0022-538X/10/$12.00 doi:10.1128/JVI.01400-10
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
The Matrix Protein of Vesicular Stomatitis Virus Binds
Dynamin for Efficient Viral Assembly
䌤
He
´le
`ne Raux,
1Linda Obiang,
1Nicolas Richard,
1Francis Harper,
2Danielle Blondel,
1and Yves Gaudin
1*
Centre de Recherche de Gif, Laboratoire de Virologie Mole´culaire et Structurale, CNRS (UPR 3296), IFR115, Alle´e de la Terrasse, 91198, Gif sur Yvette, France,1and CNRS, FRE 2937, IFR 89, Laboratoire de
Replication de l’ADN et Ultrastructure du Noyau, 94801 Villejuif, France2
Received 6 July 2010/Accepted 30 September 2010
Matrix proteins (M) direct the process of assembly and budding of viruses belonging to theMononegavirales
order. Using the two-hybrid system, the amino-terminal part of vesicular stomatitis virus (VSV) M was shown to interact with dynamin pleckstrin homology domain. This interaction was confirmed by coimmunoprecipi-tation of both proteins in cells transfected by a plasmid encoding a c-myc-tagged dynamin and infected by VSV. A role for dynamin in the viral cycle (in addition to its role in virion endocytosis) was suggested by the fact that a late stage of the viral cycle was sensitive to dynasore. By alanine scanning, we identified a single mutation of M protein that abolished this interaction and reduced virus yield. The adaptation of mutant virus (M.L4A) occurred rapidly, allowing the isolation of revertants, among which the M protein, despite having an amino acid sequence distinct from that of the wild type, recovered a significant level of interaction with dynamin. This proved that the mutant phenotype was due to the loss of interaction between M and dynamin. The infectious cycle of the mutant virus M.L4A was blocked at a late stage, resulting in a quasi-absence of bullet-shaped viruses in the process of budding at the cell membrane. This was associated with an accumulation of nucleocapsids at the periphery of the cell and a different pattern of VSV glycoprotein localization. Finally, we showed that M-dynamin interaction affects clathrin-dependent endocytosis. Our study suggests that hijacking the endocytic pathway might be an important feature for enveloped virus assembly and budding at the plasma membrane.
Vesicular stomatitis virus (VSV) is the prototype rhabdovi-rus and has been used as a model for years to study many aspects of the viral cycle. Its negative-strand RNA genome encodes successively the nucleoprotein N, the phosphoprotein P, the matrix protein M, the glycoprotein G, and the RNA-dependent RNA polymerase L. N, P, and L are associated with the RNA molecule and compose the transcriptionally active nucleocapsid. The nucleocapsid is enveloped by a lipid bilayer derived from host cell plasma membrane during the budding process. The matrix protein M and the glycoprotein G are membrane-associated proteins. M protein is located beneath the viral membrane and bridges the nucleocapsid and the lipid bilayer. G protein is an integral transmembrane protein that is involved in viral entry.
The structure of VSV is arguably the best characterized among all of the viruses belonging to the Mononegavirales
order. The structures of M (16, 21), N (23), G (43, 44), and fragments of P, either alone (12, 41) or in complex with N (22), are known at an atomic resolution. Furthermore, the organi-zation of the compacted nucleocapsid and its mode of inter-action with M in the bullet-shaped virion have been deter-mined by cryoelectron microscopy (19). Nevertheless, the
mechanisms of viral assembly in the infected cell and budding at the plasma membrane have not been elucidated.
It is known that rhabdovirus M proteins play a major role in assembly and budding processes (37). It has been demon-strated that VSV M interacts with both artificial and cellular membranes (1, 8, 33, 49) and that it binds to the viral nucleo-capsid (13, 34). It also self-associates into large multimers at physiological salt concentrations (17, 18, 36). Since these in-teractions are supposed to be important features for both viral assembly and initiation of the budding process, the domains of M involved in the formation of all of these complexes have been identified and characterized (10, 16, 21). Furthermore, the flexible amino-terminal part of VSV M contains two mo-tifs,24PPPY27and37PSAP40, that constitute potential late
do-mains. Late domains are found in the proteins of many envel-oped viruses and have the ability to recruit cellular partners that are involved in the ultimate step of the budding process (i.e., fission between viral and cellular membranes). Mutations in these motifs lead to phenotypes in which budding virions, although assembled normally, fail to be released and accumu-late as tethered particles on cellular membranes (11, 20, 29). P(S/T)AP motifs recruit the protein TSG101 (14), a compo-nent of ESCRT complexes that play a key role in the biogen-esis of multivesicular bodies (MVB). PPxY motifs have been shown to interact with WW domains of Nedd4-related E3 ubiquitin ligases (25, 52) that are supposed to be involved in cargo recruitment during MVB formation. Mutations in the PPPY but not in the PSAP motif of VSV M affect the efficiency of viral budding in BHK-21 cells (28, 29).
* Corresponding author. Mailing address: Centre de Recherche de Gif, Laboratoire de Virologie Mole´culaire et Structurale, CNRS (UPR 3296), IFR115, Alle´e de la Terrasse, 91198, Gif sur Yvette, France. Phone: 33 1 69 82 38 36. Fax: 33 1 69 82 43 08. E-mail: gaudin@vms .cnrs-gif.fr.
䌤Published ahead of print on 13 October 2010.
12609
on November 8, 2019 by guest
http://jvi.asm.org/
VSV M is not the only protein involved in viral budding. It has been demonstrated that VSV G has a budding activity (45) and that its extracellular membrane proximal stem contains a domain that is required for efficient VSV budding (42). Fur-thermore, G protein has the inherent property of partitioning into membrane microdomains (3) that have been suggested to constitute the assembly and budding sites. Remarkably, mem-brane-associated nucleocapsids colocalize with G-protein-en-riched microdomains. In contrast, M protein does not colocal-ize with these areas of nucleocapsid accumulation and seems to accumulate in other microdomains, smaller in size, located outside the virus budding sites (51). This raises many questions on how the different components of the virions are recruited to sites of virus assembly and specifically incorporated into the budding viral particle. Here again, it is likely that some viral proteins hijack or interfere with specific cellular trafficking machinery.
In order to better understand the process of VSV assembly and budding, it is necessary to identify the specific cellular factors that are responsible for the trafficking of the different components of the virions within the cell and their accumula-tion at the budding sites. Here, we report that VSV M protein interacts directly with dynamin and perturbs the clathrin-me-diated endocytosis. Disruption of this interaction affects the cellular localization of both G and N proteins. As a conse-quence, VSV assembly and budding is severely impaired. The present study suggests that hijacking the endocytic pathway might be an important feature for enveloped viruses assembly and budding at the plasma membrane.
MATERIALS AND METHODS
Plasmid constructs.Expression constructs for dynamin 2 were derived from full-length cDNA kindly provided by Alice Dautry-Varsat (Institut Pasteur,
Paris, France). cDNA encodingChandipura virus(CV) andSpring viremia carp
virus(SVCV) matrix protein were kindly provided by James E. Dahlberg (Uni-versity of Wisconsin). Full-length dynamin 2 with an amino-terminal c-Myc tag was generated by PCR cloning into pCDNA3.1 (Invitrogen) (pDyn2/c-Myc). cDNA encoding dynamin 2 and fragments were fused to the sequence encoding the GAL4 activation domain (AD) and cloned into pGAD-GE (derived from pGAD-GH [Clontech]). cDNAs encoding VSV M, CV M, the N-terminal part of SVCV M, and VSV M mutants were fused to the sequence encoding the DNA-binding domain (BD) of LexA and cloned into pLex (Clontech). The proper framing and accuracy of the sequences of all DNA constructs were confirmed by DNA sequencing.
Yeast two-hybrid assays.The M protein of VSV (Indiana, Orsay strain) was fused to the DNA-binding domain of LexA and used as a bait to screen a nerve growth factor-induced PC12 cell (rat adrenal pheochromocyta cell line) cDNA library in which each DNA was fused to the sequence encoding the GAL4 activation domain. The yeast L40 strain containing the two LexA responsive
reporter genes,HIS3andlacZ, was first transformed with the bait plasmid
pLex-M by using a lithium acetate protocol. pLex-M-expressing L40 cells se-lected and grown in Trp-deficient medium were then transformed with plasmid DNA from the PC12 cDNA library. Double transformants were grown on plates
containing medium lacking Trp and Leu (Trp⫺Leu⫺) to select for the presence
of both the bait and library plasmids and deprived of His (Trp⫺Leu⫺His⫺) to
select for protein-protein interaction. Positive clones were then assayed for
-galactosidase activity. A total of 150 of the 6.4⫻106independent
transfor-mants were isolated on the basis of their ability to activate the transcription of
bothHIS3andlacZreporter genes. These clones conferred on L40 the ability to
grow in the absence of histidine and to produce-galactosidase activity in the
presence of the LexA BD-M hybrid but not with LexA BD alone or with LexA BD-lamin.
Coimmunoprecipitation.Monolayers of HEK293T cells were grown to 90%
confluence in 60-mm-diameter dishes and transfected with 2g of plasmid
pDyn2/c-Myc using Lipofectamine 2000 as described by the manufacturer (In-vitrogen). At 20 h after transfection, cells were infected with wild-type (WT)
virus at a multiplicity of infection (MOI) of 5 PFU/cell. At 4 h after infection, the cells were harvested by scraping them into cold phosphate-buffered saline (PBS)
and lysed on ice in 200l of buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM
NaCl, 5 mM EDTA, 0.5% NP-40, and an anti-protease cocktail (2g of
leu-peptin per ml, 2g of antipain per ml, 2g of pepstatin per ml, 2g of
chymostatin per ml, and 16g of aprotinin per ml). Nuclei were eliminated from
the lysate by centrifugation at 12,000⫻gfor 2 min at 4°C. The cytoplasmic
fraction was incubated overnight at 4°C with a mouse polyclonal anti-Mvsv antibody. Immune complexes were precipitated by incubation with protein A-Sepharose for 1 h at 4°C, washed three times, and denatured in Laemmli buffer. Immunoprecipitated proteins were analyzed by Western immunoblotting with mouse monoclonal anti-dynamin antibody (BD Transduction Laboratories).
Dynasore treatment.Monolayers of BSR cells were grown to 90% confluence in six-well plates and were infected with WT virus at an MOI of 3 PFU/cell. After 1 h at 37°C, the medium was replaced with Dulbecco modified Eagle medium (DMEM), 10 mM HEPES (pH 7.4), and 0.1% bovine serum albumin (BSA)
containing or not 80M Dynasore. After 1.5 h, the same medium change was
performed. The culture supernatants were collected at 5 h postinfection (thus, some infected cells had been incubated in the presence of dynasore for 4 h, while others had been incubated for only 2.5 h). The amount of produced virus was determined by plaque assay.
Recovery of recombinant virus.pVSV-FL(⫹), pBS-N, pBS-P, and pBS-L were kindly provided by John K. Rose (Yale University) The WT or mutant M genes of the VSV Indiana serotype (Orsay strain) were inserted into the original
full-length genomic plasmid pVSV-FL(⫹) (32) using two unique sites of
pVSV-FL(⫹), XbaI in the C-terminal region of the phosphoprotein gene and MluI in
the 5⬘noncoding sequence of the glycoprotein gene, after removal of the
cor-responding VSV Indiana Mudd Summers M gene. Recombinant VSV was re-covered as described previously (48). The plasmids were transfected into the cells
with Fugene 6 (Roche Diagnostics) in the presence of 10g of AraC (Sigma)/ml.
Working stocks of recombinant virus were prepared as follows. Monolayers of BSR cells were infected at an MOI of 0.1. After 18 to 24 h, supernatants were collected and cellular debris was removed by low-speed centrifugation; titers were determined by standard plaque assay onto BSR cells, and the M gene was sequenced.
Selection of revertants.BSR cells were infected with cloned stocks of VSV-M–L4A recombinant virus (0.001 PFU/cell), and supernatants were collected 24 h later, diluted 1:100 or 1:1,000, and used to infect new BSR cells. The culture supernatants were collected 6 h postinfection, diluted, and plated onto BSR cells for plaque assay and the isolation of revertants. Well-separated clear plaques (wild-type phenotype) were picked, and small stocks of putative revertants were prepared. BSR cells were infected with the putative revertants, and the M gene was sequenced to assess the reversion.
Electron microscopy.Cells were grown to confluence in 100-mm-diameter petri dishes and infected at an MOI of 10. At 5 h postinfection, the cells were fixed for 1 h at 4°C with 1.6% glutaraldehyde in 100 mM phosphate buffer (pH 7.2). During fixation the cells were scraped and centrifuged. They were then postfixed with 2% aqueous osmium tetroxide, dehydrated in increasing concen-trations of ethanol, and embedded in Epon. Thin sections were stained with uranyl acetate (Prolabo) with counterstaining with lead citrate (Prolabo) (40).
Immunofluorescence assay.Cells were fixed with 4% paraformaldehyde in 1⫻ PBS for 15 min and then permeabilized in 0.1% Triton X-100 in PBS for 5 min. Matrix protein was detected by using a mouse polyclonal anti-M antibody. Mouse monoclonal 24A1 antibody (kindly provided by Robin Weiss) was used to detect nucleoprotein. A rabbit polyclonal anti-G serum was used to detect G. Anti-mouse coupled to Alexa 488 and anti-rabbit coupled to Alexa 568 (Invitrogen) were used as secondary antibodies. Images were acquired on a Leica SPII system
equipped with a 63⫻lens (oil) connected to a charge-coupled device camera and
a computer equipped with Leica SP software. Excitation was performed at 488 nm (Alexa 488) and 545 nm (Alexa 568). Images were processed using Leica LCS Lite and ImageJ software.
Tfn endocytosis.Cells grown on glass coverslips were infected at an MOI of 0.4 or 3. After 3.5 h at 37°C, the cells were starved for transferrin (Tfn) by incubation in depletion medium (DMEM; 10 mM HEPES [pH 7.4], 0.1% BSA) for 1 h. They were then incubated in 1 ml of prewarmed depletion medium containing 50
g of Alexa 488-conjugated Tfn (Molecular Probes)/ml for 25 min. at 37°C. At
the end of the incubation period, the cells were cooled by shifting to an ice/water bath, washed three times with ice-cold PBS, and fixed on ice for 15 min with 4% paraformaldehyde in PBS. When the cells had been infected at an MOI of 0.4, an indirect immunofluorescence staining of VSV nucleoprotein was performed in order to visualize the cells that had been effectively infected. Primary and secondary antibodies were diluted in PBS containing 0.1% saponin. Nuclei were
stained with DAPI (4⬘,6⬘-diamidino-2-phenylindole). Confocal laser microscopy
on November 8, 2019 by guest
http://jvi.asm.org/
was performed as described above. Excitation was performed at 351 nm (DAPI) and 488 nm (Alexa 488).
RESULTS
Identification of dynamin 1 and 2 as partners of VSV Indi-ana M protein.The yeast two-hybrid system was used to search for proteins interacting with VSV (Indiana, Orsay strain) matrix protein. A nerve growth factor-induced PC12 cell (rat adrenal pheochromocytoma cell line) cDNA library was screened in a two-hybrid assay with the full-length VSV M protein as bait. From 6.4⫻106yeast transformants screened,
150 positive clones were isolated, 119 of which were se-quenced. On the basis of sequence similarity, 18 of the cDNAs encode overlapping portions of the rat dynamin 1, and another one encodes truncated dynamin 2.
Dynamins 1 and 2 are large GTPases that are essential components of vesicle formation in receptor-mediated endo-cytosis and caveola internalization (for a review, see reference 26). It has also been shown that dynamin 2 is involved in the regulation of actin assembly and filament organization (38, 47). In mammals, dynamin 2 is ubiquitously expressed and has four splice variants, and dynamin 1 is found specifically in neuronal tissue and has eight splice variants (5). The complete protein is about 870 amino acids (aa) long (the precise number of amino acids depends on the splice variant) and is composed of 5 domains: an N-terminal GTP hydrolysis domain (residues 1 to 299), a middle domain (residues 300 to 520), a pleckstrin ho-mology (PH) domain (residues 521 to 622), a GTPase effector domain (GED) (residues 623 to 745), and a C-terminal pro-line-rich domain (PRD) (26).
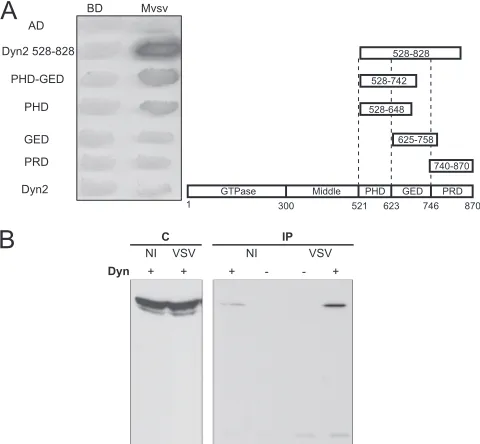
The interaction of M with one of the positive clone encoding portion of dynamin 2 (residues 528 to 828) was confirmed (Fig. 1A). We also investigated the interaction between full-length dynamin (1 and 2) and VSV M protein. However, such an interaction was not detected using the two-hybrid system (Fig. 1A, not shown for dynamin 1).
To demonstrate that M protein indeed associates with full-length dynaminin vivoindependently of the yeast genetic as-say, HEK293T cells were transfected by a plasmid expressing a Myc-tagged version of dynamin 2 (pDyn2/c-Myc) for 20 h before infection by VSV at an MOI of 5. At 4 h after infection, the cells were lysed, and M was immunoprecipitated from cell extracts by using a polyclonal anti-M antibody. The proteins present in the immune complexes were then detected on a Western blot with a mouse monoclonal antibody directed against dynamin. Dynamin was detected specifically in immu-noprecipitates from infected cells.
A late stage of VSV cycle is sensitive to dynasore.A potential role for dynamin during the viral cycle was then investigated. First, we transfected cells by a plasmid encoding the K44A dominant-negative mutant of dynamin 2 (6) before viral infec-tion. However, the cells that were expressing the dominant-negative mutant could not be infected (data not shown), most probably because VSV enters the cell via the clathrin-mediated endocytic pathway that was then inhibited (9, 30, 50).
To assess a role for dynamin at a postentry stage of viral infection, dynasore, an inhibitor of the GTPase activity of dynamin (35), was added on VSV-infected cells (at an MOI of 3) 1 h or 2.5 h postinfection. At 5 h postinfection, the culture
supernatants were collected, diluted, and plated onto BSR cells for plaque assay. The treatment reduced the number of infectious particles by about 2 orders of magnitude when dy-nasore was added at 1 h postinfection and by more than a factor of 10 when dynasore was added at 2.5 h postinfection (Fig. 2). This suggested that dynasore was able to inhibit a late stage of the viral cycle and that dynamin was thus playing a key role at this stage.
M binds the dynamin PH domain.All of the positive clones encoded only the carboxy-terminal part of the dynamin 1 cor-responding to the PH, GED, and PRD domains. The fragment of dynamin 2 contained in the single clone encoding a portion of this protein extended from aa 528 to 828. Thus, in this fragment, the PRD domain was not complete. To determine more precisely the region of the dynamin sufficient for binding to M protein, different dynamin 2 carboxy-terminal fragments were fused to the activation domain in Gal4-AD and were tested for their interactions with a LexA-M fusion (Fig. 1A).
[image:3.585.301.541.70.292.2]Neither the PRD (aa 740 to 870) nor the GED (aa 625 to 758) interacted with M, whereas a significant interaction was still observed with fragments containing either both the PH and GED domains (aa 528 to 742) or the PH domain alone (aa
FIG. 1. VSV M protein binds dynamin. (A) Mapping studies of M protein binding domain within dynamin 2. The interactions between M protein fused to the DNA-binding domain of LexA and different frag-ments of dynamin fused to Gal4 activation domain (AD) were assessed by the appearance of blue colonies in the presence of X-Gal (5-bromo-4-chloro-3-indolyl--D-galactopyranoside). The fragment of dynamin 2 extending from aa 528 to 828 corresponds to the clone isolated during the screen. (B) Detection of M-dynamin complex in cells. (A) HEK293T cells were transfected (⫹) or not (⫺) with plasmid pDyn2/c-Myc. At 24 h posttransfection, cells were mock infected (NI) or infected (VSV) by wild-type VSV at an MOI of 5 PFU/cell. At 4 h after infection, cells were harvested and lysed. Cell lysate was incu-bated with a mouse polyclonal anti-Mvsv antibody. Immune complexes were precipitated by incubation with protein A-Sepharose. Immuno-precipitated proteins (IP) were analyzed by Western immunoblotting with mouse monoclonal anti-dynamin antibody. In the left portion of the blot, direct cellular extracts (C) of transfected cells (either mock infected [NI] or infected by VSV) were also analyzed.
on November 8, 2019 by guest
http://jvi.asm.org/
528 to 648). This indicated that the binding domain was located in the PH domain of dynamin 2 (Fig. 1A).
A single mutation in the amino-terminal part of M abolishes interaction between VSV M and dynamin in the yeast two-hybrid system.VSV matrix protein is composed of two do-mains, a globular carboxy-terminal domain and a largely un-structured amino-terminal domain (comprising ca. the 57 first residues) (15, 16). In order to identify the domain involved in dynamin binding, both domains were fused to LexA and tested for their ability to bind the dynamin fragments previously found in our positive clones. These experiments indicated un-ambiguously that the domain of interaction was located in the amino-terminal part of M. Further deletion mutant analysis indicated that the presence of the first 10 aa was necessary for dynamin binding (Fig. 3A). Alanine scanning performed on residues 4 to 8 revealed that mutations of leucines 4 and 8 into alanines abolished the interaction between VSV M and dy-namins 1 and 2 (Fig. 3B, data not shown for dynamin 1).
Interactions between dynamin and other vesiculovirus M pro-teins in the yeast two-hybrid system.We investigated whether the detected interaction was conserved among other members of the vesiculovirus genus. The interaction of the isolated clones of dynamin 1 and 2 with the full-length M protein of CV and with the amino-terminal part of M protein of SVCV was investigated with the yeast two-hybrid system. Despite a low identity of sequence (ca. 25%) with VSV M protein, both CV M and the amino-terminal part of SVCV M displayed inter-actions with dynamins 1 and 2 (Fig. 3C, data not shown for dynamin 1). Moreover, when the leucine in position 4 was
replaced by an alanine in CV M, the resulting protein failed to interact with dynamin (Fig. 3C).
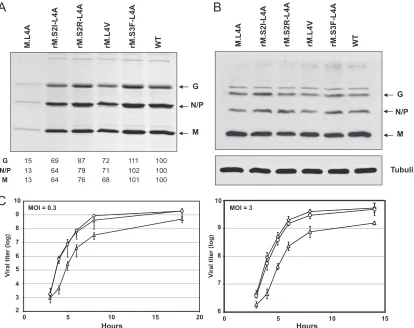
[image:4.585.316.523.63.398.2]Recovery and characterization of recombinant VSV contain-ing M.L4A.To assess the role of M-dynamin interaction during the viral cycle, a recombinant virus harboring the M.L4A mu-tant protein was generated by using a VSV reverse genetics system. The mutant was easily recovered and could be pas-saged in cell culture. To determine the effects of the mutation on virus replication and budding, cells were infected with ei-ther the WT or mutant M.L4A at an MOI of 3. The virus released in the culture medium after 6 h was purified and
FIG. 2. Dynasore inhibits VSV viral cycle at a late stage. Monolay-ers of BSR cells were infected with WT VSV at an MOI of 3 PFU/cell. After 1 h or 2.5 h of infection, the medium was changed for DMEM containing 80M dynasore. The culture supernatants were collected at 5 h postinfection. Viral production was determined by a plaque assay. The results are the average of three independent experiments.
FIG. 3. Mapping studies of the dynamin 2 binding domain within M protein and the interaction of dynamin 2 with the M proteins of other vesiculoviruses. (A) The interactions between deletion mutants of M protein fused to the DNA-binding domain of LexA and the fragment (aa 528 to 828) of dynamin 2 fused to Gal4 AD were assayed as in Fig. 1A. (B) Refinement of the analysis of dynamin 2 binding domain within M protein by alanine scanning. The interactions be-tween mutant M proteins (in which amino acids 4 to 8 have been replaced by an alanine) fused to the DNA-binding domain of LexA and the fragment (aa 528 to 828) of dynamin 2 fused to Gal4 AD were assayed as in Fig. 1A. (C) Conservation of the interaction between M and dynamin 2 among the vesiculovirus genus. Interactions between CV and SVCV M proteins and the C-terminal part of dynamin 2 (residues 528 to 828) were assayed as in Fig. 1A. Since the full-length M protein of SVCV fused to the DNA-binding domain of LexA acti-vated the expression of -galactosidase when expressed alone, we could only assess the interaction between the amino-terminal part of SVCV M protein (residues 1 to 34) and dynamin. The interaction of mutant L4A of CV M protein with dynamin was also analyzed.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.585.60.267.65.326.2]analyzed by SDS-PAGE (Fig. 4A). In parallel, the amount of viral proteins present in infected cell extracts was quantified by Western blot analysis (Fig. 4B). Similar amounts of M, N, and G proteins (L and P were not detected by the anti-VSV serum) were found in cells infected either by WT VSV or by M.L4A virus. In contrast, the amount of mutant virus released in the supernatant was strongly reduced to ca. 13% of that of WT virus. These data showed that viral protein synthesis was not affected by the mutation that rather specifically blocked a late step of the viral cycle (most probably assembly or budding). Nevertheless, the three structural proteins of VSV (M, N, and G) were present in the mutant virions in proportions similar to that found in WT virus. Consistently, when the morphology of purified mutant and WT viruses was analyzed by negative-staining electron microscopy, no difference in the typical bul-let-shape morphology of the virions was observed (data not shown).
We then performed either one-step (MOI of 3) or multiple-step (MOI of 0.3) growth curves for the mutant, as well as the WT virus. Whatever the MOI, a delay in virus release was observed at early time points with the mutant compared to the
WT virus (Fig. 4C). Either at an MOI of 0.3 after 8 h of infection or at an MOI of 3 after 6 h of infection, the titer of the mutant was still 1 order of magnitude below that of WT virus.
Taken together, all of these results indicate that the muta-tion essentially decreased the number of viral particles re-leased without affecting their morphology and the ratio of infectious virions per physical particle.
[image:5.585.84.498.68.396.2]Selection and characterization of revertant viruses. The morphology of the plaques of mutant M.L4A after staining was different from that of the WT. They appeared fuzzy, as if infection-induced cell lysis was incomplete (Fig. 5A). This plaque phenotype allowed us to identify revertants among our mutant population. These revertants were amplified, and the M gene was sequenced. In each case, a single nucleotide sub-stitution was found in the gene (Fig. 5B). Four different rever-tants were obtained. For two of them, the mutation resulted in the replacement of the serine in position 2 by an isoleucine (rM.S2I-L4A) or an arginine (rM.S2R-L4A). For another one, the mutation resulted in the replacement of the serine in po-sition 3 by a phenylalanine (rM.S3F-L4A), and for the last one
FIG. 4. Characterization of the production of recombinant virus M.L4A and its revertants. (A) Protein profiles for WT VSV, M.L4A recombinant virus, and its revertants rM.S2I-L4A, rM.S2R-L4A, rM.S3F-L4A, and rM.L4V. Virions were harvested from supernatant of infected BSR cells at 6 h postinfection. Virion proteins were analyzed by SDS-PAGE and Coomassie blue staining. The percent yield of each protein in this experiment was calculated using ImageJ software on a scan of the gel. (B) Lysates of the infected BSR cells were analyzed by Western blotting with an anti-VSV polyclonal serum and a monoclonal antibody directed against tubulin. (C) Growth kinetics of WT VSV (〫), M.L4A recombinant virus (‚), and its revertant rM.S3F-L4A (E). BSR cells were infected at an MOI of 0.3 (left side graph) or 3 (right side graph), and samples were harvested for titration at the indicated times postinfection. Virus titers represent averages from at least three independent experiments. Theyscale is not the same on both graphs.
on November 8, 2019 by guest
http://jvi.asm.org/
by the replacement of the alanine in position 4 of the mutant by a valine (rM.L4V). Leucine in position 4 was never recov-ered: such a revertant would have necessitated two nucleotide substitutions.
The amount of revertant viruses released in extracellular medium was similar to that of WT (Fig. 4A). The growth curves were also very similar to that of WT (Fig. 4C, shown for rM.S3F-L4A).
Finally, we analyzed the ability of the M proteins of the revertant viruses to bind dynamin 1 and 2 in the two-hybrid system (Fig. 5C). All of the proteins had recovered a significant level of interaction compared to M.L4A. These experiments definitively proved that the mutant phenotype was due to the loss of interaction between M and dynamin and that dynamin plays a role in the ultimate steps of the viral cycle.
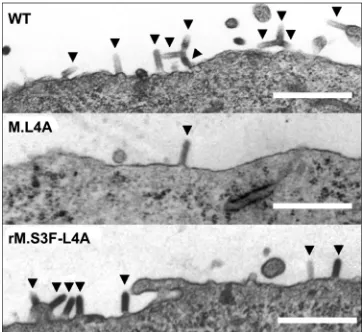
Electron microscopy analysis of viral assembly and bud-ding. To determine at which step of the assembly/budding process the viral cycle was blocked, thin sections of infected cells were examined by electron microscopy (Fig. 6). For both WT and rM.S3F-L4A, electron micrographs showed a large number of bullet-shaped virions assembled at the cell surface, apparently in the process of budding, as well as viral particles that have been released in the extracellular medium. In striking contrast, for M.L4A, very few viral particles were observed either in the process of budding or in the extracellular medium (Table 1). This result indicates that the defect in the assembly/ budding process of M.L4A occurs upstream of the one
ob-served with mutants affected in the PPPY motif for which bullet-shaped viruses in the process of budding accumulated at the viral membrane (29).
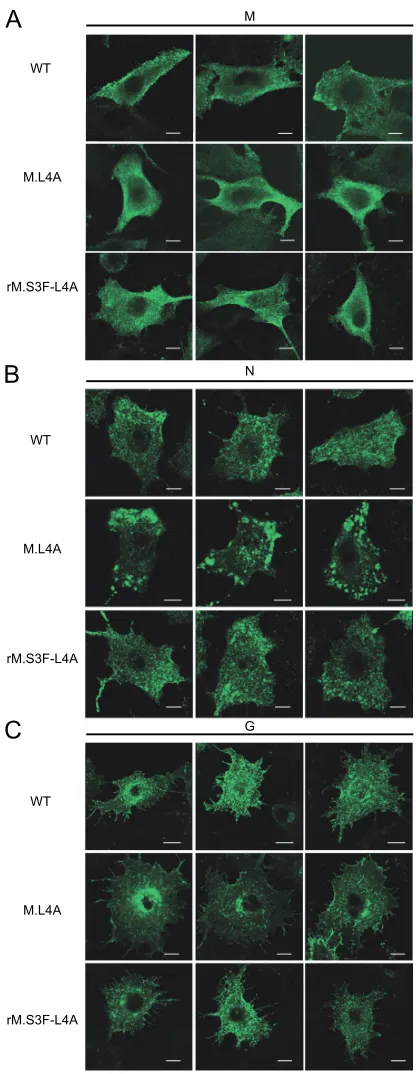
Different localization of G and N proteins in cells infected by WT VSV or mutant M.L4A.Using immunofluorescence exper-iments, we compared the cellular distribution of viral proteins M, N, and G at 4 h postinfection by wild-type or mutant viruses. It appeared that the intracellular localization of M was the same in cells infected by both wild-type virus and M.L4A mutant (Fig. 7A).
In contrast to the N protein of wild-type VSV, after 4 h of infection, the N protein of the M.L4A mutant accumulated in large aggregates (Fig. 7B) located beneath the plasma mem-brane. It has to be noted that after longer times of infection (6 h), such aggregates were also observed with WT VSV (data not shown).
The localization of the mutant G glycoprotein was also af-fected (Fig. 7C). Whereas with the WT virus, immunofluores-cence experiments indicated that G was in vesicles that were distributed throughout the cytoplasm, the G protein of the M.L4A mutant appeared to be confined to the perinuclear vesicles.
[image:6.585.71.252.64.325.2]The localization of both N and G proteins of revertant rM.S3F-L4A was the same as that of the wild type (Fig. 7B and C). This indicated that the mutant phenotypes observed were indeed due to the lack of interaction between M and dynamin.
[image:6.585.330.511.69.235.2]FIG. 5. Characterization of revertant phenotypes. (A) Comparison of the morphology of the plaques of WT VSV, M.L4A recombinant virus, and revertant rM.S3F-L4A. (B) Nucleotide sequence of the 5⬘ end of the open reading frame of M mRNA for wild-type, mutant, and revertant viruses. (C) Characterization of the binding between M pro-teins (WT, L4A, S2I-L4A, S2R-L4A, S3F-L4A, and L4V) and the C-terminal fragments of dynamins 1 (aa 523 to 850) and 2 (aa 528 to 828) using the yeast two-hybrid system.
FIG. 6. Electron micrographs of BSR cells infected by WT VSV, M.L4A recombinant virus, and rM.S3F-L4A revertant at 5 h postin-fection. Size bars, 500 nm. Virions in the process of budding at the plasma membrane of cells infected by the different viruses are indi-cated by arrowheads.
TABLE 1. Number of budding virions per unit length of plasma membranea
Strain Membrane
concn (M)
Total no. of budding
virions
No. of budding
virions/m of
membrane
WT 60 178 2.97
M.L4A 65.9 13 0.20
rMS3F-L4A 68 101 1.49
a
Bullet-shaped budding virions were counted on electron micrographs (such as those in Fig. 6).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.301.542.643.707.2]WT VSV M protein affects dynamin-dependent cell endocy-tosis. We next sought to determine whether the interaction between the M protein and dynamin has an effect on dynamin-dependent cellular endocytosis. We thus examined Tfn endo-cytosis in both WT VSV- and mutant M.L4A-infected cells.
First, cells were infected at an MOI of 0.4 for 3.5 h and then incubated with fluorescent Tfn for 25 min at 37°C and fixed. An indirect immunofluorescence staining of VSV nucleocapsid was performed to identify infected cells (Fig. 8A).
In a second experiment, cells were infected either by WT or by M.L4A virus at an MOI of 3 (so that⬎95% of the cells were infected) before incubation with fluorescent Tfn. The endocy-tosis of Tfn was compared to that in uninfected cells both by fluorescence-activated cell sorting (FACS) and confocal mi-croscopy (Fig. 8B).
The FACS data indicated that the intensity of Tfn fluores-cence associated with the cells was similar in uninfected cells and in cells infected either by WT or by M.L4A virus (data not shown). Nevertheless, the pattern of endocytosis was different (Fig. 8).
In uninfected cells, clear uptake of labeled Tfn into vesicular elements accumulating on a single side of the cell nucleus was observed (Fig. 8). In striking contrast, in cells infected by WT VSV, labeled Tfn did not accumulate near the cell nucleus and, in many cells, seemed to remain in vesicles located just beneath the plasma membrane (Fig. 8B, arrow). In cells infected by M.L4A virus, an intermediate phenotype was observed. Taken together, these data indicated that wild-type M protein af-fected Tfn endocytosis through its interaction with dynamin.
DISCUSSION
In this study, using the yeast two-hybrid system, we have identified both dynamin 1 and 2 as partners of VSV matrix protein. The binding, which is conserved all along the vesicu-lovirus genus, involves the extreme N terminus of M and the PH domain of dynamin. Using a genetic approach, we have demonstrated that M-dynamin interaction is required for effi-cient viral production. Indeed, mutant virus M.L4A for which M protein did not interact with dynamin was affected in viral particle production. It was thus genetically unstable and rever-tant viruses, producing similar amounts of viral particles as WT VSV arose quickly. The matrix proteins of all of the revertants that were obtained recovered a significant level of interaction with dynamin.
The interaction domain on M is located in the flexible amino-terminal part of the molecule (residues 1 to 57) (15, 16). Our genetic approach allows the identification of amino acids in position 2, 3, 4, and 8 as key residues for the interaction with dynamin. The amino-terminal part of the protein contains also the24PPPY27and37PSAP40motifs. The PPPY motif has been
demonstrated to have a significant late domain activity in BHK-21 cells (29), most probably by allowing M binding to ubiquitin ligase Nedd-4 (24). The 37PSAP40motif has been
[image:7.585.58.266.90.627.2]shown to bind TSG101, a component of the ESCRT complex, but the role of this interaction remains elusive (27, 28). The amino-terminal part is also involved in the inhibition of the nucleocytoplasmic transport by targeting the nucleoporin Nup98 (54). The amino-terminal domain of M is thus the major interacting domain of the molecule, as often appears to
FIG. 7. Localization of M, N, and G proteins in cells infected by WT VSV, mutant M.L4A, or revertant rM.S3F-L4A. BSR cells were infected by WT VSV, mutant M.L4A, or revertant rM.S3F-L4A. At 4 h postinfection, the cells were analyzed by immunofluorescence with specific antibodies to detect M protein (A), N protein (B), and G protein (C); the staining patterns of M, N, and G are shown for different cells. Note the accumulation of N near the plasma membrane for mutant M.L4A. Size bars, 10m.
on November 8, 2019 by guest
http://jvi.asm.org/
be the case with natively unfolded (or poorly folded) segments of viral proteins (2).
Most of the cellular partners of dynamin that have been identified interact with the carboxy-terminal PR domain of the molecule (often through a SH3 domain) (31). Thus, binding between M and the PH domain of dynamin defines a new mode of interaction with this protein. It should be noted that in the proteolipid tubes formed by dynamin (either in the constricted or in the nonconstricted state), the PH domain seems to be poorly accessible to a protein ligand (7, 56). This raises questions about the dynamin form that is targeted by M and how this interaction interferes with dynamin functions.
The viral cycle of mutant virus M.L4A is blocked at a late stage. This blockage most probably results from the confine-ment of G in perinuclear vesicles, rendering it unable to form microdomains in the plasma membrane (3, 51). As a conse-quence, G does not sustain budding at the plasma membrane, leading to an accumulation of nucleocapsids at the periph-ery of the cell. All this appears to be due to the poor ability of M.L4A virus to bind dynamin and to affect cellular en-docytosis.
In WT VSV-infected cells, the pattern of Tfn-containing endocytic vesicles was very different from the one observed in noninfected cells. Tfn did not accumulate near the cell nucleus and remained in vesicles located just beneath the plasma mem-brane. Whether these vesicles are still attached to the plasma membrane is an open question. A plausible explanation would be that M, upon dynamin binding, inhibits the constriction of
the neck of the endocytic vesicle and thus its pinching off from the parent membrane (26). Nevertheless, in our electron mi-croscopy analyses we did not observe the accumulation of vesicles tethered to the plasma membrane by a dynamin collar, suggesting that M-dynamin interaction affects endocytic vesicle trafficking at another stage.
[image:8.585.111.471.64.338.2]Dynamin 2 has been demonstrated to be a partner of HIV-1 Nef that is required for the Nef-dependent viral infectivity enhancement (39). Endophilin 2, another component of the machinery involved in clathrin-mediated endocytosis, interacts with Moloney murine leukemia virus (Mo-MuLV) Gag. Inter-estingly, in this case, overexpression of endophilin 2 resulted in a decrease of Mo-MuLV virion production (55), suggesting that the role of the interaction might be to inhibit a function of endophilin. More recently, the recycling endosomes and the Rab11 pathway have been suggested to be involved in the assembly and the budding of viruses such as respiratory syncy-tial virus (53), hantavirus (46), and influenza virus (4), which use an ESCRT-independent budding mechanism. There are thus a growing number of studies describing interactions be-tween some viral proteins and the endocytic machinery of the cell that are required for correct assembly, budding or egress of viral particles. Such interactions may prevent the internaliza-tion of neosynthesized virions or favor the accumulainternaliza-tion of transmembrane glycoproteins at the budding sites. It is there-fore probable that perturbations of the endocytic pathway are a general feature in cells infected by enveloped viruses budding at the plasma membrane.
FIG. 8. WT VSV M protein affects clathrin-dependent endocytosis. (A and B) Tfn endocytosis in cells infected by WT or M.L4A viruses. Cells were infected at an MOI of 0.4 (A) or 3 (B). At 3.5 h postinfection, Alexa 488-conjugated Tfn (green) was added to the medium for 25 min. Cells were washed and fixed. In panel A, after fixation, infected cells were labeled with monoclonal anti-N antibody (red) and, for WT virus, uninfected cells are indicated by arrowheads. In panel B, noninfected cells (NI) are shown as a positive control of endocytosis. Arrowheads indicate cells infected by WT virus with fluorescent Tfn-containing vesicles accumulated just beneath the plasma membrane. Size bars, 32m.
on November 8, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
We thank Marcel Knossow and Anne Schmidt for helpful discus-sions at different stages of the project, John K. Rose for the gift of the VSV reverse genetic system, Agathe Subtil and Alice Dautry-Varsat for the gift of dynamin 2 cDNA, James E. Dahlberg for the gift of the vesiculovirus matrix protein cDNA, and David Pasdeloup for initial help with confocal microscopy.
Confocal microscopy was performed on the Plate-Forme Imagerie et Biologie Cellulaire (IMAGIF) of the CNRS campus, supported by the ASTRE program of the Conseil Ge´ne´ral de l’Essonne. We ac-knowledge support from the CNRS and INRA. Additional support was provided by the Program de Recherche Fondamentale en Micro-biologie et Maladies Infectieuses Parasitaires (PRFMMIP) of the Min-iste`re de l’Education Nationale, de la Recherche, et de la Technologie.
REFERENCES
1.Bergmann, J. E., and P. J. Fusco.1988. The M protein of vesicular stomatitis virus associates specifically with the basolateral membranes of polarized
epithelial cells independently of the G protein. J. Cell Biol.107:1707–1715.
2.Bourhis, J. M., B. Canard, and S. Longhi.2006. Structural disorder within the replicative complex of measles virus: functional implications. Virology
344:94–110.
3.Brown, E. L., and D. S. Lyles.2003. Organization of the vesicular stomatitis virus glycoprotein into membrane microdomains occurs independently of
intracellular viral components. J. Virol.77:3985–3992.
4.Bruce, E. A., P. Digard, and A. D. Stuart.2010. The Rab11 pathway is required for influenza A virus budding and filament formation. J. Virol.
84:5848–5859.
5.Cao, H., F. Garcia, and M. A. McNiven.1998. Differential distribution of
dynamin isoforms in mammalian cells. Mol. Biol. Cell9:2595–2609.
6.Cao, H., H. M. Thompson, E. W. Krueger, and M. A. McNiven.2000. Disruption of Golgi structure and function in mammalian cells expressing a
mutant dynamin. J. Cell Sci.113(Pt. 11):1993–2002.
7.Chen, Y. J., P. Zhang, E. H. Egelman, and J. E. Hinshaw.2004. The stalk region of dynamin drives the constriction of dynamin tubes. Nat. Struct. Mol.
Biol.11:574–575.
8.Chong, L. D., and J. K. Rose.1993. Membrane association of functional
vesicular stomatitis virus matrix protein in vivo. J. Virol.67:407–414.
9.Cureton, D. K., R. H. Massol, S. Saffarian, T. L. Kirchhausen, and S. P. Whelan.2009. Vesicular stomatitis virus enters cells through vesicles incom-pletely coated with clathrin that depend upon actin for internalization. PLoS
Pathog.5:e1000394.
10.Dancho, B., M. O. McKenzie, J. H. Connor, and D. S. Lyles.2009. Vesicular stomatitis virus matrix protein mutations that affect association with host
membranes and viral nucleocapsids. J. Biol. Chem.284:4500–4509.
11.Demirov, D. G., J. M. Orenstein, and E. O. Freed.2002. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell
type-dependent manner. J. Virol.76:105–117.
12.Ding, H., T. J. Green, S. Lu, and M. Luo.2006. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus.
J. Virol.80:2808–2814.
13.Flood, E. A., and D. S. Lyles.1999. Assembly of nucleocapsids with cytosolic and membrane-derived matrix proteins of vesicular stomatitis virus. Virology
261:295–308.
14.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist.2001. Tsg101 and the vacuolar protein sorting
pathway are essential for HIV-1 budding. Cell107:55–65.
15.Gaudier, M., Y. Gaudin, and M. Knossow.2001. Cleavage of vesicular stomatitis virus matrix protein prevents self-association and leads to
crystal-lization. Virology288:308–314.
16.Gaudier, M., Y. Gaudin, and M. Knossow.2002. Crystal structure of
vesic-ular stomatitis virus matrix protein. EMBO J.21:2886–2892.
17.Gaudin, Y., A. Barge, C. Ebel, and R. W. Ruigrok.1995. Aggregation of VSV M protein is reversible and mediated by nucleation sites: implications for
viral assembly. Virology206:28–37.
18.Gaudin, Y., J. Sturgis, M. Doumith, A. Barge, B. Robert, and R. W. Ruigrok.
1997. Conformational flexibility and polymerization of vesicular stomatitis
virus matrix protein. J. Mol. Biol.274:816–825.
19.Ge, P., J. Tsao, S. Schein, T. J. Green, M. Luo, and Z. H. Zhou.2010.
Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science327:
689–693.
20.Gottlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine.1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency
virus particle release. Proc. Natl. Acad. Sci. U. S. A.88:3195–3199.
21.Graham, S. C., R. Assenberg, O. Delmas, A. Verma, A. Gholami, C. Talbi, R. J. Owens, D. I. Stuart, J. M. Grimes, and H. Bourhy.2008. Rhabdovirus matrix protein structures reveal a novel mode of self-association. PLoS
Pathog.4:e1000251.
22.Green, T. J., and M. Luo.2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small
polymerase cofactor, P. Proc. Natl. Acad. Sci. U. S. A.106:11713–11718.
23.Green, T. J., X. Zhang, G. W. Wertz, and M. Luo.2006. Structure of the
vesicular stomatitis virus nucleoprotein-RNA complex. Science313:357–360.
24.Harty, R. N., M. E. Brown, J. P. McGettigan, G. Wang, H. R. Jayakar, J. M. Huibregtse, M. A. Whitt, and M. J. Schnell.2001. Rhabdoviruses and the
cellular ubiquitin-proteasome system: a budding interaction. J. Virol.75:
10623–10629.
25.Harty, R. N., J. Paragas, M. Sudol, and P. Palese.1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus inter-acts with WW domains of cellular proteins: implications for viral budding.
J. Virol.73:2921–2929.
26.Hinshaw, J. E.2000. Dynamin and its role in membrane fission. Annu. Rev.
Cell Dev. Biol.16:483–519.
27.Irie, T., E. Carnero, A. Okumura, A. Garcia-Sastre, and R. N. Harty.2007. Modifications of the PSAP region of the matrix protein lead to attenuation
of vesicular stomatitis virus in vitro and in vivo. J. Gen. Virol.88:2559–2567.
28.Irie, T., J. M. Licata, H. R. Jayakar, M. A. Whitt, P. Bell, and R. N. Harty.
2004. Functional analysis of late-budding domain activity associated with the
PSAP motif within the vesicular stomatitis virus M protein. J. Virol.78:7823–
7827.
29.Jayakar, H. R., K. G. Murti, and M. A. Whitt.2000. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by
inhibiting a late step in virion release. J. Virol.74:9818–9827.
30.Johannsdottir, H. K., R. Mancini, J. Kartenbeck, L. Amato, and A. Helenius.
2009. Host cell factors and functions involved in vesicular stomatitis virus
entry. J. Virol.83:440–453.
31.Kim, Y., and S. Chang.2006. Ever-expanding network of
dynamin-interact-ing proteins. Mol. Neurobiol.34:129–136.
32.Lawson, N. D., E. A. Stillman, M. A. Whitt, and J. K. Rose.1995. Recom-binant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. U. S. A.
92:4477–4481.
33.Luan, P., and M. Glaser.1994. Formation of membrane domains by the
envelope proteins of vesicular stomatitis virus. Biochemistry33:4483–4489.
34.Lyles, D. S., M. O. McKenzie, and R. R. Hantgan. 1996. Stopped-flow, classical, and dynamic light scattering analysis of matrix protein binding to
nucleocapsids of vesicular stomatitis virus. Biochemistry35:6508–6518.
35.Macia, E., M. Ehrlich, R. Massol, E. Boucrot, C. Brunner, and T. Kirch-hausen.2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell
10:839–850.
36.McCreedy, B. J., Jr., K. P. McKinnon, and D. S. Lyles.1990. Solubility of vesicular stomatitis virus M protein in the cytosol of infected cells or isolated
from virions. J. Virol.64:902–906.
37.Mebatsion, T., F. Weiland, and K. K. Conzelmann.1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J.
Vi-rol.73:242–250.
38.Ochoa, G. C., V. I. Slepnev, L. Neff, N. Ringstad, K. Takei, L. Daniell, W. Kim, H. Cao, M. McNiven, R. Baron, and P. De Camilli.2000. A functional link between dynamin and the actin cytoskeleton at podosomes. J. Cell Biol.
150:377–389.
39.Pizzato, M., A. Helander, E. Popova, A. Calistri, A. Zamborlini, G. Palu, and H. G. Gottlinger.2007. Dynamin 2 is required for the enhancement of HIV-1
infectivity by Nef. Proc. Natl. Acad. Sci. U. S. A.104:6812–6817.
40.Reynolds, E. S.1963. The use of lead citrate at high pH as an
electron-opaque stain in electron microscopy. J. Cell Biol.17:208–212.
41.Ribeiro, E. A., Jr., A. Favier, F. C. Gerard, C. Leyrat, B. Brutscher, D. Blondel, R. W. Ruigrok, M. Blackledge, and M. Jamin.2008. Solution struc-ture of the C-terminal nucleoprotein-RNA binding domain of the vesicular
stomatitis virus phosphoprotein. J. Mol. Biol.382:525–538.
42.Robison, C. S., and M. A. Whitt.2000. The membrane-proximal stem region of vesicular stomatitis virus G protein confers efficient virus assembly. J.
Vi-rol.74:2239–2246.
43.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin.2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science
313:187–191.
44.Roche, S., F. A. Rey, Y. Gaudin, and S. Bressanelli.2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science
315:843–848.
45.Rolls, M. M., P. Webster, N. H. Balba, and J. K. Rose.1994. Novel infectious particles generated by expression of the vesicular stomatitis virus
glycopro-tein from a self-replicating RNA. Cell79:497–506.
46.Rowe, R. K., J. W. Suszko, and A. Pekosz.2008. Roles for the recycling endosome, Rab8, and Rab11 in hantavirus release from epithelial cells.
Virology382:239–249.
47.Schafer, D. A., S. A. Weed, D. Binns, A. V. Karginov, J. T. Parsons, and J. A. Cooper.2002. Dynamin2 and cortactin regulate actin assembly and filament
organization. Curr. Biol.12:1852–1857.
48.Schnell, M. J., L. Buonocore, M. A. Whitt, and J. K. Rose.1996. The minimal conserved transcription stop-start signal promotes stable expression of a
foreign gene in vesicular stomatitis virus. J. Virol.70:2318–2323.
on November 8, 2019 by guest
http://jvi.asm.org/
49.Solon, J., O. Gareil, P. Bassereau, and Y. Gaudin.2005. Membrane defor-mations induced by the matrix protein of vesicular stomatitis virus in a
minimal system. J. Gen. Virol.86:3357–3363.
50.Sun, X., V. K. Yau, B. J. Briggs, and G. R. Whittaker. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into
host cells. Virology338:53–60.
51.Swinteck, B. D., and D. S. Lyles.2008. Plasma membrane microdomains containing vesicular stomatitis virus M protein are separate from
microdo-mains containing G protein and nucleocapsids. J. Virol.82:5536–5547.
52.Timmins, J., G. Schoehn, S. Ricard-Blum, S. Scianimanico, T. Vernet, R. W. Ruigrok, and W. Weissenhorn.2003. Ebola virus matrix protein VP40
inter-action with human cellular factors Tsg101 and Nedd4. J. Mol. Biol.326:493–
502.
53.Utley, T. J., N. A. Ducharme, V. Varthakavi, B. E. Shepherd, P. J. Santan-gelo, M. E. Lindquist, J. R. Goldenring, and J. E. Crowe, Jr.2008. Respi-ratory syncytial virus uses a Vps4-independent budding mechanism
con-trolled by Rab11-FIP2. Proc. Natl. Acad. Sci. U. S. A.105:10209–10214.
54.von Kobbe, C., J. M. van Deursen, J. P. Rodrigues, D. Sitterlin, A. Bachi, X. Wu, M. Wilm, M. Carmo-Fonseca, and E. Izaurralde.2000. Vesicular sto-matitis virus matrix protein inhibits host cell gene expression by targeting the
nucleoporin Nup98. Mol. Cell6:1243–1252.
55.Wang, M. Q., W. Kim, G. Gao, T. A. Torrey, H. C. Morse III, P. De Camilli, and S. P. Goff.2003. Endophilins interact with Moloney murine leukemia
virus Gag and modulate virion production. J. Biol.3:4.
56.Zhang, P., and J. E. Hinshaw.2001. Three-dimensional reconstruction of
dynamin in the constricted state. Nat. Cell Biol.3:922–926.