UNIVERSITY TEKNIKAL MALAYSIA MELAKA
STRUCTURAL AND ELECTRONIC PROPERTIES
OF METAL OXIDES ATTACHED TO SINGLE-WALLED CNT
AS LI ION BATTERY ELECTRODE BY USING DFT
This report is submitted in accordance with requirement of the University Teknikal Malaysia Melaka (UTeM) for Bachelor Degree of Manufacturing Engineering
(Engineering Materials) (Hons.).
by
LIEW WENG TACK B051210142 911226-08-5391
ABSTRAK
Bateri Li-ion mempunyai gravimetrik tinggi, ketumpatan kuasa yang tinggi. Mereka telah dibuktikan sebagai penyimpanan tenaga yang paling cekap dalam telefon bimbit, komputer riba dan peranti elektronik. Nanotiub karbon berdinding tunggal dan oksida logam seperti mangan (IV) oksida (MnO2), kobalt (II, III) oksida (Co3O4),
dan nikel (II) oksida (NiO) biasanya digunakan untuk membuat bahan elektrod elektro aktif. Semua pengiraan telah dijalankan menggunakan kajian prinsip pertama berteraskan teori fungsian ketumpatan (DFT) yang telah dilaksanakan dalam kod komputer CASTEP and DMol3 untuk menjalankan simulasi. Projek ini akan memberi tumpuan terhadap kajian interaksi antara oksida logam dengan (5, 5) SWCNT dan ion Li penjerapan untuk oksida logam/SWCNT. Top C atom didapati merupakan kedudukan yang paling stabil untuk NiO dan MnO2 melampirkan pada
SWCNT dengan menunjukkan yang tinggi -3,2001 eV dan -2,8787 eV tenaga serapan. Untuk Co3O4 molekul, Co2+ itu didapati menjadi yang paling stabil
dilampirkan pada SWCNT dengan tenaga penjerapan adalah -2,0245 eV. Selepas NiO dan MnO2 melekat SWCNT itu, jurang band menurun kepada 0 eV. Selepas
Co3O4 dilampirkan pada SWCNT, Jurang band Co3O4 menurun dari 0.43 eV ke
0,091 eV Pengurangan jurang jalur menyumbang kepada peningkatan pengangkutan caj yang lebih tinggi daripada oksida logam sahaja. analisis Hirshfeld daripada NiO / SWCNT, MnO2/ SWCNT dan Co3O4 / SWCNT adalah 0.452e, 0.487e dan 0.434e Li
ion atom masing-masing. MnO2/ SWCNT adalah yang paling berkesan dalam
memindahkan caj.
ABSTRACT
Li-ion batteries have high gravimetric, high power density and low self-discharge property. They proved it as the most efficient energy storage strategy, especially in mobile phone, laptop and electronic devices. Single-walled carbon nanotubes and metal oxides such as Manganese (IV) oxide (MnO2), Cobalt (II, III) oxide (Co3O4),
and Nickel (II) oxide (NiO) are normally applied for the electro active electrode material. All the calculation was performed using a first principle study base on density functional theory that has been implemented in CASTEP and DMol3 were utilized to run the simulation for optimization. The project will focus on the investigation of the interaction between the metal oxide with (5, 5) SWCNT and Li ion adsorption to SWCNT/ metal oxides. The top C atom was found to be the most stable position for the NiO and MnO2 attach on SWCNT with an adsorption energy
of -3.2001 eV and -2.8787 eV. For Co3O4 molecular, the Co2+ was found to be the
most stable attached on SWCNT with an adsorption energy of -2.0245 eV. After NiO and MnO2 attached to the SWCNT, band gaps were drop immediately to 0 eV. The
band gap of the Co3O4 decrease from 0.43 eV to 0.091 eV after Co3O4 attached on
SWCNT. Decrease in the band gap attribute to the enhancement of the superior charge transport than metal oxides alone. Hirshfeld analysis of NiO/SWCNT, MnO2/SWCNT and Co3O4/SWCNT were 0.452e, 0.487e and 0.434e for Li ion atom
respectively. Based on the Hirshfeld analysis, MnO2/SWCNT is the most effective in
transferring charge.
DEDICATION
To everyone that contributes to this research, my family and my friends
ACKNOWLEDGEMENT
First of all I would like to express my gratitude to Prof. Dr. Mohd Asyadi’ Azam Bin Mohd Abid for his advice and encouragement throughout the duration of this research. Without his support, it would be difficult to complete the final year project.
Besides that, not to forget Raja Noor Amalina Binti Raja Seman from UTEM for giving guidance and support to me that enabled me to complete my final year project as well as my research.
Finally, I wish to thank all the parties involved in providing encouragement to me while completing this research, including both my parents and all staffs of FKP for giving me guidance to accomplish my research.
TABLE OF CONTENTS
Abstrak i
Abstract ii
Dedication iii
Acknowledgement iv
Table of Contents v
List of Tables viii
List of Figures ix
List Abbreviations, Symbols and Nomenclatures xi
List of Equations xiii
CHAPTER 1: INTRODUCTION
1.1 Background 1
1.2 Problem Statement 3
1.3 Objective 3
1.4 Scope 4
CHAPTER 2: LITERATURE REVIEW
2.1 Introduction 5
2.2 The Nature of Carbon 5
2.3 Graphene 7
2.4 Carbon Nanotubes (CNTs) 7
2.4.1 Single-Wall Carbon Nanotubes (SWCNTs) 8 2.4.2 Multi-Walled Carbon Nanotubes (MWCNTs) 9
2.4.3 Chirality of SWCNT 10
2.5 Anode for Lithium-ion Batteries. 11
2.6 Type of Metal Oxides 13
2.6.1 Manganese (IV) Dioxide (MnO2) 13
2.6.2 Cobalt (II,III) Oxide (Co3O4) 14
2.6.3 Nickel (II) Oxide (NiO) 15
2.7 Theoretical Study of Carbon Nanotube (CNT) on Electrode. 15
2.8 Computational Molecular Modeling 16
2.8.1 First Principle of Quantum Theory 16
2.8.2 Density Functional Theory (DFT) 18
2.8.2.1 The Hohenberg-Kohn Theorems 18
2.8.2.2 Kohn-Sham Method 20
2.8.2.3 Exchange-Correlation Functional 20
2.8.3 Local Density Approximation (LDA) 21
2.8.4 Generalized Gradient Approximation (GGA) 22
2.8.5 K-Point Sampling 22
2.8.6 Materials Studio Software 23
2.8.6.1 Cambridge Serial Total Energy Package (CASTEP) 24 2.8.6.2 Density Functional Calculation on Molecules (Dmol3) 25
2.9 Adsorption Energy (Ead) 25
2.10 Electronic band structure 27
2.10.1 Band Structure 27
2.10.2 Density of State (DOS) and Chemical Bonding Charges 28 Density.
2.11 Summary of Computational Theory 29
CHAPTER 3: METHODOLOGY
3.1 Introduction 30
3.2 The Computational Material Studio Software 30
3.3 Computer System Specification 34
3.4 Step 1: Building Structure of model 35
3.5 Step 2: Model Refinement 37
3.6 Step 3: Use the CASTEP and Dmol3 to Calculate the Material Properties. 38
3.7 Step 4: Analysis Structure. 40
3.8 Summary 40
CHAPTER 4: METHODOLOGY
4.1 Geometry Properties of Metal Oxides Attached on SWCNT 41
4.1.1 NiO Attached on (5, 5) SWCNT 42
4.1.2 MnO2 Attached on (5, 5) SWCNT 45
4.1.3 Co3O4 Attached on (5, 5) SWCNT 47
4.2 Absorption Analysis. 49
4.3 Electronic Properties 51
4.3.1 NiO/SWCNT Electronic Properties Analysis 51 4.3.2 MnO2/SWCNT Electronic Properties Analysis 53
4.3.3 Co3O4/SWCNT Electronic Properties Analysis 55
4.4 Li ion Absorption on Metal oxides/SWCNT 57
4.5 Population Analysis of Li ion Absorption on Metal oxides/SWCNT. 60
4.6 Summary 61
CHAPTER 5:CONCLUSIONS & RECOMMENDATIONS
5.1 Conclusion 62
5.2 Recommendations 64
5.3 Sustainability Analysis 64
REFERENCES 65
APPENDICES 66
LIST OF TABLES
3.1 3.2 3.3 3.4
Molecular Modelling by First Principle.
The Binding Distance of Metal Oxide before Optimization. The Overall Setup for DMol3 is Listed as Below.
SCF Parameters.
32 37 38 40
4.1 Comparison of Binding energy, Binding Distance and Surface Area in Difference Site NiO/SWCNT.
42
4.2 Comparison of Binding Energy, Binding Distance and Surface Area in Three Difference Site MnO2/SWCNT..
45
4.3 Comparison of Binding Energy, Binding Distance and Surface Area in Three Difference Site Co3O4/SWCNT
47
4.4 Comparison Between the Metal Oxides on the Stable Position Attached on SWNCT
49
4.5 Comparison Stable Position for Li ion Attached on Metal Oxides/SWCNT.
57
4.6 Hirshfeld analysis for Li ion Attached on Metal Oxides/SWCNT. 60
LIST OF FIGURES
2.1 Bonding Structure of Diamond, Graphite, Nanotube and Fullerene.
6
2.2 Carbon Family Tree Known Carbon Allotropes. 6
2.3 The Structure of Graphene. 7
2.4 2.5 2.6 2.7 2.8 2.9 2.10 2.11 2.12 2.13 2.14
CNT Formation by Rolling Up the Graphite Sheet. Convert of Graphene to SWCNTs.
Formation of MWCNT from Graphene.
CNT Configurations with the Chiral Vector C and Unit Vectors a and b.
Armchair. Zigzag.
Working Principle of Li-Ion Batteries.
Schematic Illustration of Active Anode Materials for the Next Generation.
Possible Applications of Metal Oxides. Homogeneous Gas Concepts.
The Time and Size in Materials Multi-Scale Characterization.
8 9 9 10 11 11 11 12 13 22 25
2.15 Position of Metal Oxide Attached on SWCNT 26
2.16 Metal Semiconductor and Semimetal 27
2.17 Electronic Energy Varies with the Wavevector.a) (5, 5) Nanotube b) (9, 0) Nanotube c) Zigzag (10, 0) Nanotube
28
2.18 DOS Plots of (a,c)C10+TiO2 + H2 (b,d) C10 + 8(TiO2 + H2) 29
3.1 3.2 3.3 3.4 3.5 3.6
General Steps for Quantum Modelling. Main Steps in Quantum Modelling. Flow Chart of Quantum Modelling. Document is Created on Material studio. Select the Structure Chirality.
Nickel Oxide (NiO).
3.7 3.8
Manganese dioxide (MnO2).
Cobalt (II, III) oxide (Co3O4).
36 36
3.9 Position of Metal Oxide Attached on SWCNT. 37
3.10
3.11
Step for Optimization by Using the Dmol3 Calculation on the Material Studio.
Parameter Setup in DMol3 and CASTEP.
37 39 4.1 4.2 4.3 4.4 4.5 4.6 4.7 4.8
Sites of Metal Oxides Attach on Pristine (5,5) SWCNT Top/Ni-C Bridge/ Ni-C Bridge/parallax Ni-C Centre/Ni-C Centroid/O-C Top/Mn-C Bridge/Mn-C 42 44 44 44 44 44 46 46 4.9 4.10 4.11 4.12 4.13 4.14 4.15 4.16 4.17 4.18 4.19 Centroid/Mn-C Co2+-C
Co3+-C O2--C
NiO/SWCNT (a) Band structure (b) DOS MnO2/SWCNT (a) Band structure (b) DOS
Co3O4/SWCNT (a) Band structure (b) DOS
The Three Possible of Li ion Attached on Metal Oxides/SWCNT.
Before Optimzation and After Optimzation of Li Adsorption on Site-B of NiO/SWCNT
Li Adsorption on MnO2/SWCNT (a) Site-U (b) Site-B:
Li Adsorption on Site-U of Co3O4/SWCNT
LIST OF ABBREVIATIONS, SYMBOLS AND
NOMENCLATURES
1D - One dimensional
2D - Two dimensional
3D - Three dimensional
µm - Micrometer
H� - Hamiltonian operator
Ψ - Wavefunction
Å - Angstrom
a - lattice vectors
BZ - Brillouin-zone
MnO2 - Manganese (IV)oxide
Co3O4 - Cobalt (II, III) oxide
NiO - Nickel (II) oxide
LIB - Lithium ion batteries
C - Carbon
CASTEP - Cambridge serial total energy package
Ch - Chiral vector
CB - Conduction band
CNT - Carbon nanotube
CO - Carbon monoxide
CVD - Chemical vapor disposition
DFT - Density functional theory
DFPT - Density functional perturbation theory DMol3 - Density functional calculation on molecules
DOS - Density of state
E - Energy
Ex - Exchange energy
e - Electron charge
Ead - Adsorption energy
KS-DFT - Kohn–sham density functional theory
eV - Electron volt
FFT - Fast Fourier transform
F(R) - Atomic force
GGA - Generalized gradient approximation
HCP - High performance computer
LDA - Local density approximation
nm - Nanometer
M - Electron mass
MS - Materials studio
MWCNT - Multi-walled carbon nanotube
N - Ion Mass
P - Momenta of ions
PC - Personal computer
PW91 - Perdew and Wang
R - ion corrdinates
r - Radius
r1 - dummy integration variable
rc - Cut-off radius
SCF - Self consistence field
SWCNT - Single-walled carbon nanotube
USP - Ultra-soft pseudopotential
VB - Valance band
Viz. - Visualizer
WDA - Weight density approximation
XC - Exchange-correlation
Z - Charge of ion
LIST OF EQUATIONS
2.1 Ch= na1+ma2 = (n,m) 10
2.2 𝜓(𝑡) = 𝐶1𝜓1 (𝑡) + 𝐶2𝜓2(𝑡) 17
2.3
2.4
2.5
2.6
2.7
2.8 2.9
2.10
2.11 2.12
H�Ψ= EΨ
𝑉𝑛𝑒[𝑃(𝑟)] = � �|𝑟 𝑍𝑘
1− 𝑟2| 𝑝(𝑟)𝑑𝑟 𝑛𝑢𝑐𝑙𝑒𝑖
𝑘
𝑉𝑒𝑒[𝑃(𝑟)] =12 �𝑝(𝑟1)𝑝(𝑟𝑟 2)
1 𝑑𝑟1𝑑𝑟2
𝐸𝑥[𝑝(𝑟)] = −9𝛼8 (𝜋)3 1/3� 𝑝4/3(𝑟)𝑑𝑟
F(R)=-∂E(R)/∂R
𝐸 𝐿𝐷𝐴𝑋𝐶 (𝑃) = � 𝑃(𝑟)ℇ𝑋𝐶ℎ𝑜𝑚[𝑝(𝑟)]𝑑𝑟
𝐸𝐿𝐷𝐴𝑋𝐶 (𝑃) = � 𝑃(𝑟)ℇ𝑋𝐶𝐺𝐺𝐴[𝑝(𝑟), 𝛻𝑝(𝑟)]𝑑𝑟
Eads= (ESWCNT+ Emetal oxides) – E metal oxides/SWCNT
Eads= (E metal oxides/SWCNT + ELi ion) – E metal oxides /SWCNT - Li ion
17
18
19
19 19
20 21
22
26 27
CHAPTER 1
INTRODUCTION
1.1 Background study
According to Goriparti et al. (2014), Li-ion batteries have high gravimetric, high power density and low self-discharge property. On the other hand, they proved it as the most efficient energy storage strategy, especially in mobile phone, laptop and electronic devices.
Nanotechnology is known as any process or product that has 1 to 100 nanometer dimensions. Since 1990s, there is a rapid growth in research interest of nanotechnology, including improved effectiveness of nanoscale electronic devices. (Pandyan, 2012).
The discovery of carbon nanotubes (CNTs) has been interested considerable attention since they have a wide range of uses in nanometre-scale application. The study of carbon nanotubes (CNTs) is greatly encouraged as to convert them into much useful application. CNT comprises of single-walled (CNTs) and multi-wall CNTs. Recently, SWCNTs become essential for most of the electronic application such as data storage, chemical sensors and reinforcing agents (Lim et al., 2008; Li et al., 2015). As a result, recent advancements in nanotechnology have provided opportunities to enhance the electronic properties in transfer charge.
Carbon nanotubes have been as a preferred used as the electrode material due wide electrochemical-stability window, a highly accessible surface area which is can improve energy storage capability in Li-ion batteries. This was proven as the large
increase in reversible capacity up to 1000 mA h/g (Goriparti et al., 2014; Ado et al., 2007).
To improve the performance, metal oxides such as Manganese (IV) oxide (MnO2),
Cobalt (II, III) oxide (Co3O4), and Nickel (II) oxide (NiO) are normally apply to the
electro active electrode material pseudocapacitors. Normally, MnO2-Based
Nanocomposites used for high-performance supercapacitors (Wang et al., 2015). The amorphous MnO2 has excellence performance pseudocapacitive in the
electrochemical field since it has an ability to make the charge storage mechanisms (Yu et al., 2013).
Quantum calculation using density functional theory (DFT) has provided a chance prescribes the initial and the fundamental properties in a material. DMol3 implement DFT to construct insight into the electronic structure molecular and structural aspects molecular. The first principle calculations is used the least amount of energy and the data with cost efficient compared to the experimental laboratory work which will help in term of satisfactory and inexpensive in large scale material design (Ahmadnezhad et al., 2015).
The project will focus on the investigation of the interaction between the metal oxide with (5, 5) SWCNT and Li ion adsorption to metal oxides/SWCNT. In this case, zigzag (5, 5) CNT is chosen since it has high stability metallic wave function. This causing the band opening or metal insulator transition. Metal-insulator transition contributes to the good electrical conductivity and makes it suitable to be use in the lithium ion batteries.
1.2 Problem statement
The limitations of typical graphite electrode are lower volumetric capacity, low Li ion diffusion and low electrical conductivity. To improve graphite electrode, many studies have been carried out so that CNTs can be implemented in lithium ion batteries application. However, the resistance of CNTs array is very high. This causes delay in the electron transport and heat generated in lithium ion batteries. The problem is believed solved when metallic oxides mixed with CNTs. This is due to its huge specific surface area and the role of supporting. Anyway, the most stable structure between CNTs and metal oxides is still an unknown until today.
Therefore, this project will explain the best way of metal oxide attaches to the surface of CNTs and how the Li+ ion attached on the metal oxides/SWCNTs. Thus, there is also investigation in the amount of Li+ ion that can be ion insertion/removal of the SWCNTs/metal oxide. CNT is preferred as electrode in lithium-ion batteries because it can increase Li+ ion insertion/removal rates. Nowadays, CNTs upon other molecules was highly recommended as to improve the electrochemical energy storage and conversion.
1.3 Objectives
1. To optimize the most stable atomic/molecule structure of pristine (5, 5) single walled carbon nanotubes (SWCNTs), metal oxides and Li ion.
2. To investigate the adsorption and electronic properties of metal oxides attached to (5, 5) SWCNTs.
3. To analyze the adsorption and electronic properties of Li ion attached to SWCNTs/metal oxide system.
1.4 Scopes
This project will discuss the structural stability, geometrical configuration and electronic properties of highly adsorption energy metal oxide on SWCNTs. To do this, CASTEP and DMol3 will be carried out based on their first principle and DFT.
The electronic property of SWCNTs are divided into band structure and density of state (DOS). The density functional theory is based on computational numerical base was used to analyse the electrical properties like density of state and band structure to find the most stable of metal oxide deposition on the surface of SWCNTs. Also, the brief explanation about the basis of using computational as to design a new material base on the quantum theory will be discussed.
Chapter 1 is to provide an overview of the project by describing the importance of CNTs as the electrode on lithium-ion batteries and explain the density functional theory (DFT) with a numerical radial function basis set to calculate the electronic properties. Subsequently, it will be used to recognized problem statements and respectively solutions will be provided.
Chapter 2 will explain about the CNTs and the first principle study of ion absorption on the electrode. Then, DFT has also been discussed which to start from the Schrodinger equation until energy cut-off, using a basis set of plane wave with Generalized Gradient Approximate (GGA) and Local Density Approximate (LDA) exchange correlation.
Chapter 3 illustrates the methodology and first principle calculation. Material Studio Visualizer will be used to build the structures of SWCNT and metal oxide deposition on SWCNTs. DMol3 and CASTEP computer modules are used to collect the information such as energy absorption and electronic properties.
CHAPTER 2
LITERATURE REVIEW
2.1 Introduction
This chapter will briefly discuss about the fundamental understanding of carbon nanotubes (CNTs) and their molecular structure followed by an overview of metal oxides attached on the SWCNTs. Next, discussion will be emphasizing on the density functional theory (DFT) with first principle study. It investigates the electronic properties of material such as band gap, density of state and rate of charge transfer.
2.2 The Nature of Carbon
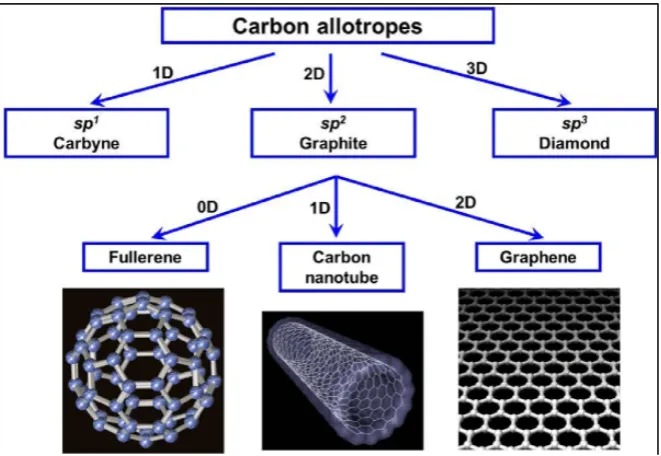
Carbon is an element having atomic number 6 and its symbol is C. From the periodic table, carbon is the most resourceful element due to the strength, type and number of bonds (O’Connell, 2006). Different bonding of carbon state will present into certain structural arrangement which is sp bonding will be growth to sp2 bonding of one dimensional chain structure (Zhang, 2012). According to Meyyappan et al.,(2004), sp2 bonding of graphite will create planar hexagonal network and sp3 bonding produce to tetrahedral structure. The Figure 2.1 and Figure 2.2 show the bonding structure of diamond, graphite, nanotube and fullerene.
Figure 2.1: Bonding Structure of Diamond, Nanotube and Fullerene (Badding, and Crespietal, 2004).
Figure 2.2: Carbon Family Tree known Carbon Allotropes (Zhou, 2015).
D'Souza and Kadish, (2012) said that allotropes are difference structural modifications of an element. Carbon can form different molecular configuration with the allotropes such as graphite, nanoform, amorphous carbon, fullerene and CNT.
[image:20.595.155.486.253.481.2]2.3 Graphene
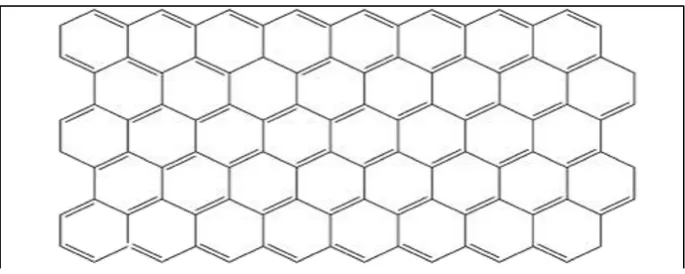
[image:21.595.149.492.443.580.2]The strong crystal built of graphene plane was identified for the first time by using the transmission electron microscopy. The strong covalent bond on the graphene is difficult to remove by using chemical substances, but it has weak van der Waals’s bond on the c-axis stacking which is easy to remove (Lagrange et al., 2015). According to Li et al. (2015) and Alfano et al. (2016), they said that graphene is a single sheet of sp2 hybridized carbon atom, which is formed in a honeycomb lattice. Graphene honeycomb lattice is shape by six member ring as shown on Figure 2.3 and it has high performance in application such as high capacity of electrode in lithium batteries and sensitive gas sensor (Rad, 2015). Li et al. (2015) stated the graphene has high performance in application is due to the unique properties of graphene such as higher mechanical properties, electronic properties and surface area higher than 2600 m2g−1. Other than that, graphene also has electronic properties like weak spin-orbit coupling and electron-hole symmetry near the charge neutrality point (Grande et al., 2013). Graphene is produced by reducing graphene oxide have low surface area because it has strong tendency to form agglomeration (Xiong et al., 2015).
Figure 2.3: The Structure of Graphene (Zhu et al., 2014).
2.4 Carbon Nanotubes (CNTs)
In 1991, CNTs were identified by Sumio Iijima at the NEC Research Laboratory by using the electric arc discharge to synthesis the fullerenes. CNTs have the sp2 and sp3 bonding structure. Based on this structure, CNTs has a stable covalent bond
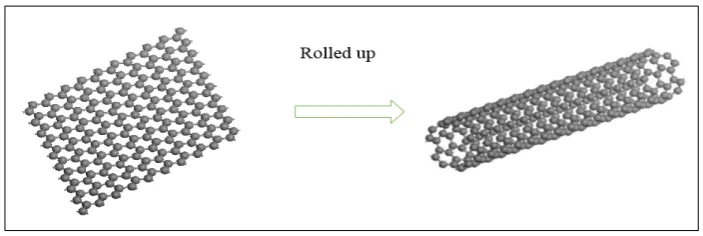
between the carbon atoms (Loiseau et al., 2006). CNTs are hollow cylinders. It is made by rolling graphite sheet into a cylinder to obtain various diameters and different microscopic structure of the tube (Reich et al., 2005). Figure 2.4 shows the formation of CNT by rolling up the graphite sheet. (Kharlamova, 2013).
[image:22.595.144.496.338.456.2]CNTs are an ideal material for essential application such as energy storage and thermal conductors due to the properties of CNTs which is good mechanical, electrical and thermal properties (Liu et al., 2008). CNT can be synthesized by a few methods such as laser ablation, arc-discharge and various chemical vapour deposition (CVD) which can affect the physical properties of CNT such as wall diameter and number of walls (Tagmatarchis, 2012). CVD can provide large quantities of CNT and low cost. SWCNTs and MWCNTs can be determined by the number of walls of CNT (Liu et al., 2011).
Figure 2.4: CNT Formation by Rolling Up the Graphite Sheet (Kharlamova, 2013).
2.4.1 Single-Walled Carbon Nanotubes (SWCNTs)
SWCNTs are unique molecular, there has different physical properties depending on the chirality of SWCNTs. The first SWCNTs were produced by the electric arc which is evaporation of cobalt catalyst and graphite (Hu et al., 2015). SWNTs typically have a diameter between 0.4 nm to 2 nm. However, the tube length can be many thousand times longer than its diameter in order of 1 µm to 100 µm (Thurakitseree et al., 2012). Each carbon atom attached with three neighbors carbon atom which is forming a hexagonal arrangement. The hexagonal arrangement will reduce the number of carbon dangling bond between each other (Tian et al., 2011).
For photovoltaic applications, SWCNTs of single chirality show better performances compared to SWCNT mixtures of metallic and semiconducting chiral species (Liu et al., 2012).
.
Figure 2.5: Convert of Graphene to SWCNTs (Visu et al. 2014).
2.4.2 Multi-Walled Carbon Nanotubes (MWCNTs)
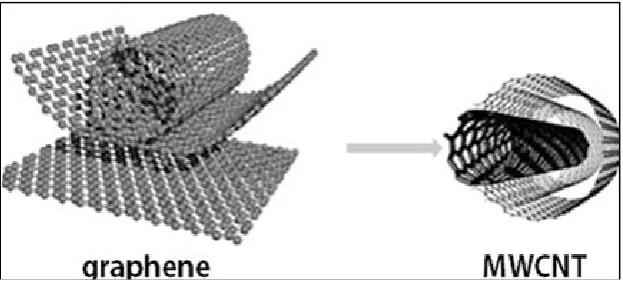
[image:23.595.165.477.578.719.2]MWCNTs contain two or more graphene layers that been wrapped into multiple layer to each CNT with van der Waals forces (Lehman et al., 2011). MWCNTs have attracted attention because of the unique electronic, mechanical properties and chemical stability (Visu et al., 2014). The MWCNT are more resistant to electrochemical oxidation and the chemical bonding of MWCNTs is sp2 bond which is same to graphite (Shao et al., 2012). The high level electron mobility and electrical conductivity of MWCNTs allow them to support on electrochemical application (Wan et al., 2015).
Figure 2.6: Formation of MWCNT from Graphene (Visu et al., 2014).
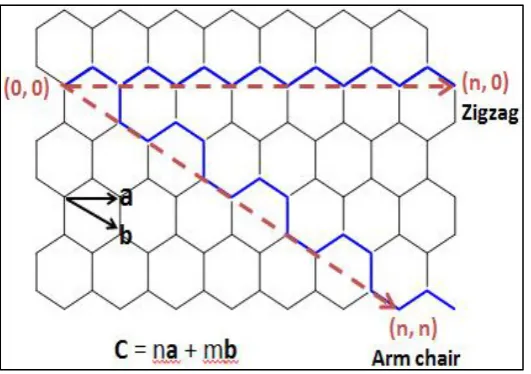
.2.4.3 Chirality of SWCNT
[image:24.595.189.455.214.401.2]SWNT has unique electronic properties which is depending on the changed chirality vector C = (n, m) of SWNT (Choi and Yong Zhang, 2012). CNTs has three different categories depending on the folded (zone-folding scheme) of the graphitic planes such as armchair, zigzag and chiral (Jasti and Bertozzi, 2010).
Figure 2.7: CNT Configurations with the Chiral Vector C and Unit Vectors a and b (Jia Choi and Yong Zhang, 2012).
According Meyyappan (2005), the chiral vector can be determining the nanotube’s electronic structure. The equation 2.1 (below) can obtain the diameter of CNT.
Ch= na1+ma2 = (n,m) Eq 2.1
Where
a1 and a2 are the lattice vectors of graphene,
m and n are integers.
When n-m of nanotube is a multiple of 3 are metallic which is known as the 1/3 rule. The CNT does not meet this condition is a semiconducting nanotubes which is inversely proportional to the nanotube’s diameter. It is mean that 2/3 of CNTs are semiconducting on Zigzag. For “Armchair” CNTs (n,n) are metallic (Lim, 2007).