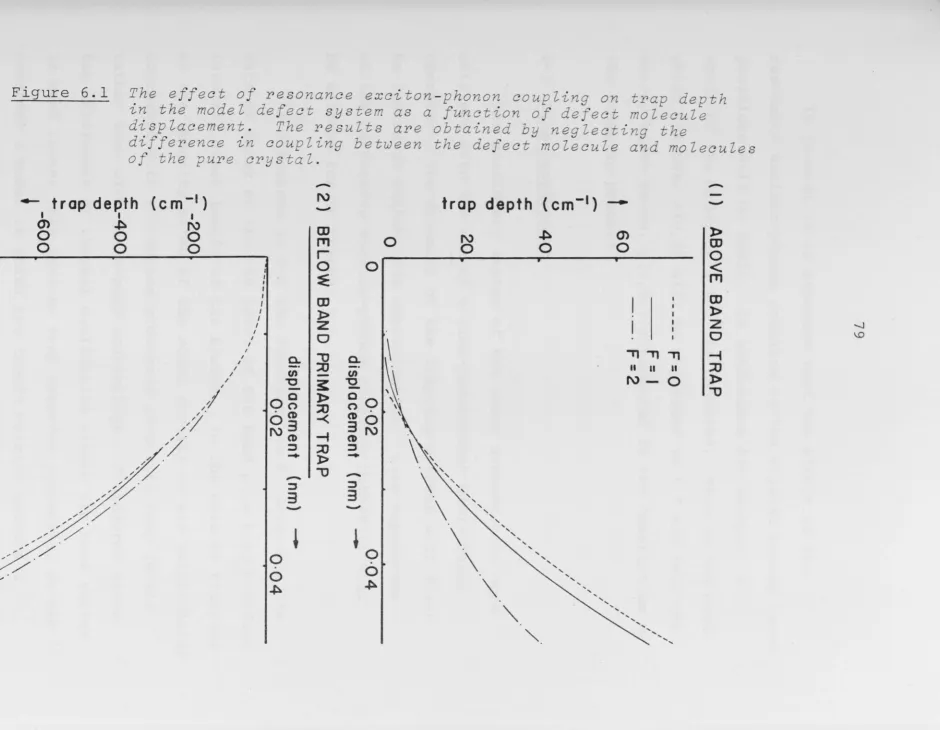
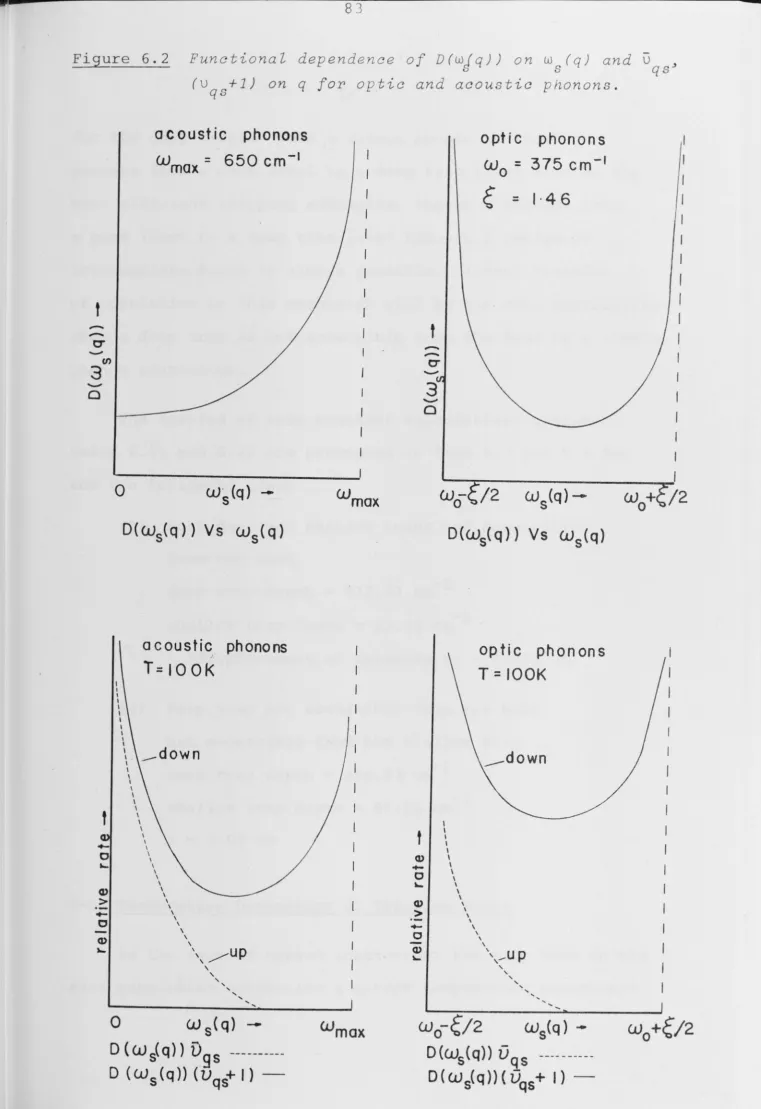
ne
dime sicnal
med
els
f
0r11
Cr'~StCI
~h0t0chemistr'~
-a thesis presented as partial requirement for the degree ofDoctor of Philosophy · at the
Australian National University by
this thesis has not been previously submitted by the author for any other degree and is to her knowledge original work, except where due reference is made in the text.
abst~act
A study of photochemical reactions is presented which is limited to the relatively simple case of photodimerisation in molecular crystals. These reactions can be classified
according to whether or not they obey the topochemical rules. Reactions which do not
1 occur at_ sites or in zones in the crystal where there are departures from perfect crystal
order. Topochemical reactions may occur in the bulk perfect lattice, or in defective zones where the local structure is similar.
A one-dimensional system consisting of a chain of anthracene molecules with cyclic boundary conditions is proposed as a model in which the energy transfer process
involved in the photodimerisation may be studied theoretically.
The non-topochemical class of reaction is investigated by displacing an arbitrary molecule from its equilibrium position in the chain to a more favourable position for
dimerisation to occur. The changed resonance and dispersive interactions in the 'crystal' result in the creation of
A Fermi Golden Rule rate expression is derived to describe the rate of energy transfer. Calculations show that the most efficient band-to-trap communication is achieved by a single phonon emission with the rate determining step
for an energy cascade process being the initial band to shallow trap transition. The time for the achievement of
thermal equilibrium between the band and trap states is
found to be less than the exciton fluorescence lifetime.
For the topochemical reactions i t is assumed that
energy localisation is effected in the bulk perfect crystal.
The mechanism of dispersive exciton-phonon coupling is
considered in this regard. The difficulty in presenting a
unified theory for the treatment of dispersive coupling over the entire range of allowed coupling strengths is discussed, and the conditions for the convergence of the existing approaches analysed. For the purposes of
photo-chemistry, dispersive coupling is considered as a single site process which allows the relaxation of an excited
molecule and hence causes i t to be 'off-resonance' with
the bulk crystal thus lowering the resonance transfer time from that expected for a non-vibrating crystal. However, relaxation can only be effected if the resonance transfer time is greater than the period of a lattice vibration and thus appears to be limited to triplet excitons or to
ac~n0wl
d;ements
I wish to express my deep gratitude to Professor D.P. Crai g for his constant kindness, guidance and understanding in the
supervision of this work.
I am also indebted to Lr L.A. Dissado for his time and patience during many beneficial discussions I have had with him concerning aspects of the work presented here.
Dr D.W.T. Griffith has also been very helpful to me by presenting the experimentalist's viewpoint in discussions on this work and by assisting in the preparation of the diagrams contained herein.
Many other members of the Research School of Chemistry have given me help and encouragement during the course of my work here. I am very grateful to them all.
The award of a Commonwealth Postgraduate Scholarship is also gratefully acknowledged.
---c0ntents
ABSTRACT
ACKNOWLEDGEMENTS
CHAPTER
,
INTRODUCTION 1-1 General
1-2 Photodimerisation and the Topochemical Hypothesis
1-3 Energy Transfer in Molecular Crystals 1-4 Model Approach
THE MODEL
2-1 Description 2-2 Phonon Modes
2-3 Second Quantisation of Phonon Modes 2-4 Exciton States
2- 5 Excitons -in
.
Second QuantisationMODEL FOR A DEFECT CRYSTAL-PHONON MODES
PAGE
1
2 5 9
10 12 19 22 27
3-1 Introduction 29
3-2 Equation of Motion for a Defect Crystal 29 3-3 Evaluation of Phonon Green's Functions 36 3-4 Phonon Modes for the Defect Crystal 40
ELECTRONIC STATES OF A CRYSTAL WITH STRUCTURAL DEFECTS
4-1 Introduction
4-2 Theory of Electronic States of Impure
43
Crystals 43
'
a
5-1 Introduction
5-2 The Exciton-Phonon Coupling Operator 5-3 Stationary States
5-4 Green's Function Approach
THE DEFECT CRYSTAL WITH RESONANCE EXCITON-PHONON COUPLING
57
57 61 67
6-1 Introduction 76
6-2 Stationary States 76
6-3 Time Development 80
6-4 Calculation of Trapping Rates 82 6-5 Temperature Dependence of Trapping Rates 84 6-6 Kinetic Analysis of Trapping Rates 90 6-7 Pseudo-localised Modes in the Trapping
Process 93
6-8 Review of Experimental Work 97
DISPERSIVE EXCITON-PHONON COUPLING
7-1 Introduction
7-2 The Dispersive Exciton-Phonon Coupling Operator
7- 3 The Canonical Transformation for Strong Coupling Region
7-4 The Canonical Transformation for Intermediate Coup ling Region
EXCITON-PHONON COUPLING-TIME DEPENDENT APPROACH
8-1 Introduction
8-2 Interaction Representation 8-3 The Time Evolution Operator 8-4 Exciton-Phonon Coupling in the
Interaction Representation 8-5 Damping of Exciton States
the
the
105
105
108
117
123 123 125
9-1 Introduction 147
9-2 A Model for Self-Trapping 150
9-3 The Dynamics of Self-Trapping 155
9-4 Self-Trapping versus Defect
Trapping in Molecular Crystals 157
,0
CONCLUSION 1591-1 General
CHAPTER 1
INTRODUCTION
Photochemistry in the solid state differs from that in solution or gas phase through the constraints of a rigid environment on the movement of reactant species and thus on implied differences in the mode of energy transport to and
from reactant sites.
The range of the photochemical reaction in the condensed
1
phase is very wide since i t can be extended to include the
photochemistry occuring in large biological molecules which
are composed of uniformly repeating units. Thus the compli-cated membrane photochemistry involved in vision and photo-synthesis and the degradation of DNA through thymine
photodimerisation are extensions of this class of process. Solid state photoreactions of economic importance include
the photodecomposition processes responsible for the degradation of plastics and the fading of dyes, and the future possibilities for an organic solar energy cell.
From a theoretical standpoint these examples are of forbidding complexity and in the present work only the
'simple' photochemical processes known to occur in the
class of reactions since they are both well documented and provide an interesting example of the effects of lattice control.
1-2 Photodimerisation and the Topochemical Hypothesis
The recognition of the influence of the crystal environ-ment on product configuration i~ photodimerisation reactions
2
in the solid state led Schmidt and co-workers to the develop-ment of the topochemical hypothesis. According to the
topochemical rules, the product dimer stereochemistry is
determined by the crystal packing geometry of monomer pairs, provided the contact distance between parallel reactant pairs is of the order of 0.4 nm or less. The application of this rule may best be illustrated by the example of the
trans-cinnamic acids. These compounds crystallise in three packing
3,4
types, labelled a, Sandy. In the a packing arrangement molecule pairs overlap in a head-to-tail fashion with a
double bond separation around 0.4 nm. Dimerisation in this case then gives the centric dimers (or a-truxillic acids) exclusively. For a
S
type crystal the dimerising pairs are stacked head-to-tail along the b crystal axis with a double bond separation of between 0.38 nm and 0.42 nm giving aresultant dimer (S-truxinic acid) of mirror symmetry. In they type crystals no double bonds are closer than 0.47 nm and in accordance with the above rule, this crystal
Figure 1.1 The photodimerisation of the trans- cinnamic 5
acids (reproduced from Cohen and Green) .
TOP VIEW OF
CARBOXYLIC ACID PAIR
- -_ _ _ _ _ X ~ - - - EDGE
ON
VIEWOF
( - =COOH X=centre of symmetry) CARBOXYLIC ACIDPAIR
(3
type
d=3·8-4
·
2A
R-_---
j --
-R
I
R-=--
X - - - --R
I
R---
X - ---R
I
y
type
d=5·2A
\
R--- X
---R
\
R- - - - X - - = - R
\
R-
- - x • - = - R
a
type
R - ~
\
d >5·5A
x--=:-R
~----hv
>
hv
>
hv
R-.:__
x ~ RR-=
-
- x -
-
-R
~
R
NO
A large number of compounds are now known to dimerise
according to these topochemical rules. However, from
observations on the photodimerisation of some of the
anthracenes i t has become evident that factors other than
the crystal structure of the perfect lattice may be important
in determining the photodimerising activity of a particular
crystal. For example, 9-cyanoanthracene, which crystallises
in the head-to-head arrangement, forms only the sterically
preferred trans dimer product. This observation led Craig
6
and Sarti-Pantoni to propose that the reaction was occurring
at a defect site at which the monomer molecules were
favour-ably aligned for this 'non-topochemical' dimerisation. The
presence of these photoactive impurity regions has subsequently
7
been demonstrated.
Again, the unfavourable orientation of monomer units in
the perfect anthracene crystal precludes dimerisation occurring
according to the topochemical rules. However, i t is well
known that dimerisation does occur and i t has been shown by
8, 13 9
Thomas and Williams and more recently by Rajikan that there
is a one-to-one correspondence between the sites of
photo-dimerisation and the sites of emergent dislocations.
The phenomenological aspects of both these topochemical
and non-topochemical reactions in organic molecular cyrstals
1,2,5, 10
have been well documented and reviewed. However, a detailed
understanding of the mechanism and energy transfer processes
present work to study in some detail the migration of energy
through the crystal to a favourable site for the reaction and the subsequent localisation of energy at that site for a sufficiently long time that reaction may proceed.
1-3 Energy Transfer in Molecular Crystals
11
The exciton theory of molecular cyrstals describes the
role of intermolecular interactions of the crystal in the
delocalisation of molecular electronic excitation energy over all sites of the perfect lattice. The excitation then
propagates through the rigid lattice as a wave packet com-posed of a linear combination of states each characterised by a wave vector k. In analogy with the derivation of the
momentum of a free particle in a uniform potential, the wave vector describes the quasi-momentum, p, of the exciton
pseudo-particle propagating in the periodic pbtential of the crystal lattice as
p hk 1.1
The time for the excitation to reside on each site in the periodic lattice is in the order
where I is the intermolecular resonance interaction energy giving Tel typically in the range 10-12 - 10-14 seconds.
The dynamics of exciton motion through the crystal
vibrations. The periodicity of the lattice confers a wave
like property on these vibrations and allows them to be
characterised by a wave vector
g.
are known as phonons.
Such quantised vibrations
It is the presence of the phonons in the crystal which
allows all the levels of the exciton band, each of which is
associated with one momentum state of the system, to be
populated. The initial absorption of a photon by a crystal
leads to the population of the exciton band levels associated
with momentum k ~ 0 only. This is known as the optical
selection rule and is due simply to the conservation of
momentum. However, the subsequent emission and absorption
of phonons through momentum conserving exciton-phonon
scattering processes allows this population to be redis-.
tributed through levels associated with all k values.
Eventu~lly a Boltzmann population distribution will be built
up over the band states with the time taken for the band to
reach thermal equilibrium depending on the strength of the
coupling between the excitons and phonons. In particular,
below-band electronic energy levels which are associated
with impurities in the crystal structure will be populated in this way.
The frequency of these exciton-phonon scattering events
is dependent on the thermal population of the various phonon
modes. Thus at low temperatures, the number of phonon
may sample many crystal sites between scattering events. The exciton is then said to propagate through the crystal in a coherent manner. At high temperatures the average time between scattering events may be less than Tel so that the excitation has a high probability of being scattered at every site i t encounters. The wave like nature of exciton propaga-tion is then lost and the excitapropaga-tion moves through the crystal by a random hopping motion known as incoherent transport.
In the present work only coherent exciton transport will be considered since most experimental results for molecular crystals are obtained in a temperature region in which this applies.
There are two types of exciton-phonon coupling which should be considered, both of which arise through the linear
1 2 dependence of the exciton energy on molecular displacement. Firstly, resonance exciton-phonon coupling involving inter-site scattering of the two quasi-particles is responsible for energy transfer from the bulk perfect crystal to sites of physical or chemical defects which are formed during crystal growth. This will then provide a mechanism for
energy localisation at sites which may be favourably aligned for a non-topochemical reaction.
Secondly, dispersive exciton-phonon coupling is a single site coupling process which results in a shift of the excited state molecule from its ground state equilibrium position. This process, along with the accompanying relaxation of the
effective if the exciton resonance transfer time is of the order of, or less than, the period of a lattice vibration
-13
(~10 seconds). If we could then view the dispersive exciton-phonon coupling process on the time scale of
lattice frequencies we would see the creation of a more
stable environment for the excited molecule as the coupling and relaxation processes moved to completion. The effect of this stabilisation is to decrease the probability of resonance transfer away from the excited molecule. This of course can occur on any site in the bulk perfect crystal
but there is a statistical probability that excitation
will reside longer on some sites than others with the quoted
resonance transfer time being only an averaged value. The
longer excitation resides on any such randomly selected site, the greater will be the barrier to energy transfer
from that site. Such an energy localising process is 4 7
referred to as 'self trapping' since by this mechanism
energy is not transferred to a preformed defect but resides preferentially on a site stabilised by the presence of the excitation itself. Although there is as yet no experimental evidence to support the occurrence of self trapping of
singlet excitons in molecular crystals i t is thought that this may be the energy localising step preceeding
1-4 Model Approach
In the following chapters a theory for energy transfer
in molecular crystals through the mechanism of
exciton-phonon coupling will be examined for the case of a
mathe-matically tractable linear chain model. This model is
acceptable for certain of the systems discussed at the
beginning of this chapter and for certain molecular crystals
in which, for one particular crystal direction, the monomer
unit separation is considerably less (and the associated
coupling strength correspondingly greater) than for any
other. Different approximate solutions will be examined and
contrasted for varying types of simulated defects in the
model. An analysis of the rates of energy transfer to and
from defects will be made with the aim of establishing a
time scale for a sequence of processes following optical
excitation. Finally, a brief discussion will be presented
on the nature of possible steps in the photodimerisation
2-1 Description
THE MODEL
The model which has been chosen for the present work
is that of a linear chain of anthracene molecules aligned
with molecular planes perpendicular to the intermolecular
axis (fig. 2.1). For mathematical convenience the number
of molecules in the chain has been limited to 101 with end
effects eliminated through the use of cyclic boundary
conditions.
The equilibrium intermolecular spacing (R
0) for the ground state system is determined through minimisation of
a potential function V(R) with respect to molecular
dis-placements along the chain axis.
i.e. 8V (R)
8R RO 0 2.1
In organic molecular crystals, the weak van der Waals
binding forces are often described by a sum of potentials
over all non-bonded intermolecular atom pairs. In particular,
the '6-exp' potential function has been used in this way in
1 4
calculations on molecular crystal dynamics. Here, the
atom-atom potential is given by
a-a
V (R) + Be -aR 2.2
where A, B, and a are constants for particular types of atom
Figure 2.1 Relationship of molecular and crystal
axes for the linear chain model
MOLECULAR AXES
M
2
> - - - ~ 1 - - - + - ~
L
6 3
N
LINEAR CHAIN MODEL
L
H-H non-bonded contacts are given in Table 2-1. Using this
potential function and the anthracene molecular co-ordinates 16
obtained by Cruickshank an equilibrium intermolecular spacing of 0.34 nm was obtained for the model system. This spacing
is much less than in the three dimensional anthracene crystal but is nearer the closest C-C contact distance of 0.37 nm
in that crystal. Also, in the pseudo one-dimensional stack structures reported for 9-cyanoanthracene and pyrimidine
1 7
the intermolecular spacing along the stack is 0.39 nm and 1 8
0.38 nm respectively.
This calculated spacing of 0.34 nm is actually within the range of interchromophore separations (vis. 0.30 nm
-19
0.35 nm) which give rise to excimers in molecular crystals. These complexes, which are formed between an electronically excited chromophore and a second identical chromophore in
its ·ground state are stabilised by both resonance and charge
1 9
transfer interactions. In the following work, excimer
formation will be neglected in order that attention may be focussed on exciton-phonon coupling. This point will
however be taken up later in Chapter 10 where excimer
formation as a _possible precursor to dimerisation will be discussed.
2 0
2-2 Phonon Modes
A discussion of the phonon ~odes of a linear chain of
Table 2-1
Values of Atom-Atom Potential Constantst
Atom pair A (Kcal/mole A6) B (Kcal/mole) a(A-1 )
C ... C 358 4.2 X 104 3.58
C ... H 154 4. 2 X 104 4.12
H ... H 57 4.2 X 104 4.86
t taken from reference 14.
lattice dynamics. In the present model both translational
and librational motions should be considered. However,
within the harmonic approximation we are only interested in
those displacements which give rise to a nett linear restoring
2 1
force. It can be shown that the '6-exp' potential reduces to
a form with terms quadratic in R, only for displacement
along the chain axis and for rotations about the a and b
crystal axes. For these cases the equation of motion for
the set
where:
of coupled molecular oscillators can be written as
M tm - -I: ~n tn . µ '\) - 1,2,3 2.3
µ µ µv \)
nv
the indices n and m label molecules
the indices µ - 1,2 refer to librational motion
about the a and b axes respectively
the indexµ - 3 refers to longitudinal translational
motion
[image:22.773.14.753.21.986.2]vmn
µv
t ; is the translational displacement of the centre
of mass of molecule m.
M
3 is the total molecular mass
M
1, M2 are the moments of inertia
vnm is the intermolecular force constant to be
µv
determined in this case by summation over all
atom-atom pairs, i j , of the interatomic '6-exp'
force constant [ ~ ] . _-µv lJ
I: [~-] .. µv ..
l ] l ]
I: [8
a
V(R~J)] ~m "'tn nm l ] 0 tµ 0 \)2.4
where: RlJ is the separation of atom pair ij belonging to nm
molecules n and m respectively.
Certain symmetry properties of the lattice allow the
2 2 calculation of the force constant matrix to be simplified.
Translational symmetry of the 'infinite' lattice gives
v111+h n+h
µ \) 2.5
where: h i s any integral number of molecular spacings, a.
Also,
~
-µv
and I: vmn - 0
n µ3
From inversion symmetry
~ µµ
~
vµ
µ
-m -n V
µ µ
-v
-m-n µ \)2. 6
- 1,2,3 2.7
2.8
Finally, if reflection is also a symmetry operation of the
system
-m -n
V
µ \) 2.10
Thus for any lattice in which both a reflection plane and
an inversion centre are present
0 if µ
t- \)
2.11and there can be no coupling between modes of vibration.
The simple harmonic displacements of each molecule may
be expressed as a plane waveform over the chain.
1
IM
N e exp[-i(wt - qn)]µ µ
where: N is the number of molecules in the chain
e is the amplitude of the wave
µ
w and q denote the angular frequency and wave number of the wave respectively.
2.12
With this substitution for tn the equation of motion 2.4
µ
reduces to
2
w e - L D (q) e
µ \) µ\J \) 2.13
where
Dµ\J ( q) - 1 L vho e -iqh /M M h µ\J
µ \)
Thus the frequencies for the three vibrational modes at a
solution to the 3x3 determinantal equation
0 2.14
Consideration being limited to nearest neighbour
interactions only and with the neglect of intermode coupling,
i t can be shown, with the use of the above symmetry relations
that the translational and librational frequencies are of
the form
translational: w ( q) w
I
sin ( q/ 2)I
0 2.15
where w 2 - -4 VlO
0 M 33
2 ~
librational: w ( q) - [p
+
p'. sin ( q/2) ] 2 2.16where p -
-
1 [2Vl0+
voo
]Mµ µµ µµ
} µ - 1,2
p' _.. -4 - VlO
t1µ µµ
Equations 2.15 and 2.lb, which express phonon frequency
as a function of q, are known as dispersion relations. I t
can be seen that these equations will give the same value of
w(q) for q and any
2TTm
q +
a m ±1, ±2, ...
Thus in order to have only one lattice vibration corresponding
to each value of q, the range of q must be limited to
- TT < q a
TT
The wave number dependence of the librational modes is such
that
w(q) -+ p
½
as q -+ 0Modes having this type of limiting behaviour with q are
known as optical modes since they can be optically excited.
For translational vibrations
w(q) -+ 0 as q -+ 0
which behaviour corresponds to acoustic vibrations.
The dispersion curves obtained in the absence of
intermode coupling are shown by the dashed lines in fig. 2.2. The maximum acoustic phonon frequency,
w
=
650 cm-l is0
considerably higher than that for any of the acoustic branches of the anthracene crystal where
w
0 ~ 50
-1
cm • It
is not surprising that in view of the absence of two thirds of the force field, the linear chain vibration frequencies do not agree with those typical of real molecular crystals. They are however of the same order of magnitude as the
accordian mode longitudinal acoustic vibrations of the 2 3
short chain polymethylenes.
If the molecules in the linear chain are rotated about
the a and b crystal axes so that reflection symmetry is ,lost, intermode coupling must be considered. The dispersion
Curves labelled (1) ., (2) and (3) correspond to
translational motion and motion for rotation
about crys tal band a axes r es~ecti vely .
600
No intermode coupling
lntermode coupling
/ I
/
/
I /
I I I
I
I I I I I
I
I I
I I I
I I I
/ I
( I )
-/ (2J / / / , / (3)/ /
/ /
/ / (3)
f
400 I I I I I II I I
I; (2)
-
I E 0-
>-(!) 0:: wz
w 200?I
11, I/ II; I
I
I 1 I I I I I
I I I
I I I
I I I I I I
I I I /
/ I I I
I I
I I
/ I
I I
I
I I I I
I; '1
I I
t11
,,
( I )0·57r
the coupling between the optic and acoustic vibrations is
to eliminate degeneracies of these modes at values of q
2 2
where they occur. However, i t should be noted that as
with the anthracene crystal, there is still no band gap
between the optic and acoustic frequencies.
2 4 2-3 Second Quantisation of Phonon Modes
It will be found convenient to present much of the work
which follows in the formalism of second quantisation theory.
Therefore, the second quantised operators describing the
phonon modes of the crystal will be introduced here for use
in later sections.
The set of coupled simple harmonic oscillator equations
2.3 can be formally decoupled by writing tm as a Fourier
µ
expansion in terms of a new set of generalised co-ordinates
Q of the system. qs
l I Q exp(iq .n)
/MN qs qs s
µ
2.17
where: s labels the vibrational branch corresponding to
molecular displacement alongµ
Since the displacement tm must be real µ
The kinetic energy of the system_can be written as
1 ~
M
2 0-y L .V ,
2.18
1 • •
- - I: Qqs Q~s 2 qs
and the potential energy
".E.
1I: I: vnm tn tm 2.20
-2 µv \)
nm µv µ
where the solution for
w
(q) from 2.13 and 2.14 has been sused.
The total Hamiltonian for the system can then be written as
where: p
qs
T + VP.E. 1
2 I: qs (P qs P* qs +
P*
-qs
are the conjugate momenta to the generalised co-ordinates.
2.21
It can be seen that in Hamiltonian 2.21 there is no coupling between terms of different q. Transformation to these normal co-ordinates has therefore allowed the system to be described by a set of q independent harmonic oscillators for each
vibrational branch, s.
[ p q' SI f p qS] 0 2.22
i
n
O qq I Q S SI 2.23Operators b qs' b;s are now introduced such that
b - 1 k [ws(q)Qqs + i P -qs] 2.24
qs ( 2nw ( q) ) 2
s
bt - 1 k [ws(q)Q_qs i P qs] 2.25
qs ( 2fiw ( q) ) 2
s
Using 2.22 and 2.23 i t can be shown that these operators
satisfy the following commutation rules.
[ b b
t
]
qs' q's'
·
o
qq' 0ss'[ b
t
bt
]
qs' q's'2.26
0 2.27
By then substituting expressions obtained for Q and P
qs qs
in terms of b and bt into 2.21, the Hamiltohian operator
qs qs
for the system can be expressed as
I fl w ( q) s qs
[b
t
bqs qs + 2.28
These operators, b and b t , are respectively the creation
qs qs
and annihilation operators of second quantisation. Thus if
lu
> represents the state with u phonons of wave vector qqs qs
then
b
lu
> -;u-qs qs qs
-u - l>
qs
b
t
I
u
>qs qs /(-uqs +l)
-u + l>
qs
2.29
so that
lu
>qs
(u
+
1
)
n
w (q)qs 2 s u qs >
Expression 2.31 clearly shows the quantised nature of the lattice vibrations as phonons of wave number q and the commutation relations of the phonon operators show that these particles formally behave as bosons. They thus obey
2.31
Bose-Einstein statistics and hence have occupation numbers,
u , not restricted to O and 1. qs
l l 2-4 Exciton States
Th e zero th or er wave d f unc ion o t . f t e r h th excite . d sta e t of the linear chain can be represented in the basis of a set of either localised or delocalised functions. The former is obtained as a product function of the ground and excited state molecular wave functions 1))0 and 1/Jr respectively as
n n
o r
1/)2 • • • ljJn • • • 1/JN 2.32
The delocalised set, ~r (k) is then formed through taking modulated sums of the localised functions so that they
trans-form as irreducible representations of the translation group of the crystal.
~r ( k) 11 2.33
Since the intermolecular forces are weak, they can be
molecules in an oriented gas. The system Hamiltonian can therefore be written as
where: and
H 0
V
H H + V
0 2.34
L H is the sum of free molecule Hamiltonians m m
f. V is the sum of pairwise intermolecular n<m nm
interaction terms.
The potential V is commonly expanded as a series of multi-nm
pole interactions. Usually, the dipole-dipole interaction is dominant and this is the only term in the expansion which will be considered in the present work.
If the eigenvalues of the system Hamiltonian are to correspond to transition energies, then the ground state energy, EG, of the crystal must be subtracted from the
solution to 2.34. The energy EG is determined by
2.35
where: 2.36
Then, for the linear chain with one exciton present the Hamiltonian 2. 3* is diagonalised in the basis of the orthogonal set of delocalised functions ¢r(k) as
E (k) ¢r (k) 2.37
E (k) + + I (k) 2.38
where: ~wr is the free molecule transition energy
I(k) is the modulated sum of resonance, 'exciton
exchange' interactions
I- ( k) L
n<m eik(n-m) 1 nm 2.39
where: I nm
r
=
< ... 1/Jn 1/J ·m o · ...
I
V nmI
••• 1/J o n 1/J m r ... > 2.40Dr is a sum of first and second order terms giving
the difference in electrostatic and dispersion
energies between ground and excited states.
1st order electrostatic:
L
n<m
2nd
r o < ... 1/J 1/J ...
n m
order electrostatic:
I
< . . . 1/J 1/J r o.
. .
L L n m
n<m s,t Er + Eo
:/
0 n mI
o oL L < . . . 1/J 1/J n rn
...
n<m s,t
Eo +
:/
0 nL 0 0
I
I
0 0< ~- .. 1/J 1/J . . . V ••• 1/J 1/J •.. >
nm nm nm nm
Iv nm
I
•••
1/Js1/Jt nm ••• > I 2--
Es-
Etn m
1vnml . . . 1/J 1/J nm s t ••• > I 2 Eo
-
Es-
Etm n m
where: Er is
n the energy of the rth excited state of molecule
n. In the dipole approximation, the above matrix elements
of V have the form
nm
A A • '
3 ( R. i) ( R.
i)
}
(
µ i) ( µ J) 2.41where: the i,j index refers to the axis of polarisation (along L, M, or N) for the transitions on
molecules n and m respectively, and
µi is the value of the transition dipole moment
along i .
R is the vector of length R joining molecular nm
centres n and m.
Thus for the linear chain geometries of fig. 2.1 with
an angle 8 between the molecular L axis and the crystal a
axis
IMM
nm
ILL nm
INN nm
- l (1 -R3
nm
l
(1 -R3
nm
2.42
3 sin 2 8) (µL)2 2.43
3 cos 2 8) ( µN) 2 2.44
The calculation of I(k) from these molecular interaction
terms is further simplified for the linear chain model where 'shape dependence' of the sum over n and mis not important.
This difficulty arises in three dimensional systems where, -3
as the dipole interaction decreases as R , the number of
. t t ' d' l · 3
in erac ing ipo es increases as R In the linear chain however, the sum converges rapidly with nearest neighbour interactions dominating. Then, since
the exciton band levels (in the nearest neighbour
approximation) can be written as
E (k) 6W r + D r +2 S cos (k) 2.46
where: S i s the nearest neighbour resonance interaction.
I t can be seen that the greatest density of exciton
states will be at the two band edges where the minima of
2.47 arise
E (k)
S
sin ( k) 2.47If 8 is small, the resonance interaction energy is
negative only for the N polarised transition. This will result in an exciton band structure, as for the three
dimensional anthracene crystal, in which the k
=
0 energy1 1
level is at the bottom of the band. However, the Land M
polarised transitions (which are responsible for the first
two excited states of the anthracene crystal) will give an
inverted band structure for the linear chain.
For the purposes of the present model calculations the
following parameters for the first singlet excited state
have been used.
v\..M
I Q1
M
µ
25000
-1000
-1
cm
-1
cm
0.
-
o
61 nmn.l~ C ".)·. , ~ ....
As this is the only transition which will be considered, the superscript r will be dropped from further notation.
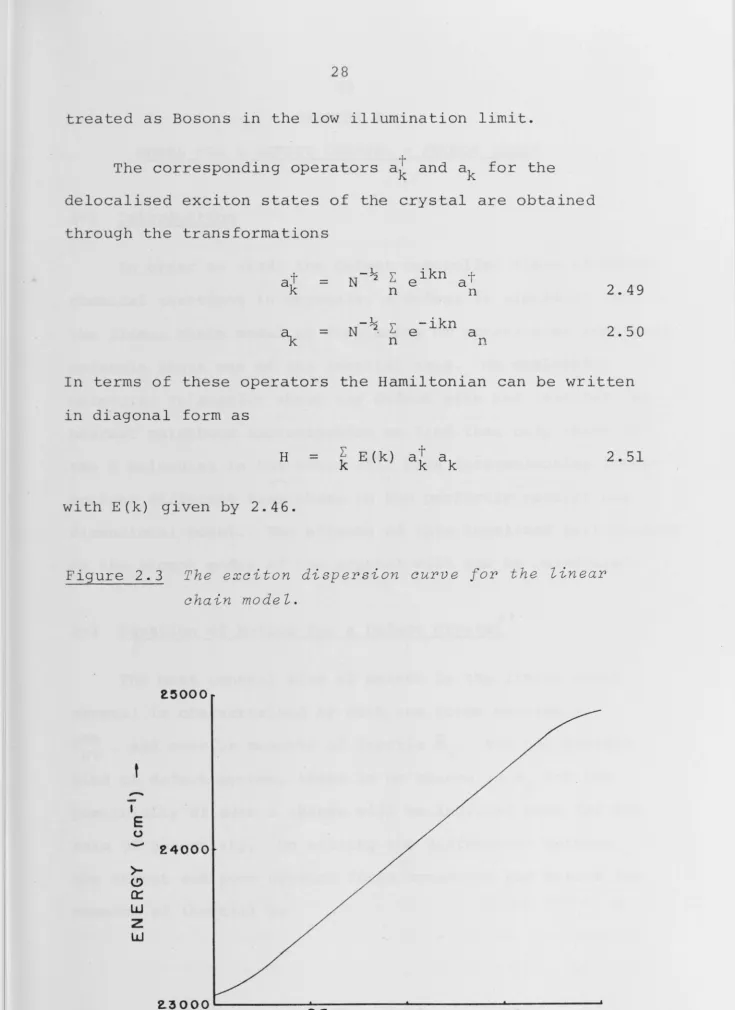
The above parameters give the exciton dispersion curve of fig. 2.3. The relatively close intermolecular spacing in this model is responsible for the large bandwidth of 1500 cm-1. However, the ratio of the maximum acoustic
phonon frequency to ·the exciton .bandwidth is approximately that found in many real crystals.
2 5 2-5 Excitons in Second Quantisation
For the treatment of excitons in second quantisation we introduce the creation and annihilation operators at
n and a which are site transition operators describing the
n
processes of excitation and de-excitation between the ground
and excited states of molecule n. The state of the molecule at site n can be described by an occupation number N which
n
is unity if the molecule n is excited and zero otherwise. The restriction on the number of states to which a molecule may belong, means that the operators at, a should obey the
n n Fermi commutation rules
[a I at] 1
-
2N 2.48n n n
However, if the number of excitations in the crystal is
small the average of N for the crystal can be approximated n
by zero. Then the commutator 2.48 will be that of Bose
treated as Bosons in the low illumination limit.
t
The corresponding operators ak and ak for the
delocalised exciton states of the crystal are obtained
through the transformations
at - N -1: 2 I: e ikn at
k n n
-k I: -ikn
~ - N 2_ n e a n
2.49
2.50
In terms of these operators the Hamiltonian can be written
in diagonal form as
H
[image:37.768.11.746.25.1035.2]with E(k) given by 2.46.
Figure 2.3 The exciton dispersion curve for the linear
chain model.
25000
t
-'
E 0-
24000>-(!) 0::
w
z
w
~ ~ 0 0 0 ' - - - ' - - - ~
0
0·25
7T 0·507T 0·757TWAVENUMBER {k)
CHAPTER 3
MODEL FOR A DEFECT CRYSTAL - PHONON MODES
3-1 Introduction
In order to study the defect controlled class of
photo-chemical reactions in crystals, a defect is simulated in
the linear chain model by displacing or rotating an arbitrary
molecule about one of the inertial axes. On neglecting
molecular relaxation about the defect site and invoking the
nearest neighbour approximation we find that only three of
the N molecules in the chain will have intermolecular
inter-actions different from those in the perfectly regular one
dimensional model. The effects of this localised perturbation
on the phonon modes of the crystal will now be considered.
2 2
-3-2 Equation of Motion for a ·Defect Crystal
The most general kind of defect in the linear chain
crystal is characterised by both new force constants,
~ , and mass or moments of inertia
Mµ.
For the presentkind of defect system, there is no change in M but the
µ
possibility of such a change will be included here for the
sake of generality . . On writing the differences between the defect and pure crystal force constants and masses (or
and M
-µ
~
- v111n
µv µv - (l - s) M
µ
we obtain the equation of motion for the defect system (corresponding to 2.3 for the perfect chain) as
3.1
__ L [v111n T,;nn]
nv
µv - tµv µv - 1,2,3where the defect ts assumed to be at molecule m
=
O. Since the presence of the defect has destroyed the periodicity of the lattice, the simple harmonic molecular displacements cannot in this case be given as a function of wavenumber q as in 2.12. Instead, we write for tm the wave solutionµ
l
1M
µm
s
µ
-iwt
e 3.3
Substitution of 3.3 into the equation of motion 3.2
.
gives
Drnn n 2 m I rnn n 3.4
I s w s -
7µv
sµv \) µ \)
nv
nv
where
~
Drnn b! \) 3.5
µv /M M
µ \)
as before, and
rnn
~
20 0mo 0no 3.6
7µv
- ~£i
+
w E:µv µ \)
When E: and all 7rnn are zero, the w 2 correspond to the . µv
squared phonon fre_quencies of the pure chain. The effect
can be treated by the classical Green function technique.
We firstly write 3.4 in matrix notation as
2
Ds -
w
I s = ,/s 3.7Similarly, the equation of motion for the unperturbed crystal
0
can be written in terms of solutions s as
0 2 0
Ds - w I s == . 0 3.8
Equations 3.7 and 3.8 may then be combined so that the
solutions for the perturbed system may be given in terms
of those for the unperturbed case as
8
=
~ + ,/ 8 2 (D - w I)A Green function matrix G is then defined such that i t
satisfies the equation
2
DG -
w
I G=
6.where 6. represents the matrix of Dirac delta functions
{ ornn c5 } _.
µv
3.9
3.10
2
G is thus the reciprocal of the matrix [D -
w
I] andhence may be substituted into 3.9 to give
0
+
G ,1 3.118 - 8 8
The matrix elements of G are given by
2 rr,n
Grnn (w2) ornn
-z::
[w-
Dµv]-
c5 3.12Since the pure crystal eigenvalue solutions w (q) and the s
corresponding eigenvectors eµ (qs) have already been
obtained i t is convenient to express G~ (w2) in terms of
these functions. We have
ws 2 (q) e (qs)
µ 3.13
Further, the functions
N-½
eµ (qs) eiq.n form a completeorthonormal set in the space of the perfect crystal so that
I: nµ
I: qs
0 ss'
Grnn ( w 2 ) may b e expan e d d . in a d ou b 1 e F ourier series . . t ogive .
µv
3.14
3.15
1 * 2 iqn iq' m
NI: I: eµ(qs)G(qs;q's';w )ev(q's')e e · qs q's'
On using the orthonormality relations 3.14 and 3.15 in
conjunction with 3.12 and 3.13 we obtain G(qs;q's';w2)
the form
Thus
2
G(qs;q's';w)
-o
o
qq'
s s'2 2
[w (q)-w ] s
*
e (qs) e (qs)
µ \)
2 2
[w (q)-w
l
siq(m-n) e
3.16
in
3.17
I t is then necessary to evaluate the matrix elements of G
and/ explicitly in order to determine the solutions to
3.11. The type of solutions to 3.11 which can be expected
with Gin the form of 3.18 can be found by exarru.ning
the simpler case of an isotopic defect in a chain of atoms
2 6
having only one phonon branch. In this case
and
1
0011
1 E 1
N q [w2(q)
2
s w
3.19
3.20
If we seek the particular solutions to 3.11 for which
~=Owe ensure that there are nci zeros in the denominator
of 3.19. This requires that the summation in G00 2
11 (w) be
restricted to odd values of q. The perturbed frequencies
of the system are then determined by the equation
l (N+l)/2 l
E [ - - - ]
N q(odd) w2 (q) - w2
2
w s
=
1 3.21If equation 3.21 is multiplied out, an algebraic equation
of degree (N + 1)/2 (for a chain of N atoms) in . W 2 is .
obtained. The graphical solution for these (N + 1)/2 roots
is shown in fig. 3.1.
I t can be seen that where O < s < 1, (N - 1)/2 of the
roots interleave the frequencies of the unperturbed chain
while the remaining root lies above this phonon band.
When the number of molecules is so large that the band of
Figure 3.1 Graphical solution for the roots of equation
3. 21 representing the phonon frequencies for
~
L
w
2w2-w:
k
a chain of atoms containing an isotopic impurity
(reproduced f r om Ref. 26) .
u.
I
I
I-
--I
EI
- , - I
0
continuum, the (N - 1)/2 states of the perturbed system
will be effectively coincident with this continuum and only
the frequency of the above band mode has to be determined.
This corresponds to a localised phonon mode since i t does
not overlap with and hence cannot decay into the continuum
of essentially delocalised states. The amplitude of the
localised mode thus falls off rapidly with distance from the
impurity centre. This type of state only ari ses when
~
=
O so that there is no wave like character associatedwith i t and thus the condition for the occurrence of
localised modes is
IGI - II
0 3.22For the more general case of multiple vibrational branches localised modes may occur both above the maximum frequency of the highest vibrational branch and in energy gaps between vibrational branches. Further, the creation by a defect, of a mode above the continuum of acoustic phonon states say, may result
in
a state which is equi-energetic with a mode.of an optical branch. This then constitutes a pseudo-localised mode since i t is a mode which can decay into a delocalised
phonon manifold and thus has essentially delocalised character at large distances from the defect but which has an enhanced amplitude at the defect sites. Similarly such
pseudo-localised modes can arise when a mode falls below the levels of an optic branch and into the acoustic manifold. The
occurrence of such a mode causes a sharp peak in the density of states function for the particular phonon branch.
On returning to the Green's function for the present system we see that in cases for which the phonon modes form
an energy continuum 3.18 may be replaced by
1
2n I:
r
s* iq(m-n)
eµ (qs)
ev
(qs) edq 3.23
-TT
For the localised modes, there will be no singularities
by integration in the complex plane. This integration is
considerably simplified if the vibrational motions are
decoupled, which for the linear chain model, requires that
the chain and molecular N axes be co-incident. As
2 2
discussed by Dettmann and Ludwig, this restriction does not
detract from the essential features of the problem. We
will thus consider below the evaluation of Gmn (w2) for µv
µ
=
v=
1,2,3 only.3-3 Evaluation of Phonon Green's Functions
(i) Translational Motions
In the perfect crystal the translational phonon
frequencies are obtained in the nearest neighbour
approxima-tion as
Thus
w
2 (q) -w~
sin2 (q/2)2TT
I
TT
-TT
iq(m-n) e
[ w
~
s in 2 (q/
2 )2 ,dq
- w ]
3.24
This integral may be evaluated by chan~ing the line integral
to a contour integral in the complex plane on substitution
for Z
=
e 1 q. Thenmn -4
Gll
-2 TTlWO . 2
Z (m-n)
dZ
(Z
+
U) (Z+
l:_)u
where
u
is chosen suchw2
-The path of integration
I
z
I
- 1, in the complexFigure 3.2
Path of integration .
for equation 3.25.
_,
V
We see that simple poles
z
- -uz
---
1u
for which 0 < u < 1
that
U) 2 2
(1 + WO
u
4 3.26is now on the unit circle,
z
plane (fig. 3. 2) •@
plane.
the real at
arise on axis
(inside
I
z
I
-
1) 3.27On writing
q TT+ ix
we see that at the poles either
or
u -
-z
u
=
1z
-x
- e
X
- e
3.29
3.30
3.31
so that only the pole inside the unit circle will allow G to decrease (exponentially) with increasing (m-n) and hence correspond to a localised mode. Evaluating the residue at
z
=
-u
gives( -u)
I
m-nI
+ 1 1 - u23.32
The phase factor between neighbouring molecules in this
2
case is -1 so that
w
corresponds to a translational opticlocalised mode.
(ii) Librational Motions
In this case localised states may be either above or below the librational band and hence be localised in either
the optic or acoustic region. For a particular phonon
branch, the librational phonon frequencies are obtained in
the nearest neighbour approximation for the perfect lattice as
where p 2 D10 µµ + Doo µµ
p' - -4 DlO µµ
Then, on again substituting for Z
=
eiq in 3.23 we havewhere
giving
Gmn _ -4 l
J
µµ p' 2TI
J
p + p' ( l + u) 2
4 u
z(m-n) dZ
(Z +· U) (Z + 1)
u
2
- w
4 (-u) jm-nl + l
Gmn - 0
µµ p'
l
-
u2< u < 1
3. 34
3.35
3.36
3.37
3.38
This case again corresponds to an optic localised mode
with a phase factor between neighbouring molecules of -1.
In order that O < u < 1, i t is necessary that the 2
following conditions on w , p and p' be satisfied
2
w > p + p' if PI > 0
or 0 < w 2 < p + p' if -p <
p'
< 03.39
3.40
Thus w2 must lie above the maximum frequency of the
pure crystal optic branch.
Alternatively, the substitution for Z - eiq permits
Gmn _. -4
µµ p' l
2TI
Z (m-n) dZ
(Z - U) (Z - 1)
u
where p
-
p' (1- U)2 - w 24 u 3.42
giving 4 (u) lm-nl
+ l
Gron - 0 < u < l
p' 2
µµ
l
-
u3.43
In this case the phase factor between molecules m and n is
+l so that w corresponds to a librational motion having the
same symmetry as the acoustic modes. Thus this type of mode
can couple with the acoustic manifold. Then for O <
u
< lwe require
0 <
2 w
2
w
< p> p
if p I > Q
if -p < p' < 0
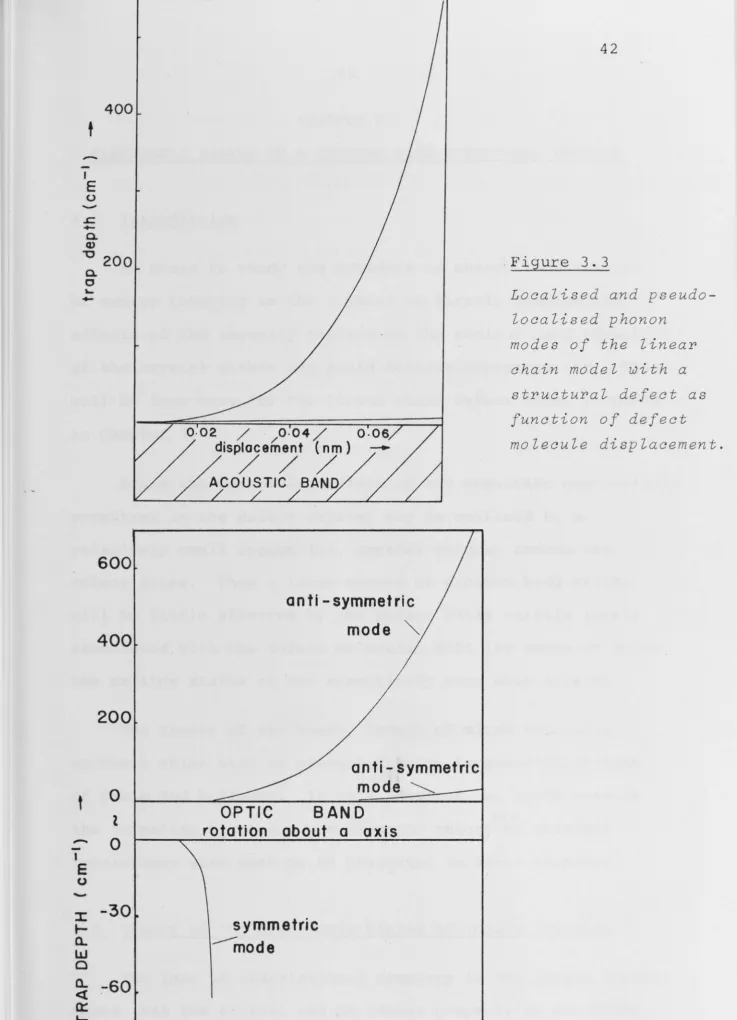
3-4 Phonon Modes for the Defect Crystal
3.44
3.45
Expressions 3.3Z~ 3.38 and 3.43 for the matrix elements
of G may now be used to obtain solutions to 3.11. The case
in which the defect takes the form of a molecular
displace-ment along the chain is of. particular interest for the
present work since this may allow parallel molecules to
approach a more favourable separation for a photodimerisation
reaction to occur. The localised and pseudo-localised mode
energies obtained for the linear chain model with a
trans-lational defect are shown in fig. 3.3 as a function of
molecular displacement for an optic and acoustic branch.
molecule, the force constants of the molecules
perturbed by the defect may be either increased or decreased
from their values in the pure crystal depending on whether
the nearest neighbour spacing is decreased or increased
respectively. For each phonon branch, an increase in force constant results in the creation of an above band state.
Conversely, for a decrease in force constant, states will
emerge from and lie below the optic branches.
A completely localised phonon mode which lies beyond
the range of all phonon frequencies of the perfect lattice
is unlikely to be important in the photochemical process
since i t cannot be responsible for the dissipation of excess
energy from the reaction site, nor can i t participate in any
processes involving the long range transfer of energy to
the reaction site. The pseudo-localised modes, however,
can participate in both these kinds of processes since they
can decay into a continuum of delocalised states through
t
--
IE u
-
.c....
a. Q)
400
,:, 200
a. 0 '-
+-600
400
200
t
0t
-
0
-
I E 0-:I::
-30
....
a..w
Qa.
:-60
<l a::
t-0
OPTIC rotation anti-symmetricmode ""'
anti-symmetric
mode ~
BAND
about a axis
symmetric ---mode
0
·
04
0
·
08
DISPLACEMEN-T (nm)
-Figure 3.3
Localised and
pseudo-localised phonon
modes of the linear
chain model with a
structural defect as
function of defect
[image:51.764.6.743.19.1039.2]CHAPTER 4
ELECTRONIC STATES OF A CRYSTAL WITH STRUCTURAL DEFECTS
4-1 Introduction
In order to study the dynamics of energy localisation or energy trapping in the crystal we firstly examine the
effects of the impurity centres -on the exciton band structure of the crystal within the rigid lattice approximation. This
will be done here for the linear chain defect model proposed
in Chapter 3.
Again the perturbing effect of the resultant non-periodic potential in the defect crystal may be confined to a
relatively small region (cf. crystal volume) around the
defect sites. Thus a large number of exciton band states will be little affected by the defect while certain levels
associated with the defect molecules will lie above or below
the exciton states of the essentially pure bulk crystal.
The theory of the energy levels of mixed molecular
crystals which will be presented below is essentially that 2 7
of Craig and Philpott. It will, however, be given here in
2 8,3 0
the formalism of second quantisation theory to maintain
consistency with work to be presented in later chapters.
4-2 Theory of the Electronic States of Impure Crystals
in terms of the periodic wave functions ¢(k) (or by the
t
corresponding creation and annihilation operators ak, ak).
Therefore, a new set of basis states (labelled by subscript
2 8
v say) must be found which will enable the Hamiltonian
M
operator, H, for the perturbed system to be written in
diagonal form as
E E at a
\) \) \) \) 4.1
where: the E are the new energy levels of the imperfect
\)
crystal, and the new mixed crystal state creation
and annihilation operators at , a can be formed
\) \)
ai
linear combinations of the pure crystaloperators a t , ak
a \)
E
k
.
,
"
*
atL, ukv k
k
4.2
Alternatively at and a can be related to the mixed crystal
\) \)
site operators by the transformation
a
\) E u
nv
n
a
n
.
,
*
E u
nv
n
4.3
with the two expansion co-efficients ukv and unv related by
the Fourier transformation
u
nv
4.4If the impurity site in the present model is labelled
by the subscript x, then the Hamiltonian for the defect
a
X
+ E ,:i
ntx )llxn
t
an a n 4.5
where: HP is the Hamiltonian of the pure crystal and will
be used here in the form
1
E E
N k nn' eik(n-n') E(k) a~ an'
~ is the difference in transition energies between
the impurity and host molecules.
~ is the difference in dispersion energy between
the defect and pure crystal.
¢
andt
are respectively the differences inxn xn
dispersive energy and ·resonance interaction
between the molecule pair x and n in the defect
and pure crystal.
The operators a
\)
t
, av
will be assumed to obey Bosecommutation relations
t
, a ' ] \)
=
cS\) \)
'
4.6Applying these commutation properties to 4.1 gives
[a
\) E \) a \)
The corresponding commutator evaluated with HM given by 4.5
gives