose tolerance in newborn CF ferrets might be due to altered insulin secretion, we quantified circulating insulin follow-ing a glucose or l-arginine challenge. l-Arginine stimulation tests
(ASTs) have classically been used to evaluate first-phase insulin secretion (45). In this context, l-arginine leads to depolarization
[image:5.585.47.285.80.437.2]of the β cell membrane and the immediate release of docked insu-lin granules at the membrane. l-Arginine challenge of newborn
Figure 3
CF ferrets develop impaired glycemic regulation at 1–2 months of life. (A) Blood glucose was measured on nonfasted, randomly fed CF and non-CF sibling pairs during the first 3 months of life. Each sibling pair was reared by the same jill, with blood draws taken simultaneously from each animal in the pair. Three independent CF and non-CF sib-ling pairs are shown (Non-CF-1/CF-1, Non-CF-2/CF-2, and Non-CF-3/ CF-3). The percent of n total readings above 200 mg/dl glucose for each animal was: Non-CF-1 (4%, n = 23), CF-1 (44%, n = 23), Non-CF-2 (5%, n = 19), CF-2 (42%, n = 19), Non-CF-3 (0%, n = 17), CF-3 (47%, n = 17). Urine glucose in CF animals during episodes of hyper-glycemia ranged from 100 to 1,500 mg/dl (CF-1: 8 of 9 urine samples tested positive; CF-2: 6 of 12 urine samples tested positive, CF-3: 1 of 12 urine samples tested positive). No glucose was found in any non-CF control urine measurements. (B) The ratio of CF to non-CF blood glucose for each sibling pair at each time point measured in A. Age-dependent trends in CF/non-CF blood glucose ratios were evaluating using the log of the ratio of blood glucose and a mixed-effects model as described in Methods. There was a significant effect of age on the log of this ratio (P < 0.0152). (C) Average blood glucose levels for spe-cific age windows prior to and following the first hyperglycemic episode (>200 mg/dl glucose) in the CF animal of each sibling pair. Values are mean ± SEM. *P < 0.0007, **P < 0.0016, †P < 0.0137, CF versus non-CF
To investigate other potential reasons for the varia-tion in islet sizes between genotypes, we examined the expression of two transcription factors involved in islet neogenesis (PDX1 and SOX9). PDX1 and SOX9 are expressed in multipotent pancreatic cells but maintain selective expression in differentiated β cells (PDX1), δ cells (PDX1), and duct cells (SOX9) (46). Although this is a highly debated topic, β cells of the adult pancreas may originate from multiple sources, including self-replication of β cells within islets, con-version of α cells to β cells within islets, or transdiffer-entiation of PDX1-positive acinar and/or ducts cells (46). Analysis of PDX1 expression in insulin-positive cells demonstrated no differences in the distribution of these cells between various sizes of islets and geno-type (Supplemental Figure 2, A and B). Furthermore, the frequency of PDX1+insulin– and PDX1+insulin+
cells within acinar/duct regions was also not different between genotypes (Supplemental Figure 2, C and D). As expected, SOX9 was highly expressed in cells of the ducts, with no discernible differences between geno-types, and expression was absent within islets of both genotypes (Supplemental Figure 2E). Thus, using these two markers, we could find no evidence for dif-ferences in expression patterns between genotypes that might suggest developmental abnormalities in the formation of islets.
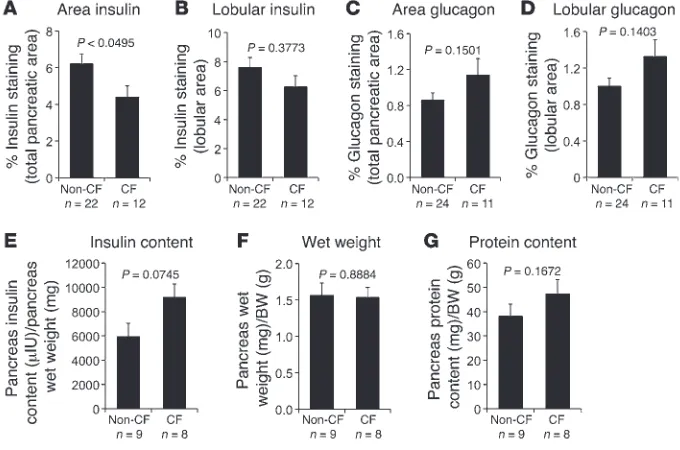
We quantified the portion of the pancreas staining for insulin (β cells) and glucagon (α cells), as their abundance might explain CF-associated alterations in glucose regulation. There was a small, but signifi-cant, reduction in insulin staining in samples from CF newborns when insulin staining was normalized to the total pancreatic area (Figure 7A), which by defi-nition included acellular acinar and ductal lumenal contents. However, there was no significant differ-ence when insulin staining was normalized to the total lobular area, which included only cellular areas of the pancreas (Figure 7B). The decrease in insulin staining when the data were normalized to total area was most likely a result of mild dilation of acini and ducts in the CF pancreas, which increased the relative pancreatic area. Supporting this hypothesis, the ratio of total lobular area to total pancreas area was signifi-cantly lower (P < 0.0004) for CF (0.705 ± 0.027, n = 12) as compared with non-CF (0.830 ± 0.015, n = 22) new-born samples. There was no significant difference in glucagon staining between newborn CF and non-CF pancreata using either total or lobular area normalization (Figure 7, C and D). Since his-tochemical staining indexes the spatial amount of cells as opposed to the quantity of the antigen, we also evaluated the total insulin content in newborn ferret pancreata. There was no significant dif-ference in the insulin content, wet weight, or total protein content between CF and non-CF pancreata (Figure 7, E–G); however, CF pancreata demonstrated a trend toward higher insulin content.
To determine the effect of postnatal exocrine destruction on the loss of endocrine pancreas, we examined pancreata from older animals for insulin and glucagon staining. Similar to newborn CF pancreata, P2–P5 CF pancreata demonstrated a significant reduc-tion in insulin staining normalized to total pancreas area, but not had significantly more small islets and β cell clusters (0.1 to less
than 1.0 mm2) compared with non-CF pancreata (Figure 6, A–C).
CF pancreata also had significantly fewer large islets (>2 mm2), but
no difference in medium sized islets (1–2 mm2) (Figure 6, A, B, D,
[image:6.585.43.331.85.268.2]and E). To determine whether the proliferation of insulin-positive cells in CF animals was altered in a manner that might impact islet size, we co-immunostained pancreatic tissue for proliferating cell nuclear antigen (PCNA, a proliferation marker) and insulin to determine the percentage of proliferating β cells. The average per-cent proliferation for non-CF pancreatic β cells was 22.6% ± 1.3%, and the average for CF pancreatic β cells was 20.3% ± 1.9%, with no significant difference (Supplemental Figure 1, E and F). There were also no significant differences in PCNA staining observed for the various sizes of islets between genotypes (Supplemental Figure 1F).
Figure 4
GTTs are abnormal in neonatal and juvenile CF ferrets. (A and B) GTTs were performed on 3-hour-fasted kits following i.p. injection of 2 g/kg BW glucose of (A) newborn and (B) 5- to 11-day-old CF and non-CF kits. (A) AUC0–120min was CF
28,507 ± 2221 mg/dl ∙ min versus non-CF 17,940 ± 2,011 mg/dl ∙ min (P < 0.0037). (B) AUC0–60min was CF 16,872 ± 1,518 mg/dl ∙ min versus non-CF 10,688 ± 956
mg/dl · min (P < 0.0087); AUC0–120min was CF 29,160 ± 3,903 mg/dl ∙ min versus
non-CF 18,113 ± 1,629 mg/dl ∙ min and not significantly different (P < 0.0823). Values in A and B represent mean ± SEM for n independent animals. (C and D) MMTTs were performed following a 3-hour fast on 3 groups of age-matched CF and non-CF siblings at 45–90 days of age. Each of 3 independent CF animals shown in Figure 3 was paired with 1–2 non-CF animals. (C) Individual MMTT assays demonstrating the diversity in blood glucose profiles from 2 independent pairs. (D) Average MMTT glucose values for the 3 paired CF/non-CF groups. Each paired group had 3–6 MMTT experiments performed, and the averages for CF and non-CF animals in each paired group were used to calculate the mean ± SEM (n = 3). The average AUC0–120min for paired group 1 was CF 29,245 ± 5,920
of interest. Under nonfasted conditions, a relationship between glucose and insulin levels is expected. Indeed, in non-CF nonfast-ed newborns, glucose and insulin levels were positively correlatnonfast-ed (P < 0.0468), such that lower glucose levels predicted lower insu-lin levels (Supplemental Figure 3A). By contrast, in CF nonfasted newborns, insulin did not vary with glucose (P = 0.7743), such that high insulin levels were observed even when glucose was low (Supplemental Figure 3A). These findings support the notion that insulin secretion by the newborn CF ferret pancreas is not properly regulated in a manner that extends beyond IFPI.
One potential reason for elevated and widely variant insulin lev-els in nonfasted CF kits could be increased glucocorticoids due to the physiological stress of CF, thus promoting hepatic glucose production and insulin resistance. Since the major glucocorticoid in humans and ferrets is cortisol (47), we evaluated plasma corti-sol levels in newborn randomly nursing CF and non-CF animals. Results from this analysis demonstrated no significant difference in plasma glucose, cortisol, or cortisol/glucose ratios between nonfasted CF and non-CF animals (Figure 9, D–F), demonstrat-ing that changes in stress-induced glucocorticoid cortisol is likely not the cause of variant insulin levels in CF newborns.
While insulin is primarily cleared through the liver via an insulin receptor–mediated process (48), the proteolytic C-peptide fragment produced during insulin processing is primarily cleared through the kidney. C-peptide and insulin are secreted at a 1:1 molar ratio. Severe hepatic insulin resistance can be accompanied by reduced insulin receptor–mediated clearance and resultant hyperinsu-when normalized to total lobular area of the pancreas (Figure 8, A
and B). There was no significant change in glucagon staining in 2- to 5-day-old CF kits by either normalization method (Figure 8, C and D), as was the case in newborn kits. By P9–P32, there was a significant reduction in both insulin and glucagon staining in CF pancreata compared with non-CF pancreata (Figure 8, E–G). Of note, in the older animals, it was not possible to normalize stain-ing to lobular area given the loss of lobular tissue and ensustain-ing fibrosis. This marked reduction in insulin and glucagon content in older CF animals preceded or coincided with the onset of glyce-mic instability in the nonfasted state (Figure 3).
[image:7.585.44.342.83.427.2]Newborn CF ferrets have elevated and widely variant blood insulin and C-peptide in the nonfasted state. Because newborn CF ferrets demon-strated relative glucose intolerance, IFPI, and a heightened peak in later-phase insulin, it was important to evaluate how tightly kits could regulate their blood glucose and insulin levels during nonfasted conditions (i.e., intermittent spontaneous nursing). Interestingly, nonfasted CF newborn kits demonstrated signifi-cantly higher plasma insulin levels and insulin/glucose ratios as compared with non-CF controls, despite blood glucose levels that were indistinguishable between these two groups (Figure 9, A–C). Notably, CF plasma insulin levels and insulin/glucose ratios both demonstrated significantly increased variances by F test (P < 0.0001), in comparison to non-CF controls. By contrast, there was no significant difference in the variance of blood glucose val-ues between genotypes (F test, P = 0.3609). The cause of the highly variable and elevated insulin levels in nonfasted CF newborns is
Figure 5
First-phase insulin secretion is impaired in newborn fasted CF kits, while second-phase insulin secretion is accentuated. Glucose stimulation tests (GST) and L-arginine stimulation tests (AST) were performed on fasted newborn animals. (A–C) Blood insulin and glucose were measured in 3-hour-fasted newborn animals at 2 minutes following i.p. injection of l
insulin secretion (5.1-fold), while the percent insulin secretion for CF islets was not significant different at low (4.1% ± 0.9%) and high (6.2% ± 1.0%) glucose (Figure 9, J–L). These alterations were reflected in a 4-fold-reduced insulin secretory index (SI) in response to glu-cose challenge for CF islets as compared with non-CF islets (Fig-ure 9, J and K). Similar to in vivo findings in nursing CF kits, the variance in values for percent insulin secretion by CF islets at low glucose was significantly greater than in their non-CF controls (P < 0.0001), suggesting dysregulated insulin secretion by the CF islet. Such findings provide strong support that islet-intrinsic CFTR defects alter glucose-responsive insulin secretion by the β cell.
Insulin-mediated AKT activation is not altered in newborn CF ferret liver, skeletal muscle, or fat. Studies in cultured isolated islets suggest that a component of altered insulin secretion in CF may be intrinsic to the islet. However, it is also possible that insulin resistance could contribute to both the abnormal glucose tolerance and hyperin-sulinemia observed in CF animals. The status of insulin resistance in CFRD is debated, with insulin sensitivity found to be reduced (15, 23, 24), enhanced (15, 25, 26), or normal (12, 27–29). Addition-ally, hepatic insulin resistance with enhanced peripheral insulin sensitivity is observed in a subset of CF patients that are exocrine-insufficient but lacking diabetes (15). To assess the state of insulin sensitivity in newborn CF and non-CF animals, we evaluated insu-lin-stimulated AKT phosphorylation in the liver, skeletal muscle, and adipose tissue as an indirect measure of insulin sensitivity, because hyperinsulinemic clamps are not feasible in newborn fer-rets due to their size. Although altered AKT signaling has yet to be validated in the ferret as a marker of insulin resistance, reduction in insulin-stimulated AKT phosphorylation is a common feature of insulin resistance states in humans (51), pigs (52), and mice (53, 54). Results from these experiments demonstrated that newborn CF ferrets had levels of insulin-stimulated AKT phosphorylation linemia that exceeds C-peptide levels (49). Thus, in the absence
of significant alterations of insulin clearance in CF animals, one would expect both plasma insulin and C-peptide levels to be simi-larly elevated in nursing CF as compared with non-CF kits. Indeed nonfasted CF newborn kits demonstrated significantly higher plas-ma C-peptide levels and C-peptide/glucose ratios as compared with non-CF controls (Figure 9, G and H), despite similar blood glucose levels in these two groups (Supplemental Figure 3B). These results indicate that the elevated insulin levels in CF kits represents bona fide insulin hypersecretion. Importantly, the molar ratio of plasma C-peptide/insulin was not significantly different between CF and non-CF animals (Figure 9I), although there was a trend toward lower molar ratios in the CF group. Like plasma insulin levels, C-peptide levels and C-peptide/glucose ratios both demonstrated significantly higher variances in CF animals (P < 0.0001). However, variances in plasma glucose or the molar ratio of C-peptide/insulin were not significantly different between groups.
[image:8.585.41.367.81.370.2]Isolated cultured CF islets improperly regulate insulin secretion. In vivo studies in newborn animals suggest that insulin secretion by the pancreas is improperly regulated. To evaluate whether islet-intrin-sic CFTR defects influence insulin secretion, we utilized a previous method developed for neonatal pigs (50) to isolate and culture new-born islets from CF and non-CF ferrets. Insulin secretion over the course of 1 hour was then assayed under conditions of 2 mM and 30 mM glucose exposure. Similar to the trend toward higher levels of insulin found in the CF pancreas at birth (Figure 7E), isolated 9-day-cultured CF islets contained approximately 1.5-fold more total insulin on average then non-CF islets (see Figure 9 legend for details). Interestingly, CF islets had a significantly higher (5.3-fold) percent insulin secretion at 2 mM glucose in comparison to non-CF controls (Figure 9, J–L). Following stimulation with 30 mM glu-cose, non-CF islets demonstrated a significant induction in percent
Figure 6
Differences in islet size between CF and non-CF newborn kits. (A) Representative images from insulin-stained pancreas sections from newborn non-CF and CF ferrets. Scale bar: 50 μm. (B) High magnification of boxed area in A demonstrating the size classification of small (0.1 to <1 mm2), medium (1–2 mm2),
and large islets (>3 mm2). Scale bar: 50 μm.
content suggest no significant differences between genotypes. By contrast, increased canalicular and bile duct plugging in CF ani-mals suggests that early liver disease could in part contribute to alterations in insulin metabolism.
Discussion
The multiorgan nature of CF disease has made it difficult to dis-sect the early pathogenesis of CFRD. Glucose abnormalities and altered kinetics of insulin secretion are common in CF patients, occurring as early as in childhood. However, studies in infants and children younger than 6 years of age have yet to be performed. Our findings in the CFTR-knockout ferret demonstrate that insulin secretion is abnormally regulated very early during the course of CF disease and that islet-intrinsic CFTR defects cause dysregulat-ed glucose-dependent insulin secretion. These abnormalities and other contributing factors, such as insulin resistance and pancre-atic destruction, are likely important to the emergence of altered glucose metabolism in CF and the development of CFRD.
Most CF patients with altered and normal glucose tolerance demonstrate IFPI with delayed time to peak insulin (9–12). Such findings were similar in newborn CF ferrets. First-phase insulin secretion is critical for maintaining normal glycemia in response to glucose load, and IFPI is considered a key component of dia-betes physiology (60). Our data suggest that IFPI is present very early in CF and may in part be facilitated by islet-intrinsic defects. The abnormalities in glucose tolerance that manifest at birth in fasted CF ferrets are most likely due to IFPI (Figure 4A and Figure 5, A–F). As expected with IFPI, delayed insulin responses follow-ing glucose stimulation of CF kits were accentuated (Figure 5G) at intermediate time points when hyperglycemia was observed dur-ing GTT. Whether the 10.5-fold rise in insulin/glucose ratios at 60 minutes (Figure 5H) at the peak of GTT insulin represents a pathologic overshoot is difficult to determine. However, the mag-nitude of overshoot was far greater for CF kits than that observed in CFRD adults (20) and children with abnormal glucose metabo-lism (61); insulin/glucose ratios at the time of delayed peak insulin in CF patients are similar or slightly lower than in control patients (20, 61, 62). Thus, the magnitude of insulin overshoot observed in that were not significantly different from those in non-CF controls
in all 3 tissues analyzed (Supplemental Figure 4). Although these findings do not rule out that potential CF-associated changes in hepatic or peripheral insulin sensitivity may impact insulin secre-tion or glucose tolerance, they do support the nosecre-tion that impaired insulin clearance due to insulin resistance is not likely the cause of elevated insulin levels observed in fasted and fed CF animals.
CF liver disease predisposes patients to glucose abnormalities and diabetes (55, 56). However, it remains unclear whether neo-natal liver pathologies in CF influence glucose and insulin regula-tion. Elevated levels in liver function tests (LFTs: alanine transami-nase, aspartate transamitransami-nase, γ-glutamyltransferase, and bilirubin) are commonly observed in the majority of newborn CF infants (41, 42, 57) and newborn CF ferrets (40). Neonatal elevations in LFTs observed in CF infants are generally thought to reflect cholestasis due to thickening of bile (42, 57, 58). However, when liver biop-sies were evaluated in CF neonates, there was no direct correlation between LFTs and histologic cholestasis (41). Whether this is a consequence of the focal nature of disease or an inherent compo-nent of the early liver CF pathobiology remains unclear (41). To further probe for potential hepatic pathologies in the CF ferret model that might impact early glucose and insulin abnormalities, we quantified bile duct plugging in canaliculi and bile ducts of CF and non-CF newborn animals. Results from these studies demon-strated significant increases in canalicular and bile duct plugging in CF animals as compared with controls (Supplemental Figure 5, A–D). Such findings correlate with elevations in plasma bilirubin observed in neonatal CF ferrets (40).
[image:9.585.41.381.85.310.2]Fatty liver disease is not uncommon in human CF (41, 58), and liver dysfunction (including fatty liver) is often associated with insulin resistance and abnormal glucose metabolism in humans and mice (49, 59). Fatty liver (or steatosis) is characterized by exces-sive triglyceride deposition. Therefore, as a second index for poten-tial alterations in liver metabolism, we assessed triglyceride con-tent in the liver between CF and non-CF newborn ferrets. However, no significant difference in hepatic triglyceride content between genotypes was observed (Supplemental Figure 5E). Cumulatively, our finding on insulin/AKT signaling and hepatic triglyceride
Figure 7
much higher levels and those with normal or near-normal levels. We speculate that this clustering may reflect differences in the postprandial timing of the blood sampling in this randomly nurs-ing population. For example, those animals sampled shortly after feeding may mirror the hyper-insulin secretory state observed at 30–60 minutes during a GTT (Figure 5G). By contrast, animals that had not fed within 2–3 hours prior to sampling would be expected to have much lower insulin levels and normal insulin/ glucose ratios (Figure 5, G and H). Because of the high insulin lev-els, we were interested in whether these randomly fed (nursed) CF kits would demonstrate a tendency toward lower blood glucose, which they did, but not to a statistically significant degree (Figure 9A, P = 0.0513). This suggests that the degree of hyperinsulinemia may tend toward being excessive. Consistent with this, insulin levels failed to decrease with lower glucose levels in nursing CF kits, while glucose and insulin levels were positively correlated in nursing non-CF kits (Supplemental Figure 3A). These tendencies observed in CF kits toward insulin oversecretion might play a role in the hypoglycemia that is observed in humans with CF (63, 64).
Abnormalities intrinsic to the CF pancreas are another impor-tant factor that may influence insulin secretion. Although the new-born CF pancreas and CF cultured islets had approximately 50% more insulin content than non-CF controls, there was no signifi-cant difference in percent lobular insulin and glucagon staining CF kits may be specific to the early neonatal period and
physio-logically appropriate, since hypoglycemia was not observed at 120 minutes, when insulin/glucose ratios had normalized to the level seen in non-CF animals (Figure 5H). One explanation for the high insulin levels would be systemic insulin resistance often observed in older CF patients (15, 23, 24). However, we found no evidence to support this hypothesis through the evaluation of plasma cortisol levels (Figure 9, D–F) or insulin/AKT signaling in liver, muscle, and adipose tissue (Supplemental Figure 4). Nonetheless, the observed significantly enhanced bile plugging in hepatic canaliculi and bile ducts and non-significant trend toward reduced C-peptide/insu-lin molar ratios of CF animals suggest that hepatic abnormalities could impact insulin action and clearance in newborn CF ferrets.
[image:10.585.92.491.86.381.2]The abnormal regulation of insulin in fasted glucose-challenged CF animals prompted us to evaluate how insulin secretion was regulated in randomly nursing kits. Given the IFPI observed in newborn CF kits, we hypothesized that glucose and insulin levels would be poorly regulated in fed animals. Indeed this was partly the case: plasma insulin, C-peptide, insulin/glucose, and C-pep-tide/glucose values were highly variable and on average much higher (8.5-, 6.5-, 14-, and 10-fold, respectively) in nonfasted CF kits compared with non-CF controls (Figure 9, A–C and G–I). Interestingly, CF animals appeared to partition into two popula-tions in terms of plasma insulin and C-peptide levels, those with
Figure 8
between genotypes at birth (Figure 7). The observed differences in islet sizes between genotypes (Figure 6), however, indicate sub-tle changes in β cell organization within CF pancreata that could impact insulin regula-tion. However, no differences in the distribu-tion of two pancreas developmental markers (SOX9 and PDX1) were observed between genotypes, leaving the etiology of this find-ing still unknown. Whether this morpho-logic difference in islet size exists in CF human newborns is also unknown; however, previous reports have documented in utero and neonatal abnormalities in the develop-ment of the exocrine pancreas in CF indi-viduals (65, 66). For example, exocrine acinar and duct lumens demonstrate a 10-fold-increased volume in CF fetuses at 32–38 weeks after conception (65), a number that is strikingly similar to the 14-fold increase seen in newborn CF ferrets (Figure 1). Given the lower lobular/total area ratio in the CF ferret pancreas at birth and the simi-lar pancreas wet weight/BW ratios of the genotypes, it is possible that CF pancreata have reduced total cellular mass at birth. Such a possibility in the CF pancreas could be due to loss of acinar cells and/or acinar cell compression. Additionally, the observed enhanced apoptosis in newborn CF ferret pancreatic acinar/duct cells demonstrates that early damage to the exocrine pancreas does occur, and this could potentially influ-ence functions of the neonatal CF endocrine pancreas. These pathologies likely initiate prior to birth, based on CF human fetal stud-ies, and could impact development of islets. While we observed no significant differences in β cell apoptosis of CF and non-CF islets at birth, alternative mechanisms of β cell injury not detected by activated caspase-3 could indeed impact insulin secretion and glucose tolerance in CF neonates. Although we cannot rule out that small differences in β cell apop-tosis or replication at birth (<1%–4%) might contribute to altered islet mass, the major con-tributing factor to reduction in islet mass in CF animals is likely the progressive pancreatic exocrine damage that occurs with age.
[image:11.585.52.358.82.470.2]Although mild insulin resistance and early exocrine pancreatic damage may be islet-extrinsic factors that contribute to altered insulin secretion in CF kits, studies in isolat-ed culturisolat-ed islets suggest that islet-intrinsic abnormalities alter glucose-responsiveness of the CF islet. As in nursing CF kits (Figure 9, A–C, and Supplemental Figure 3A), insulin secretion by CF islets is highly variable and less responsive to glucose stimulation in compari-son to controls (Figure 9, J–L). Although IFPI
Figure 9
Abnormalities in islet insulin and C-peptide secretion occur in newborn CF ferrets. (A–I) Plasma collected from nursing newborn kits (6–12 hours after birth) was used to measure glucose, insulin, cortisol, and C-peptide. Since insufficient quantities of plasma could be obtained from 1 animal for all these measures, different cohorts were evaluated in
A–C, D–F, and G–I. However, in a subset of animals in A–C there was sufficient plasma for C-peptide measurements, thus the cohort of animals in G–I is included within A–C
(for glucose, insulin, and insulin/glucose ratios for the cohort in G–I, see Supplemental Figure 3). (J and K) Insulin secretion by isolated cultured islets from (J) non-CF and (K) CF newborns. For each data point, pooled 9-day cultured islets were split into 2 groups of equally sized islets and incubated in the presence of 2 or 30 mM glucose for 1 hour. The percent insulin secretion for each experiment and the average SI for all experiments were calculated as described in Methods. (L) Combined data from J and K. Supplemental Fig-ure 6 shows representative insulin and glucagon immunofluorescence staining patterns for CF and non-CF islets. Values are mean ± SEM for n independent animals/experiments. Differences in A–I were assessed using a nonparametric 2-tailed Mann-Whitney U test. Significantly higher variances in CF compared with non-CF values were observed for datasets in B, C, G, and H (F test, P < 0.0001). No significant differences in variances were observed for A, D–F, and I. Differences in percent insulin secretion of J and K were assessed using a nonparametric 2-tailed paired Wilcoxon test, while differences in the secretory index (SI) of CF and non-CF islets were assessed using the nonparametric 2-tailed Mann-Whitney test (†P < 0.0111 comparing SI values in J and K. For L, #P < 0.001,
*P < 0.01, †P < 0.05, Kruskal-Wallis nonparametric 1-way ANOVA with a Dunn’s post-test.
The average total insulin content of islets (secreted plus that remaining in the islet over the 1-hour period) was not significantly different between in CF (1,198 ± 302 μU insulin,
Methods
Ferret breeding and rearing. The previously described CFTR exon 10–dis-rupted ferret model was used for all studies (39). CFTR+/– hobs and jills were bred at Marshall Farms and shipped to the University of Iowa for birth at 28 days gestation (2 weeks prior to whelping at 42 days gestation). Litters of CFTR+/+, CFTR+/–, and CFTR–/– kits were rapidly genotyped at birth by PCR using modifications to a previously described method (40). A small piece of tail tissue (0.1–0.2 cm in length) from each kit was lysed by heating at 95°C in 50 μl lysis buffer (25 mM NaOH, 0.2 mM EDTA) for 40 minutes. The lysate was then neutralized by adding 50 μl of 40 mM Tris-HCl pH 7.4 and clarified by centrifuging at 10,000 g for 10 minutes. One microliter of supernatant was then used directly for the PCR using the genotyping reaction and primers previously described (40). When litters were reared, a protocol previously described was used with minor modifi-cation (40). This involved gavage feeding with 150–200 μl Elecare (Abbott Laboratories) containing 10% GoLYTELY, ursodeoxycholic acid (5 mg/kg), and omeprazole (5 mg/kg) 4 times daily. Kits also were free to nurse and, depending on weight gain, were supplemented with 2–4 additional Elec-are/10% GoLYTELY gavages daily. All animals were reared from birth on the antibiotics metronidazole (5 mg/kg, 4 times daily) and Zosyn (4.0 mg/kg, twice daily) with each injection given in 100 μl saline s.c. As animals gained weight, the amount of saline was increased proportionally with BW for each injection. Because CF kits are highly susceptible to lung infection, an additional antibiotic was used if kits began to lose weight during a 2-day period. These two antibiotics included enrofloxacin (1.5 mg/kg, twice daily) and ceftazidime (10 mg/kg, 4 times daily). Control non-CF siblings (CFTR+/– or CFTR+/+) were treated identically during the rearing process.
Histology and immunohistochemistry. At necropsy, pancreata were removed and fixed in 10% neutral buffered formalin. Tissues were routinely pro-cessed, embedded in paraffin, and sectioned at 4 μm. Sections were stained with hematoxylin and eosin for light microscopy. Formalin-fixed, paraffin-embedded pancreata were stained by immunohistochemistry for insulin (MP Biomedicals), glucagon (MP Biomedicals), PCNA (Dako), PDX1 (Mil-lipore), and SOX9 (Millipore). For single stains, tissue sections were depa-raffinized in xylene and rehydrated through graded alcohols. Slides were treated with proteinase K for 5 minutes. Endogenous peroxidase activity was quenched by immersing slides in 3% hydrogen peroxide for 8 minutes. After blocking in Background Buster (Innovex Biosciences) for 30 minutes, tissues were incubated for 1 hour at room temperature with the primary antibody: guinea pig anti-insulin (1:4,000, 60 minutes) or rabbit anti-gluca-gon (1:50, 60 minutes). Rabbit anti–guinea pig HRP (Dako) was applied to the insulin slides at 1:500 for 30 minutes. Rabbit EnVision (Dako) second-ary antibody was applied for 30 minutes to all slides. Slides were developed with DAB Plus (Dako) for 5 minutes, followed by DAB Enhancer (Dako) for 3 minutes. Slides were then counterstained and coverslipped. Negative control slides were prepared by substituting primary antibody with buffer. Dual staining for insulin (1:4,000, 60 minutes)/PCNA (1:4,500, 15 min-utes), insulin (1:4,000, 60 minutes)/PDX1 (1:4,000, 60 minmin-utes), and insu-lin (1:4,000, 60 minutes)/SOX9(1:4,000, 60 minutes) was performed using the EnVision G2 Doublestain System (Dako). Following deparaffinization, slides were retrieved in citrate buffer (pH 6.0) in a Decloaker (Biocare) for 20 minutes. The dual staining kit directions were then followed.
Morphometric analysis. Whole-section images of insulin- and glucagon-stained pancreas were obtained using a BX-51 microscope with CellSens digital imaging software (Olympus) at ×200. Images were analyzed using ImageJ software (http://rsbweb.nih.gov/ij/). Insulin- and glucagon-immu-noreactive cells had diffuse cytoplasmic staining. Images were converted to an RGB stack, and the staining was thresholded in the blue channel identically for all samples for each stain. The percent parenchymal stain-ing of insulin and glucagon was determined by dividstain-ing the stained area by in randomly nursing kits would be expected to lead to compensatory
insulin secretion to maintain normal glycemia, abnormal glucose responsiveness of the CF β cell may also contribute to the inability of CF kits to properly regulate insulin secretion in the fed state. Inter-estingly, the intrinsic predisposition of isolated cultured CF islet to hypersecrete insulin at low glucose is not predicted by insulin levels in fasted CF kits, suggesting that the mechanistic basis of altered insulin secretion in CF kits is likely more complex than just IFPI. At low glucose, in vitro cultured CF islets secrete 5.2-fold more of their insulin content relative to non-CF controls, while also having impaired induction of insulin secretion to high glucose challenge. By contrast, the induction of insulin in fasted CF kits is accentuated at later time points, most likely due in large part to compensation for IFPI — CF newborn kits demonstrate a 38-fold rise in insulin/ glucose ratios between 2 and 60 minutes after glucose challenge as compared with a 1.9-fold decline in this ratio for non-CF animals for the same time frame. We hypothesize that an islet-extrinsic factor(s) may allow for normalization of insulin in fasted CF ani-mals, suppressing the intrinsic islet tendency to over-secrete insulin at low glucose, and that such factor(s) are not present in the isolated CF islet. Excess abundance of such an insulin secretion–suppressive factor(s) in the fasted state could also account for the delayed insu-lin responses during transition to a fed state in CF animals.
Glucose abnormalities progressed with age in CF ferrets, dem-onstrating significant alterations in glucose excursion following MMTT. When these results are compared with CFRD clinical clas-sifications using oral GTTs, older CF ferrets demonstrated two MMTT phenotypes, including impaired glucose tolerance and indeterminate glycemia (Figure 4C). Although MMTTs are not a pure measure of glucose tolerance, being influenced by entero-insular axis hormones that could be altered in a CF-specific man-ner, the MMTT glucose excursion curves were similar to those observed in CFRD patients not only following oral GTTs but also following a meal (20, 61). Interestingly, progressive destruction and inflammation of the exocrine pancreas in older CF ferrets (Figure 2) were associated with the appearance of spontaneous hyperglycemia and glycosuria (Figure 3). These older CF ferrets demonstrated a 52% loss of insulin-positive cells (Figure 8E), which is very simi-lar to the approximately 50% loss in β cells seen at autopsy in CF patients, including those with overt diabetes and glucose intoler-ance (18). Relatively little is know about how early phases of pan-creatic inflammation and exocrine decline impact glycemic regula-tion in CF patients. In non-CF children, a single episode of acute pancreatitis can lead to insulin requiring diabetes mellitus (67). Pancreatic-sufficient CF patients are most susceptible to acute pan-creatitis during adolescence and young adulthood (68–70), while pancreatic-insufficient patients rarely develop acute pancreatitis. Based on the CF ferret, inflammation association with the onset of exocrine pancreas decline appears closely linked to spontaneous glucose abnormalities and worsening of glucose tolerance.
Measurements of plasma insulin, C-peptide, and cortisol. Plasma insulin was measured using an ELISA kit that is specific for bovine and porcine fully processed insulin and does not cross-react with proinsulin or C-peptide (Calbiotech). Plasma C-peptide was measured using a human/rat/mouse C-peptide–specific Enzyme Immunoassay Kit (RayBio) based on the prin-ciple of competitive enzyme immunoassay. Plasma cortisol was measured using an ELISA kit (Calbiotech). To evaluate the total insulin content of the pancreas, newborn kits were weighed and immediately sacrificed after birth, and the pancreas was dissected. Whole pancreas wet weight was recorded, and then pancreas protein extracts were prepared by acetic acid extraction as previously described (50). Extracts were used for insulin ELISA measurements and total protein measurements. Protein concentra-tions were determined using the Bio-Rad Protein Assay.
Measurements of acute and late-phase insulin secretion. Acute insulin secre-tion tests were performed on 6- to 12-hour-old kits that had been fasted for 3 hours using l-arginine and glucose stimulation tests as previously described, with minor modifications (71). Kits were injected (i.p.) with glu-cose (3 g/kg BW) or l-arginine (0.3 g/kg BW). Blood glucose levels were measured from a pin prick to the tail immediately before and 2 minutes after the injection. Plasma samples were then immediately collected into chilled heparinized tubes for insulin ELISA measurements. Plasma sam-ples were immediately frozen and stored at –80°C prior to analysis. For analysis of late-phase insulin secretion, 3-hour-fasted 6- to 12-hour-old kits were injected (i.p.) with glucose (2 g/kg BW), and blood was harvested at 30, 60, and 120 minutes after injection for blood glucose and plasma insu-lin determinations as described above.
Neonatal islet culture and insulin secretion studies. CF and non-CF islets were isolated and cultured using a previously described protocol for neonatal pig islets (50). Briefly, pancreata from 3–5 CF or non-CF kits were minced into 1- to 2-mm3 pieces and incubated in HBSS containing 0.5% BSA, 1%
penicillin/streptomycin, and 2.5 mg/ml collagenase for 4 minutes at 37°C. Following digestion, the tissue fragments were washed 3 times in medium without collagenase and then cultured in 24-well plates with 2 ml Ham’s F12 medium (containing 10 mM glucose) supplemented with 0.5% BSA, 50 μM 3-isobutyl-1-methylxanthine (IBMX), 10 mM nicotinamide, 2 mM l-glutamine, and 100 U/ml penicillin and 100 μg/ml streptomycin. The medium was changed on the second day after isolation and every other day thereafter. On the ninth day of culture, approximately 500–600 islets were selectively removed with a small glass pipette, washed, and divided into 2 equal-sized matched groups for each genotype and then incubated in RPMI 1640 containing 2 mM glucose, 2 mM l-glutamine, and 0.5% BSA for 1 hour at 37°C. To initiate the insulin secretion experiment, islets were then transferred into 1 ml of fresh RPMI 1640 medium containing 2 mM or 30 mM glucose for 1 hour at 37°C. The media and islets were then collected, and total insulin was quantified in both by ELISA. The amount of insulin secreted into the media during the 1-hour period and that remaining in the islets was used to calculate the percent insulin secretion (% insulin secretion = secreted insulin in the media/total insulin in the media and islets at the end of the experiment). SI was calculated as percent insulin secretion at 30 mM glucose divided by percent insulin secretion at 2 mM glucose.
Insulin-mediated AKT signaling. Newborn 6- to 12-hour-old ferrets were fasted for 3 hours and then anesthetized with isofluorane. The hepatic por-tal vein was surgically exposed and 1.5 U/10 g BW insulin was injected in a volume of 60–100 μl over approximately 10 seconds. Animals were then euthanized at 5 minutes after insulin injection, and skeletal muscle, liver, and perirenal fat were rapidly excised. The tissue samples were immedi-ately snap-frozen in liquid nitrogen and stored at –80°C until use. Tissues were processed in lysis reagent (C2978, Sigma-Aldrich) containing 1% (v/v) protease (P8340, Sigma-Aldrich) and phosphatase (524625, Calbiochem) inhibitor cocktails. Lysates (70 μg protein) were blotted using antibodies (a) the total pancreatic area, including acellular duct and acinar lumens,
and (b) the total lobular area, including endocrine and exocrine parenchy-ma (which includes only the cellular area and excludes acellular duct and acinar lumens). For measurement of the lobular tissue area, the section was thresholded and area measured in the red channel. For islet morphometry analysis, the insulin-stained sections were thresholded in the blue channel, and particles were counted as defined by particle size restrictions. Islets were designated by area: small (0.1 to less than 1.0 mm2, medium (1.0–2.0
mm2), and large (>2.0 mm2). Islets sizes and insulin/glucagon staining were
quantified from one section per animal taken from a similar plane of sec-tion and digitally scanned in its entirety. The entire secsec-tion was quantified from each animal. On average, approximately 550 islets were quantified per section/animal. The percentage of the islet sizes was then determined based on the total number of islets counted in the section, and the per-cent insulin or glucagon staining (lobular and total area) was calculated as described above. For quantification of immunohistochemical staining patterns (PCNA/insulin, activated caspase-3/insulin, and PDX1/insulin), sections were imaged at ×400, and 5 images per section (1 section per ani-mal) were quantified. For each section, approximately 300 insulin-positive islet cells were counted, and islet size was documented. The insulin-positive islet cells that were also PCNA, activated caspase-3, or PDX1 positive were counted and converted to a percent of total cells. To quantify acinar/duct apoptosis, caspase-3–positive cells were counted in five ×400 fields (~1,200 total acinar/duct cells per section). For quantification of PDX1 staining in acinar/duct regions of the pancreas, PDX1-positive or PDX1/insulin dual-positive cells were counted for each section as a fraction of total nuclei (~500 acinar/duct cells per section) and values converted to a percentage. For quantification of the acinar/duct dilation score, images were taken at ×200 and all lumen diameters measured. Two images per section were quantified. Inflammation was scored on a scale of 0 to 3 based on the num-ber of interstitial inflammatory cells per five ×400 fields: 0 (no inflam-matory cells), 1 (<10 inflaminflam-matory cells), 2 (10–30 inflaminflam-matory cells), and 3 (>30 inflammatory cells). Degenerative neutrophils, occasionally found within dilated acinoductular lumens of younger animals and always seen in the acinoductular lumens of older animals, were not included in the inflammation scoring.
GTTs. GTTs were carried out on 3-hour-fasted newborn (6–12 hours after birth) and 5- to 11-day-old kits. CF (CFTR–/–) and non-CF (CFTR+/+ and CFTR+/–) kits are designated as experimental and control groups, respec-tively. Kits were injected (i.p.) with glucose (2 g/kg BW), and blood glucose levels were measured from tail pin prick immediately before and 5, 10, 30, 60, 90, and 120 minutes after the glucose injection using an automatic blood glucose monitor (One Touch LifeScan). AUC was calculated using the trapezoid rule.
MMTTs in older animals. After weaning at 5–6 weeks, both CF and control animals were maintained on a slurry of canned food (Halo) and Elecare (Abbott Laboratories) supplemented with pancreatic enzymes. MMTTs were performed in weaned animals 45–90 days of age using the follow-ing protocol optimized for reproducibility in juvenile ferrets. After befollow-ing allowed to feed until satiated, animals were fasted for 3 hours prior to the MMTT. Blood glucose was measured during the fast at –90, –60, –30, and 0 minutes before the MMTT. The MMTT was initiated by feeding the ani-mals a quantity of Elecare and canned food normalized to a body surface area estimate (BSAE, in cm2) of each animal: (BW [g] × body length [cm])0.5.
Each MMTT was performed in pairs with aged-matched CF and non-CF animals using the same ratio of food to BSAE (typically 0.2 ml Elecare per cm2 BSAE and 0.27 g solid food per cm2 BSAE was used). Postprandial
measures analysis for changes in blood glucose with age and MMTT blood glucose values after feeding. These analyses were conducted using the lme4 package for the R statistical computing environment. In the analysis of the age-dependent trends in CF/non-CF blood glucose ratios of older animals (3 paired groups), the log of the ratio of blood glucose of a CF animal to that of its matched non-CF animal was computed, and a mixed-effects model was then fitted, with age the fixed effect and animal pair the ran-dom effect. For MMTT data on paired CF/non-CF groups, we conducted two analyses on the log of blood glucose using a mixed-effects model. In both analyses, the effect of animals was treated as random, and the group effect of each experiment was treated as fixed. In the first analysis, time after feeding was included as a fixed effect with a random slope. This analysis assessed whether post-feeding blood glucose was significantly different between CF and non-CF paired groups using aggregate data from the MMTT experiments. In the second analysis, the focus was on the effect of CF versus non-CF status at every time point during the MMTT assay using aggregate data from the MMTT experiments for the paired groups. Morphometric analysis assessing 3 or more datasets was evaluated using Kruskal-Wallis nonparametric 1-way ANOVA with a Dunn’s post-test. Morphometric analysis assessing 2 datasets was evaluated using the nonparametric 2-tailed Mann-Whitney U test. Percent insulin secretion from islets of a single genotype was evaluated using the nonparametric 2-tailed paired Wilcoxon test, while differences in the SI of insulin between genotypes was evaluated using the nonparametric 2-tailed Mann-Whitney
U test. Differences in percent insulin secretion of islets between genotypes at low and high glucose were evaluated using Kruskal-Wallis nonparamet-ric 1-way ANOVA with a Dunn’s post-test. In all statistical analyses, P val-ues less than 0.05 were deemed significant.
Study approval. This study was performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Iowa.
Acknowledgments
This work was supported by NIH grants DK047967, DK091211, HL108902, HL099516 (to J.F. Engelhardt), DK092284 (to Z.A. Stewart), DK081548 and Fraternal Order of Eagles Diabetes Research Center grant (to A.W. Norris), and the University of Iowa Center for Gene Therapy (DK54759). We also gratefully acknowl-edge Scott Blackman (Johns Hopkins University) for helpful dis-cussions during the course of this work.
Received for publication September 1, 2011, and accepted in revised form July 26, 2012.
Address correspondence to: John F. Engelhardt, Room 1-111 BSB, Department of Anatomy and Cell Biology, College of Medicine, University of Iowa, 51 Newton Road, Iowa City, Iowa 52242, USA. Phone: 319.335.7744; Fax: 319.335.6581; E-mail: john-engelhardt@ uiowa.edu. Or to: Andrew W. Norris, 1270 CBRB, 285 Newton Road, University of Iowa, Iowa City, Iowa 52242, USA. Phone: 319. 356.4443; Fax: 319.356.8170; E-mail: andrew-norris@uiowa.edu. against AKT and phospho-AKT Ser473 (9272, 9271, Cell Signaling
Tech-nology). Filters were first probed with the phospho-AKT Ser473 antibody, then stripped and reprobed with the total AKT antibody. The ratio of phospho-AKT to total AKT immunoreactivity was assessed for each animal using NIH Image. To avoid subtle differences in exposure times between experiments, tissues from 8 CF and 8 non-CF animals were equally divided for two Western blot experiments (4 CF and 4 non-CF animals) that probed for phospho-AKT and total AKT in each organ at the same time. An inter-nal insulin-treated reference control from each tissue in the first experi-ment was loaded onto gels of the second experiexperi-ment and used to normalize subtle differences in exposure times between the two experiments.
Bile plug quantification in the liver. Liver sections from newborn 6- to 12-hour-old non-CF and CF ferrets were scored for bile plugs within hepatic canaliculi and bile ducts. Scoring for bile canalicular plugs was performed on two liver sections per animal by counting the total number of canalicular plugs (scored as any visible accumulation of bile) in five ×400 fields per section. The average number of canalicular plugs per tissue sec-tion was used to determine the canalicular bile plug score for each animal. The percentage of bile duct plugs was determined by counting the bile ducts containing bile plugs (any visible accumulation of bile) and dividing by the total number of bile ducts per section. Two sections were counted in their entirety, and the percentages were averaged for each animal. To minimize counting multiple profiles of the same bile duct, each portal area was counted as one bile duct.
Hepatic triglyceride analysis. Total triglyceride content of the liver was mea-sured using a previously described protocol (72). Newborn 6- to 12-hour-old ferrets were fasted for 3 hours and euthanized, and the liver was rap-idly excised and immediately snap-frozen in liquid nitrogen and stored at –80°C until use. Briefly, a defined mass of liver tissue (~200 mg) was homogenized in 350 μl ethanolic potassium hydroxide, followed by incu-bation at 55°C overnight until no oil layer was visible. The sample was brought up to 1 ml with 50% ethanol, votexed, and then centrifuged at 4°C for 5 minutes. The supernatant was removed to a new tube, topped off to 1.2 ml with 50% ethanol, and then vortexed. Two hundred micro-liters of that sample was then mixed with 215 μl of 1 M MgCl2, vortexed,
and incubated on ice for 10 minutes prior to centrifugation at 4°C for 5 minutes. The supernatant was then removed to a new tube and assayed for triglycerides using Free Glycerol Reagent (F6428, Sigma-Aldrich) and Glycerol Standards (G7793, Sigma-Aldrich) to construct standard curves. The total triglyceride (mg) per gram liver was then calculated.
Statistics. Data are expressed as mean ± SEM of n independent obser-vations or animals. Individual GTT time points and GTT AUC, insulin, C-peptide, and cortisol comparisons between CF and non-CF animals were evaluated using the nonparametric 2-tailed Mann-Whitney U test. A 2-tailed F test was used to compare variances in values of CF and non-CF datasets. Statistical analysis of regression slopes and intercepts for log[insulin] versus glucose plots used the linear model function in R (http://www.R-project.org). For studies involving older animals with CF and non-CF paired groups, multiple variance components were modeled in a mixed-effects model evaluating longitudinal analysis and
1. Moran A, et al. Epidemiology, pathophysiol-ogy, and prognostic implications of cystic fibrosis-related diabetes: a technical review. Diabetes Care. 2010;33(12):2677–2683.
2. Hameed S, Jaffe A, Verge CF. Cystic fibrosis related diabetes (CFRD) — the end stage of progressive insu-lin deficiency. Pediatr Pulmonol. 2011;46(8):747–760. 3. Lanng S, Thorsteinsson B, Nerup J, Koch C. Influ-ence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. Eur J Pediatr. 1992;151(9):684–687.
4. Bismuth E, et al. Glucose tolerance and insulin
secretion, morbidity, and death in patients with cys-tic fibrosis. J Pediatr. 2008;152(4):540–545.e1. 5. Koch C, et al. Presence of cystic fibrosis-related
dia-betes mellitus is tightly linked to poor lung func-tion in patients with cystic fibrosis: data from the European Epidemiologic Registry of Cystic Fibro-sis. Pediatr Pulmonol. 2001;32(5):343–350. 6. Brodsky J, Dougherty S, Makani R, Rubenstein RC,
Kelly A. Elevation of 1-hour plasma glucose dur-ing oral glucose tolerance testdur-ing is associated with worse pulmonary function in cystic fibrosis. Diabe-tes Care. 2011;34(2):292–295.
7. Cheung MS, et al. Growth in children with cystic fibrosis-related diabetes. Pediatr Pulmonol. 2009; 44(12):1223–1225.
8. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Dia-betes Care. 2009;32(9):1626–1631.