JOURNAL OF VIROLOGY, Oct.1976,p.86-95
Copyright©1976 American Society for Microbiology
Vol. 20, No. 1 Printed inU.S.A.
Isolation
and Structural Characterization
of
Monomeric
and
Dimeric Forms of Replicative Intermediates
of
Kilham
Rat
Virus
DNA
M. GUNTHER* ANDP. MAY
Unite deBiophysique, Institut de Recherches ScientifiquessurleCancer, B.P. No.8-94,800Villejuif,France
Received for publication19February1976
Two virus-specific species of newly synthesized DNAwere isolated fromrat fibroblast cellcultures infected with the Kilhamratvirus (RV).ThesetwoDNA specieswere purified; their behaviouron hydroxyapatitechromatography and
theirsedimentation coefficients insucrose gradientswere determined. One of
thetwospeciescorrespondstothe lineardouble-strandedform of the RV DNA, and the other corresponds to the dimeric duplex form. After denaturation, a
fractionof both species showedanintramolecularrenaturation; these molecules arecomposed of viral strandcovalently linkedtocomplementary strand. Models
forthe structure of both speciesareproposed. Both speciesmaybeconsideredas
double-stranded replicative intermediates of the single-strandedRV DNA.
Kilhamrat virus(RV),oneof the small icosa-hedric single-stranded DNA (ssDNA)virusesof theparvovirus group, wasfirstisolated froma rat sarcoma by Kilham (6). (The recognized notationRV will beusedhere, althoughthere areatleast three other knownparvovirusesof therat.) RVDNAhas beendemonstratedtobe linear(15) and singlestranded (8, 11, 13), and
tohave amolecular weight of about 1.6 x 106 (8, 11, 13).
Vertebrate parvoviruses fall into two
subgroups: (i) parvoviruses, including RV, H,
virus, and minute virus ofmice, which have been shown to replicate autonomously in rap-idly dividing cells (21)andtobe dependentfor theirreplicationupon acellular event(s)inthe late S phase (9, 16); and (ii) adeno-associated
viruses, whichcannot replicate in cells unless anadenovirus"helper"ispresent(12). Plus and minusstrands ofadeno-associated virus DNA havebeen showntobecoatedseparately(2).
Thestudy ofparvovirusreplication may pro-videamodel for thereplication of ssDNA ina
mammaliansystemand for thereplicationofa
linearDNAmolecule. It isknown thatssDNA ofseveralparvoviruses isconvertedintoa dou-ble-stranded formafter infection(10, 14,20).
Inthispaper, twospecies ofdouble-stranded forms of RV DNA found in infected cells are
described. Thelength andthe structureof these molecules werestudied, andtheirpossiblerole intheRV DNAreplicationisbrieflydiscussed.
MATERIALS AND METHODS
Cell strains and virus. The KilhamRV used in
these experiments wasobtained from R. Tournier
and originally derived from W. Rowe's strain. RV was produced in Wistar AF rat embryo primary
culture. Rat embryos (17 daysold)weretrypsinized
andseeded inbottles of a tissue culture Rollacell
(New Brunswick Scientific Co.) in 120 ml of Eagle
medium supplementedwith10%tryptose phosphate
broth and 10% calf serum (Sorga). Infected cells
(multiplicity of infection, 0.1 to 1 PFU/cell) were
collected 2 days after infection and frozen and
thawed three times. Cellular debris was then
treatedtwice inhypotonicmedium(double-distilled H20). The supernatantscollectedbylow-speed
cen-trifugation were combined and made isotonic by
addition of double-concentrated culture medium.
This viral suspension wasused as a stock virus after
titrationby the plaque assay technique on
second-arycultures of ratembryocells.
Intracellular synthesis ofviral DNA wasstudied
in aratfibroblast strain(RTcells)obtainedfrom a
rat embryo cell culture. (Some RT cells have been
found [41tobecontaminated byanewparvovirus,
the "RT virus." For the experimentsdescribedhere,
different passages ofroutinely cultured RT cells
weretested by the technique of Hallauer et al. [4;
HAtiter ofglycine buffer extracts] and foundfree of
virus. Insome rare cases a contaminant viruswas
foundindegenerating cultures, but this was always
identified by inhibition hemagglutination tests as
theRVvirus.We cannotruleoutthepossibilitythat
this contaminant virus comes from the RV virus
usedinthislaboratory.) RT cellsweremaintained
intheculture mediumsupplemented with 5% calf
serum inFalconplasticbottles(75 cm2).Studies of
viral DNAsynthesis werecarriedout inpetri dishes
(60-mmdiameter)with thesamemedium.
Viruspurification.Infected cellswerelysedwith
1%sodium deoxycholate and treated for18hat37°C
with50,ug of DNase I per mland5 ,ug of RNaseIll
(Sigma)per ml in10mMMgCl2. Themixture was
adjustedto0.01%trypsin(Choay)andincubated for 86
on November 10, 2019 by guest
http://jvi.asm.org/
REPLICATIVE INTERMEDIATES OF RAT VIRUS DNA
30 min at 370C. The virus was then purified by
isopycniccentrifugationinCsCl (1.4g/cm3)at35,000
rpm for 18h at 25°C in aSpinco SW50.1rotor.
Viral DNA extraction. Viral DNA was extracted
by sodium dodecyl sulfate (SDS) and purified
ac-cordingtotheprocedureofMayandMay (8). Viral
DNA was alsoeasilyprepared byextraction in 0.1 M
NaOH for 20 min at room temperature.
Purification of DNA from infected cells.Growing
RT cells (1.5 x 105 to 1.8 x 105 cells/cm2) were
infected with RV at amultiplicityof infection of 5
PFU/cell. At various timespostinfection (p.i.),cells were labeled with tritiated thymidine ([3H]Tdr, 5
,uCi/ml, 20Ci/mM; Saclay) forperiods of 1 to 3 h.
Low-molecular-weight 3H-labeled DNA was then
extracted byan SDS-selective procedure according
to Hirt(5),exceptthat thelysatewasincubatedin
the presence of 100 yg of pronase per ml for 30 min at
370C. Similar results were obtained when pronase wasaddedbefore or afterremovingthe high-molec-ular-weightDNAbycentrifugation. The 3H-labeled
DNAfrom the Hirt supernatantwasdeproteinized
bytwochloroform treatmentsandthenpurifiedin an equilibrium cesium chloride density gradient
(CsCl,1.72g/cm3)at35,000 rpm at250C for48h in a
Spinco65rotor, eitherdirectlyor afteran ethanol
precipitationovernightat-20°C.Fractionsfrom the
majorpeak ofradioactivity (Fig. 1) were collected
and dialyzed against
tris(hydroxymethyl)amino-methane (Tris)-EDTA buffer (0.01 MTris,0.001 M
EDTA,pH 8). The purification wascompletedbya
phenol extraction at room temperature, and DNA wasdialyzedagainst the samebuffer.
Alkaline denaturation of DNA. The 3H-labeled
DNA wasincubatedfor 10 min at roomtemperature
in 0.1NNaOHandthenneutralizedwith 0.1 NNa
H2PO4 unlessspecifiedotherwise.
Hydroxyapatite chromatography.
Hydroxyapa-tite chromatography of purified 3H-labeled DNA,
either native ordenatured,wasperformedaccording
tothemethodofTapieroetal. (19). Portions of
3H-labeledDNA preparations weredilutedto4ml with 0.01Mphosphate buffer, pH7.85, and mixed with 0.25g ofhydroxyapatite (DNAgrade;Bio-Rad) and
incubated for 5 min at room temperature.
Alkali-denaturedDNA (100 ,ul) wasdiluted with4 ml of
0.01Mphosphate buffer before neutralization with
NaH2PO4. The hydroxyapatitewaspelletedby
low-speed centrifugation,and the supernatantwas
col-lected. Hydroxyapatite was then washed by the
sameprocedure with 0.01 Mphosphatebuffer, pH
7.85.Stepwise elutionofthe DNA wasperformed by
resuspending the hydroxyapatite pelletin 4 ml of
phosphate bufferof increasingmolarities (from 0.1
to0.7M). The tubes were incubated in a water bath
at560Cfor 8 min with intermittent agitation.
Hy-droxyapatite was againpelleted by low-speed
cen-trifugation at room temperature, and the
superna-tants were collected. Carrier DNA (calf thymus;
Choay)wasadded to eachstep-elutionfraction, and
coldtrichloroacetic acid was added to a final
concen-tration of 5%. After an incubation for 10 min in an
ice-waterbath, the acid-precipitable material was
collected on glass fiber filters (Whatman GF/C).
Eachtube was washed with 4 ml of 5%
trichloroace-tic acid, which was poured onto the filters. The
filters weredried, andtheradioactivity was
deter-minedin anIntertechniqueliquid scintillation spec-trometer.
DNA-DNA hybridization. Purified 3H-labeled
DNA wasdenaturedandfragmented into piecesby
boilingfor 15 min in 0.01 x SSC(SSCis 0.15 MNaCl
plus0.015 sodiumcitrate). At this stage, the DNA
sedimented in an alkaline sucrose gradient as a
uniformbandcorresponding toabout 9S. The DNA
wasthenreannealed in 100 ul ofTris-EDTAbuffer
containing 0.3 M NaCl at650Cwithdifferent
quanti-ties ofunlabeledRVDNA (up to 880 ng).After48h
ofincubation,two40-ul portionsdesignatedAand B
were taken and treated separately. Portion A was
diluted to 0.4 ml with Tris-EDTAbuffer, and the
total radioactivity of A was counted in the cold
trichloroacetic acid-precipitable material. For
por-tionB, onlythe radioactivity remaining in a
double-strandedform was counted in the cold
trichloroace-tic acid-precipitable material after digestion of
ssDNAby theS, nuclease fromAspergillus oryzae
preparedaccording to Vogt (22). Portion B was
di-luted to 0.4 ml in abuffer containing 0.03 Msodium
acetate, pH 4.6, 0.3 MNaCl, 0.001 M ZnSO4,and5%
glycerol. Heat-denaturedcalfthymus DNA (20 ,ug/
ml) and 5 U ofS, nuclease per ml were added.The
mixture was then incubated for 1 h at450C. The
results are given as the percentage of the
radioactiv-ityremaining in portion B as compared to that in
portionA.
Velocity sedimentationinsucrose gradients.
Por-tions (up to 0.2 ml) of the purified 3H-labeled DNA
sampleswere layered on 4 ml of a 5 to 20% linear
sucrosegradient. Neutral sucrose gradients in 1 M
NaCl and Tris-EDTA buffer were centrifuged at 200C for 150 min at 54,000 rpm in a Spinco SW56
rotor. Foralkaline sucrose gradients the 5% sucrose
solution was made pH 13 in 0.3 N NaOH, 0.7 M
NaCl, and Tris-EDTA buffer, and the 20% sucrose
solution was made pH 13 in 0.6 N NaOH, 0.4 M
NaCl, and Tris-EDTA buffer. The alkaline sucrose
gradients were thencentrifuged at200Cfor 180 min
at54,000 rpm. Gradientswere fractionated by
col-lectingdrops from the bottom of the tube directly
ontoglass fiber filters. Filters were washed twice
with cold 5% trichloroacetic acid and then with
ethanol. Radioactivitywasdetermined by counting
driedfilters in a liquid scintillation spectrometer.
RESULTS
Presence of two new species of 3H-labeled DNA in RV-infected cells. RV-infected rat fi-broblasts (RT cells) werelabeled with [3H]Tdr (5
ACi/ml)
for1hp.i.
tovariouslengths
of time. After the labeling period, they were lysed by SDS.Theradioactivitywasthencountedinthe trichloroacetic acid-precipitablematerial of the SDS-pronase Hirtsupernatants (see Materials andMethods). Undertheseconditions, incorpo-ration of[3H]Tdr was first detected at 6 h p.i. and thenbecame linear as a function of time between 9and 18 h p.i. and reached a plateau (data not shown). Under the same conditions, incorporation of[3H]Tdr in the trichloroacetic 87VOL. 20, 1976
on November 10, 2019 by guest
http://jvi.asm.org/
88 GUNTHER AND MAY
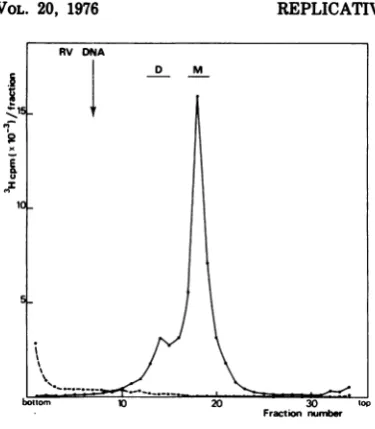
acid-precipitablematerial of the Hirtpelletsof infected cells reached a plateau at 6 h p.i. In most ofthe experiments reported here, DNA was labeled at 12 h p.i. for theindicated period, then extracted, and purified as described in Materials and Methods. As shown later, the DNA of Hirt supernatant labeled at 12 h p.i. was essentially virus specific. The last step of the DNA purification was an isopycnic centrifu-gationgradientinCsCl(Fig. 1).The bulk of the radioactivity banded as a single peak slightly
heavier than that of cellular DNA (the
differ-ence in densities was about 0.005 g/cm3),
whereas verylittle radioactivity was found in the region ofthe RVssDNA. Thestudy ofthe
3H-labeled DNA contained in the major peak
will be reported in this paper. Purification of 3H-labeledDNAbyadensitygradient centrifu-gationreducedcontamination
by
cellularDNA. The radioactivity remaining at the top of the gradient corresponded to 3H-labeled DNA linked to viral proteins in spite of the SDS-pronase treatment performed as described inMaterials and Methods. (Progeny ssDNA was
always foundinthisform.)The
study
ofthese DNA-protein complexeswillbedescribed in asubsequentpaper.
The3H-labeledDNA thuscollected from the preparativedensity
gradient
(Fig. 1) wasana-FRACTION NUMBlER
FIG. 1. Preparative isopycnic centrifugation of
newly synthesized DNA in RV-infected cells.
RV-infectedRT cells (multiplicity of infection, 5PFU/
cell) werelabeled with [3H]Tdr (5 pACilml) at12 h
p.i. foraperiod of1h. 3H-labeled DNAfromtheHirt
supernatant(purifiedasdescribed)wassubmittedto
anequilibrium density gradient centrifugation in 6
mlofCsCl (1.72 g/cm3) at35,000 rpmfor48 h at
20°C in aSpincorotor65. Fractions werecollected
fromthe tube by bottompuncture. Portions(10 d)
fromeachfraction werespottedonfilters,andtheir
radioactivitywas counted. Portionswere also taken
tomeasuretherefractiveindex. Arrows indicate the
positions ofcellularDNA and RV DNA added as
markers inaparallel gradient.
lyzed by velocity sedimentation in a 5 to 20%
neutral sucrose gradient. The pattern of sedi-mentation illustrated in Fig. 2 shows twopeaks ofradioactivity that were not found in mock-infected cells. The major peak (M)accounts for 80% of theradioactivity, and the minor (D) for 20%. To study these two species of DNA, frac-tions M and D were separately pooled and puri-fied by an additional sucrose centrifugation.
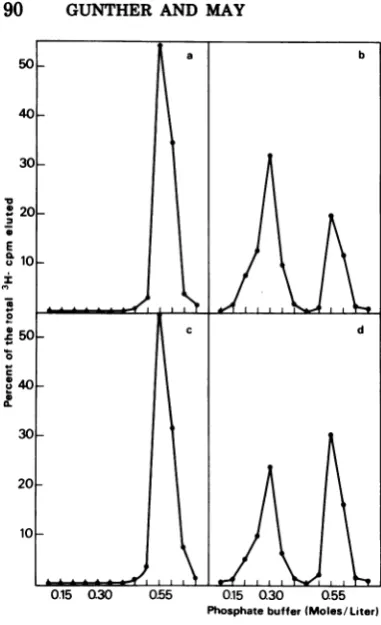
Hydroxyapatite chromatography. Elution pattems fromhydroxyapatite chromatography of RV DNA and of DNA species M (DNA-M) andD(DNA-D) were studied. Under the exper-imental conditions used (see Materials and Methods), native dsDNA was eluted from
hy-droxyapatitewith 0.55 M phosphate buffer, pH 7.85, and denatured DNA with 0.15 M phos-phate buffer. These characteristics were illus-trated in our experiments with rat cellular DNA (Fig. 3a, b). By contrast, RV 3H-labeled DNA was eluted with 0.3 M phosphate, which wasunexpected for ssDNA (Fig. 3c). Consider-ing that by increasConsider-ing concentrations of phos-phate buffer the elution from hydroxyapatite
mainly depends on thesecondary structure of the DNA (1), this result strongly suggests that RVDNA has aspecialstructure. A short
palin-dromic region could account for the value ob-tained for the molarity of elution. Since minute virus of miceDNA has been found to contain a
small duplex region (2 to 5%) ofthe viral ge-nome (G. J. Bourguignon, P. J. Tattersall, and D. C. Ward, Abstr. Int. Congr. Virol., 3rd, p. 181, 1975), we also studied the behavior of this DNA in hydroxyapatite chromatography and observedanelutionpattern similartothat ob-tained with RV DNA (data not shown). More-over, apalindromicsequence hasalso been
sug-gestedfor the terminal segments of
adeno-asso-ciated virus DNA strands on the basis of the results of Koczot et al. (7) and Gerry et al. (3).
The DNA species M and D were elutedfrom hydroxyapatite with 0.55 M phosphate,
indicat-ing a double-stranded structure, whereas no radioactivity was found at either 0.15 or 0.3 M phosphate (Fig. 4a and c). Alkaline-denatured DNAofspecies MandDhavesimilarpatterns
ofelution fromhydroxyapatiteasshowninFig. 4 (b and d). Both DNA species, M and D, are resolved into two different peaks. One peak
elutes at 0.3 Mphosphate, and the otherpeak
at 0.55 M phosphate (about 30and 50%of the totaleluted radioactivityforspeciesMandD, respectively). Similar results were obtained with DNA that was heat denatured in Tris-EDTA buffer at 100°C for 3 min and then quickly cooled. It isimportantto stressthat0.3 Mphosphateisthetypicalmolarityof elution of
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.508.70.257.390.528.2]REPLICATIVE INTERMEDIATES OF RAT VIRUS DNA
RV DNA RV ssDNA and that 0.55 M phosphate is the
J
ffi D M molarity of elution of dsDNA. These results
J1
4
suggest that3H-labeled DNA of both species M.~,15_IF and D is doublestranded. A denaturation
treat-ment converts a fraction of both species to ssDNA that is eluted at 0.3 Mphosphate as RV
0fi
l DNA, whereas the remaining molecules areelutedatthemolarity correspondingtodsDNA.
10-_
lThe
experimental conditions of denaturationused are assumed to prevent intermolecular renaturation of DNA (low ionic strength, low DNA concentration, and room temperatures).
s-
lWe
willshowinthefollowing
experiments
that the DNA which is eluted at 0.55 M phosphate afterdenaturationderivesfromintramolecular"snap-back" renaturation.
I6y
\Hybridization.
To examine the viralspecific-bo,tom
ity
of3H-labeled DNA extracted fromRV-in-nwn,mFractinr
fected rat cells, species M and D were heat denatured in 0.01x SSC and reannealed in 0.3 FIG. 2. Sedimentation in a neutral sucrose gra- MNaCl at 65°C with increasing quantities of dientofaHirtsupernatant DNAfrom RV-infected or unlabeled RV DNA. After 48 h the DNA re-mock-infectedcells. 3H-labeled DNA was layered onthe top of a 5 to20%sucrose gradient in 1 MNaCI
matring
ss was degraded bySp
nuclease, andand Tris-EDTA buffer and centrifuged at20°Cfor trichloroacetic acid-precipitable radioactivity 150 min at 54,000 rpm in a Spinco SW56 rotor. wasdetermined (see Materials and Methods). If
Fractions were collectedfrom thebottomof the tube we assumethatspeciesM and Darecompletely
directly on glass fiber filters, and the radioactivity of homologous to RV DNA, which contains only
dried filters was counted. The arrow indicates the the"viral strand," saturatingconcentrations of
sedimentation position ofRV DNA addedasmarker unlabeled RV DNA would be expected to
dis-(24S under the conditions used). Symbols:
*,
3H- place all viral strands from species M and Dlabeled DNA collectedfrom the major peak, corre-
(i.e.,
50%
oftheradioactivity,
assuming thatsponding to the horizontal bar ofFig. 1; 0, 3H-
3H-labeled
DNA is equally labeled on both labeledDNA of a Hirt supernatantfirom
mock-in- 3fected cells labeled for 1 h andextracted asdescribed strands). The 3H-labeled DNA extracted from
for infectedcells.FractionscorrespondingtoDNA-M RV-infected cells could thus be considered as
and -D werecollected separatelyfrom a similargra- 100% virus specific. As already indicated, a
dient. fraction of the alkaline-denaturedDNA species
a b c
E
06
X,
2030, ,Q15 Q30 0.55 Q.15 030 0.55 0.15 0.30 Q55
[image:4.508.53.241.60.275.2]Phosphate buffer)Moes/Liter)
FIG. 3. Hydroxyapatitechromatographyofpurified3H-labeledcellular and RV DNA. 3H-labeled DNA in
0.01 Mphosphate buffer, pH 7.85, was adsorbed at room temperature to 0 25 g ofhydroxyapatite.Stepwise
elution wasperformed at56Cbyincreasing molarities ofphosphate buffer, pH 7.85 (from01 to0.7 M), as
described inMaterials and Methods, and the radioactivity of thetrichloroacetic acid-precipitable material of
each eluate was counted. (a) NativecellularDNA; (b)alkali-denaturedcellular DNA; (c) RV DNA.
VOL. 20, 1976
89
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.508.104.394.460.612.2]90 GUNTHER AND MAY
Phosphatebuffer(Moles/Liter)
FIG. 4. Hydroxyapatite chromatography of
3H-la-beled DNA-Mand-D, by the method described in
Fig. 3. (a) Native DNA-M; (b) alkali-denatured
DNA-M; (c) native DNA-D; (d) alkali-denatured
DNA-D.
M and D undergoes intramolecular renatur-ation. The denaturingconditions used for hy-bridization experimentswerechosen toreduce this intramolecular renaturation by reducing themolecular weight. The DNAspeciesMand D were converted to ssDNA of a molecular
weight of4 x 105 (25%of thegenomelength) by
boilingDNA 15minin0.01x SSC. Denatured
DNAspeciesMand D showedanS1 resistance
of 9% in the absence ofreannealingtreatment. After reannealing, 3H-labeled DNA species Mshowedan
S,
resistanceof 92.2 + 7.5%(mean value + standard deviation forseven independ-entdeterminations). When increasing quanti-ties ofunlabeled RV DNA were added, this percentage first decreased and finally leveledoffat47%with 200ngof unlabeled DNA.This
valueremainedessentially unchangedwith
ad-dition ofincreasingamountsof RV DNAupto
880ng(Fig. 5).WhenexcessheterologousDNA was added to the incubation medium, the
amount ofreannealing of 3H-labeled DNA-M
was about 80% (Fig. 5). Thismean value was
obtained with unlabeledDNAfrom RT(rat)or
CV1 (monkey) cells and from SV40. To take intoaccountthisnonspecific hybridization, the experimental value ofrenaturationofDNA-M obtained with excess unlabeled RV DNA was
multiplied byacorrectingfactor of100/80,and we thus obtained a corrected value of 60%.
Thus,40%ofthelabeled DNA-Mwasdisplaced
by the unlabeled RV DNA, corresponding to
80% homology for this species. This value is underestimated ifwe consider that 9% of the
DNA is resistant to an
S,
nuclease digestionwithout reannealingtreatmentandisprobably counted with the DNA remaining double stranded after renaturation. Similar results
wereobtained with 3H-labeled DNAspeciesD,
except that 3H-labeled DNA-D reannealed to theextentofonly75.6 + 14.7%(meanvalue +
standard deviation obtained with seven
inde-pendent determinations). Purified 3H-labeled DNA-Dpreparationswerealwaysinlower
con-centrations than 3H-labeled DNA-M
prepa-tions.Thus, reassociation conditionswere
prob-ablynotoptimal, since the standarddeviation
wasrelatively high. When unlabeledRVDNA was added in excess to DNA-D, better
condi-Unlabelled DNA added(ngl
FIG. 5. Variations of the percentage of
renatur-ationof3H1-labeledDNA-M as a functionof
increas-ing quantities of unlabeled RV DNA. 3H-labeled
DNA-M labeled from 12 to 15 hp.i. was denatured in
0.01x SSC fori5min at 100°C and then reannealed
in 100 ,ul of 0.3 MNaCI for 48 h at 65°C in the
presence of varying quantities of unlabeled DNA.
Each assay wasperformed with 10 ng ofDNA-M. At
the end of thereannealing period two equal portions,
designated A and B, were taken; the radioactivity
was counted directly in the trichloroacetic
acid-pre-cipitable material ofportion A, and the radioactivity
ofportion B was counted in thetrichloroacetic
acid-precipitable material after anS1nuclease digestion.
All results are expressed as the percentage of the
countsper minute remaining indsDNAafter theS,
nuclease digestion (portion B) as compared to the
total counts per minute(portion A). Unlabeled DNAs
added: *, RVDNA; A\,RTDNA;*,CV1 DNA; 0,
SV40 DNA.
J.VMOL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.508.67.258.60.373.2] [image:5.508.273.465.346.487.2]REPLICATIVE INTERMEDIATES OF RAT VIRUS DNA
tions of concentration wereobtained. The per-centageof renaturation ofDNA-D inthe pres-enceof 100 and 200 ng ofunlabeled RVDNA was 46 and45%, respectively. These valuesare similar to the corresponding values obtained with DNA-M. Both species appear to possess the same extent of virusspecificity(over80%).
Sucrose sedimentation. Species M and D of
3H-labeledDNAwerestudiedseparatelyin su-crose gradients (Fig. 6). Sedimentation coeffi-cients were 14.5S for DNA-M and 18.3S for DNA-D ascompared to the 16Ssedimentation coefficient of SV40 form II DNA added as a marker (Fig. 6a, d).Since the species M andD have been observed as linear molecules with
the electron microscope (M. Gunther, H. Bu-jard, and G. Hayward, manuscript in prepara-tion), it waspossible tocalculate themolecular weight of each species. To obtain an accurate evaluation of the molecular weight of DNA-M, we compared, in the same gradient, the sedi-mentationproperties of DNA-Mandthelinear form ofSV40 DNA obtained after Hpa II endo-nuclease digestion (data not shown). DNA-M sedimentedslightlymoreslowly: the difference in sedimentation coefficients was0.4S, which
corresponded to a molecular weight of 3.15 x 106for DNA-M, with a molecular weight of 3.4 x 10c for SV40 DNA. The ratio (molecular
weightofDNA-D/molecular weight of DNA-M)
Fractionnumber
FIG. 6. Sucrose sedimentations of 3H-labeled DNA-M and -D. Neutral sucrose sedimentations were
per-formedasdescribedinthe legend ofFig. 2. Alkaline sucrose gradients (pH 13) were centrifuged at 20°C for
180minat54,000 rpm in a SpincoSW56 rotor. The 3H-labeled DNA-M sedimented in (a) neutral, (b) neutral
(after an alkaline denaturation), and (c) alkaline 5 to 20% sucrose gradients. The 3H-labeled DNA-D
sedimentedin(d) neutral, (e) neutral (after an alkaline denaturation), and (f)alkaline 5 to20%sucrose
gradients. In gradients b, c, e, and f, RV"4C-labeledDNA was added as a marker.SV40
"4C-labeled
DNAform IIwasadded as a marker in parallel gradients. 16S arrows in neutral sucrose gradients represent the
sedimentation position of SV40 form II, and in alkaline sucrose gradients arrows16S and 18S represent the
positionsof the circular and linear strands derived fromSV40 DNA formI.
VOL. 20, 1976
91
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.508.132.362.239.579.2]92 GUNTHER AND MAY
wascalculated from the experimental
sedimen-tation coefficients 18.38 and 14.5S by using Studier's formula (18). This ratiowas 2
(calcu-lated value, 1.94). Thus, the DNA of species M
appearstocorrespondtothemonomeric ds form
of RV DNA, and the DNA of species D to a
dimer of species M.
Both DNA-M and -D were alkaline
dena-tured and studied separately in alkaline and neutral sucrose gradients (Fig. 6b, c, e, f). An
alkaline sucrose gradient of DNA-D (Fig. 6f)
showedtwopeaks whose sedimentation coeffi-cientscorrespondedto20.38 and 16S. The16S peak co-migrated with RV 14C-labeled DNA
markerand the20.3Ssedimentation coefficient found for the secondpeakwasclosetothe
coeffi-cientexpected (21S)for thecorresponding sin-gle-stranded dimer. Thus an alkaline sucrose
gradient of DNA-Dgaveessentiallytwospecies
of ssDNA inamolecularweight ratio of 2
(cal-culated value, 1.86). An alkaline sucrose
gra-dientof DNA-M showed thesametwopeaks of sedimentation(Fig. 6c), whichwillbe discussed indetail later. Inaneutralsucrosegradient of
denatured DNA-M, two peaks were obtained
withcoefficients of24Sand 14.5S (Fig. 6b). As
shown later, the 24S peak, which co-migrates with RV DNA, corresponds to the single-stranded monomer form, and the 14.5S to a
double-stranded snap-back DNA of monomer
length. Under the same sedimentation
condi-tions, DNA-Dgaveamajor peakat14.5Sanda
shoulder ofradioactivity at 24S, i.e., the posi-tion of RV ssDNA (Fig. 6e). Radioactivitywas notfoundatthe position for thesingle-stranded dimer (whose expected sedimentation
coeffi-cient would beranging about5OS). Thus, inan
alkaline sucrose gradient (Fig. 6c, f),the
sedi-mentation patterns of denatured DNA from
both DNA-M and -D showedtwo bands
corre-sponding to the single-stranded monomer and
thesingle-stranded dimer;inaneutralsucrose
gradient (Fig. 6b and e) the sedimentation
pat-ternsofdenaturedDNAfrom both DNA-M and
-Dshowedtwobandscorrespondingtothe
sin-gle-stranded monomer and to an additional
componentsedimentingat14.5S.
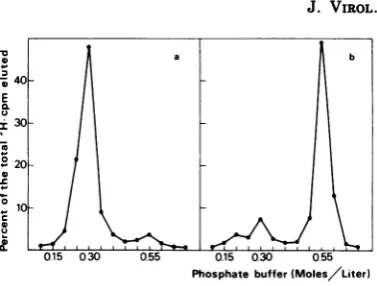
The fractions(A, B, B', C, and C') indicated
inFig. 6werepooled and examinedon
hydroxy-apatite and in sucrose gradients. The DNA
frompool Awasessentially single stranded,as
indicatedby its molarity ofelution(0.3M phos-phate)inhydroxyapatitechromatography (Fig. 7a) and resedimented asthe RV DNA in
neu-tral or alkaline sucrose gradients (data not
shown). The DNA from pools B and B' was
elutedwith 0.55Mphosphatefrom hydroxyapa-tite (Fig. 7b); i.e., it behaved as dsDNA and
sedimentedat20.3Sinanalkalinesucrose
gra-~4C
,,, 40 E
3CL
I30
20
010
- 10
D
0.15 030 0.55 0.15 0.30 0.55 Phosphate buffer (Moles/Liter)
FIG. 7. Profile of elution from hydroxyapatite of
3H-labeled DNA from pools A and B corresponding
to horizontal bars inFig. 6. The method isas
de-scribedinthe legend ofFig. 3. (a) PoolA; (b) pool B.
dient (Fig. 8a). Thus the DNA from pools B and B'appearedtobe doublestranded; this DNA in neutral sucrosebehaved as adouble-stranded
monomer and in alkaline sucrose as a
single-stranded dimer. DNA from pools C and C', i.e.,
an"alkaline"single-stranded dimer, renatured
when neutralized, and then it was eluted at
0.55 M phosphate from hydroxyapatite (data
notshown) and sedimented at 14.5S in a
neu-tralsucrosegradient, asexpectedfora
double-stranded monomer (Fig. 8b). These results
show that the rapidly renatured DNA arose
fromanalkaline single-stranded dimer. To
ac-countfor theseobservations,we supposethat in
such a single-stranded dimer, both strands,
viral andcomplementary, arelinked byan al-kali-stable covalent link, giving rise, when neutralized, to a hairpin structure. Molecules whichsnapbacktothis hairpinstructurehave the characteristic monomer length. On the
basis of thisobservation, we proposestructures
for themonomeric and dimeric species (see Dis-cussion).
DISCUSSION
In this work,we have shown thepresenceof
two species of DNA in RV-infected rat cells afterlabeling with[3H]Tdr. The DNAobtained by an SDS-pronase selective extraction from
RV-infectedratcells andcentrifuged inaCsCl
gradient formed a band slightly heavier than
that of theratcellular DNA (Fig. 1).The DNA in this band, when sedimented in a neutral
sucrosegradient,wasresolved intotwodistinct
peaks, M and D (Fig. 2). Both species were
shownto bedoublestranded by their molarity ofelution (0.55 M phosphate buffer) during hy-droxyapatite chromatography (Fig. 4) andtobe
atleast 80%virusspecific by displacement hy-bridization experiments with saturating
con-centrations ofunlabeled RV DNA. Analysis of
a b
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.508.272.461.58.201.2]REPLICATIVE INTERMEDIATES OF RAT VIRUS DNA
10 20 30 40 50 60 70 10 20 30
Fractionnumber
FIG. 8. Sedimentationof3H-labeledDNAfrompoolsBand C(correspondingtohorizontal bars inFig.6).
(a)Alkalinesucrosegradient of3H-labeled DNAfrom poolB (a); RV "4C-labeled DNA was added as a
marker(0);(b) neutral sucrosegradientof3H-labeledDNAfrom pool C (0); "4C-labeledDNA-Mwasadded
as amarker(0).
thetwo speciesbysucrosegradient
centrifuga-tion with various DNA markers (SV40 DNA form II, SV40 DNA linear form III, and RV DNA) showed that species M of 3H-labeled
DNAcorresponded to the linear duplex form of RVDNA,with amolecularweightofabout3 x 106, whereas species Dcorrespondedtothe lin-eardimeric duplexof species M.
The resultsfrom sucrosesedimentation anal-ysis (Fig. 6and 8), whencombinedwith those
obtainedbyhydroxyapatiteexperiments (Fig.4
and7), haveshown that a fraction of both spe-cies snaps back to a hairpin monomer after
denaturation. This propertyleadsus to propose
thefollowingstructuresfor the DNAspeciesM
and D(seeFig. 9 and 10).
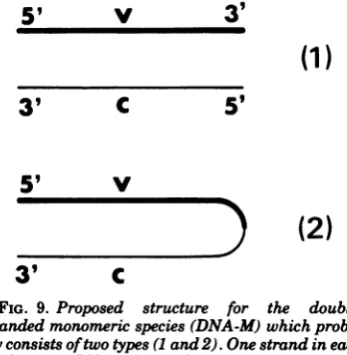
Monomeric DNA preparations probably con-sistof a mixture of two types (1and2) of
double-stranded molecules (Fig. 9). These structures are consistent withthe behavior ofdenatured
monomericdsDNA. Themonomergives riseto twokindsofsingle-strandedmolecules in
alka-line sucrosegradients:one(of monomer length) comes from structure (1) and the other (of di-merlength) comes from structure(2); the latter snaps back when neutralized into a hairpin monomer.
A large proportion (about 50%) of dimeric moleculesspontaneously renatured, after dena-turation, to hairpin monomeric molecules. Di-meric molecules must have a structure such that viral and complementary strands are
5'
V
3
'
(1)
3'C
59
5'V~) (2)
[image:8.508.45.452.59.285.2]3'
C
FIG. 9. Proposed structure for the
double-strandedmonomericspecies(DNA-M)which
proba-blyconsistsoftwotypes (Iand2).Onestrand in each
duplexin anRV viral strand (v), and the other is the
complementary strand (c). In type 2, v and c are
covalently linked,forming a hairpinmolecule.
5'
V C3'
39 C V
5'
FIG. 10.Proposed structure for the
double-stranded dimeric molecules (species D). Each linear
strand in the duplex is composed of one RV viral
strand and one complementary strand, which are
covalently linked end to end.
93
VOL. 20, 1976
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.508.267.441.344.522.2] [image:8.508.259.450.566.663.2]94 GUNTHER AND MAY
linked by an alkali-stable covalent link, as rep-resented in the scheme in Fig. 10.
This is inaccordance with the fact that radio-activity was not detected in neutral sucrose gradients in the region theoretically
corre-spondingto asingle-strandeddimer. After neu-tralization single-stranded dimers were always found in the region of monomeric dsDNA. A fraction of denatured dimeric DNA did not snap
back and behaved as single-stranded mono-meric molecules in neutral or alkaline sucrose
gradients. Thus, it is likely that a fraction of
dimeric moleculesmusthaveanickat(ornear)
the middleofthestrand(s).
There is another possible structure for the
dimer. Dimeric molecules might arise by the joiningbyhydrogen bondsoftwohairpin mono-mers, occurring either ininfected cells or dur-ingthe DNAextraction-purificationprocedure.
Suchastructureneedsaterminalredundancy
if the joining occurs between a viral and a
complementary strandandapalindromic struc-ture at the ends of the strands ifthejoining
occursbetween twoviral-ortwocomplementary strands. The existence of the latter structure cannotberuledout,althoughitseems
unlikely
if we assumethat dimericmoleculesplayarole inthe RV DNAreplication.
In conclusion, two new species of virus-spe-cificdsDNAwerefoundinRV-infectedratcells: onespecies(the more
abundant)
ismonomeric (a one-unitlength);theother isdimeric (adou-ble-unit length). Both species contain
cova-lently linkedviraland
complementary
strands (Fig. 9 and 10), whichstrongly
suggests thattheyare synthesizedby a
self-priming
mecha-nism. A similar hypothesis wassuggested by
Tattersalletal., whoreportedthe existence ofahairpinmonomerform of intracellular minute virusof mice DNA(20).
Recently,
Strausetal. (17)identifiedreplicative intermediatesincellsinfected with adeno-associated virus type
2,
whichconsist ofcovalently
linkedplus
and mi-nus DNAstrands.These authors haveproposed
areplicationscheme that involvesa
self-prim-ingmechanism. Itappearslikelythat the
repli-cation ofdifferent parvovirus DNAisa unidi-rectional and self-priming process. Further studies are necessary to obtain a detailed scheme of RV DNA
replication.
We are pres-ently studying the relationship between the replicative intermediates andtheirrole inthe synthesis of progeny ssDNA.RV DNAreplica-tionappearstoinclude differentsteps:
conver-sion of parental ssDNA to a double-stranded form then synthesis of some other double-stranded replicative intermediates
(of
mono-meric and dimeric length), one
replicative
in-termediate species acting as a template to prog-eny ssDNA. If every step requires a
self-prim-ing process, RV DNA must have a hairpin du-plex structure at both ends of the viral strand.
The behavior of RV DNA on hydroxyapatite (Fig. 3c) and preliminary results obtained with
S1 nuclease digestion suggest that RV DNA contains a short duplex region(s), which is in accordance with recent results from L. Salzman (Virology, in press). Further studies on RV DNAconcerning the structure of the ends of the molecules and the ability of RV DNA to be utilized as a primer template, as has been shown with minute virus of mice DNA (Bour-guignon etal.,Abstr. Int. Congr. Virol., 3rd, p. 181, 1975), arerequired.
ACKNOWLEDGMENTS
We wish to thank G. Hayward for his helpful discus-sions, and F. Koczot, N. Salzman, and P. Sheldrick for a critical reading of the manuscript. We also thank J. Rose forthe preprint ofreference 17 (Straus et al.), L. Salzman for thepreprint of her article inVirology, P. Nardeux for a gift of Hpa II endonuclease-cleaved SV40 DNA, and P. Tattersall for a gift of14C-labeled minute virus of mice DNA. We acknowledge M. 0. Blondel for her excellent technical assistance.
LITERATURE CITED
1. Bernardi, G. 1965. Chromatography of nucleic acids on hydroxyapatite. Nature (London)206:779-783. 2. Berns, K.I.,andJ. A. Rose. 1970.Evidencefor a
single-strandedadenovirus-associatedvirus genome: isola-tionand separationofcomplementary singlestrands. J. Virol. 5:693-699.
3. Gerry,H.W.,T.J. Kelly,and K. I. Berns. 1973. Ar-rangementofnucleotidesequences in adeno-associ-atedvirusDNA.J. Mol.Biol.79:207-225.
4. Hallauer, C., G. Kronauer, and G. Siegi.1971. Parvovi-ruses as contaminant of permanenthuman cell lines. I.Virusisolationfrom 1960-1970.Arch.Gesamte Vi-rusforsch.35:80-90.
5. Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369.
6. Kilham,L., and L. J. Olivier. 1959. A latent virus of ratsisolatedin tissueculture. Virology 7:428-437. 7. Koczot,F.J., B.J. Carter,C. F.Garon, and J. A. Rose.
1973. Self complementary of terminal sequences within plus andminusstrandsofadeno-associated virusDNA. Proc.Natl. Acad. Sci.U.S.A. 70:215-219. 8. May,P., andE.May. 1970.The DNA of Kilham rat
virus. J.Gen. Virol. 6:437-439.
9. Rhode,S. L., III. 1973. Replication process ofthe parvo-virus H-1. I. Kinetics in aparasynchronous cell sys-tem.J.Virol. 11:856-861.
10. Rhode, S. L., III. 1974. Replication process ofthe parvo-virusH-1. II. Isolation and characterization of H-1 replicative form DNA. J. Virol. 13:400-410. 11. Robinson, D. M., and F. M. Hetrick. 1969.
Single-strandedDNA from theKilhamrat virus.J. Gen. Virol. 4:269-283.
12. Rose,J. 1974. Parvovirus reproduction, p. 1-51. In H. Fraenkel-Conrat and R. Wagner (ed.), Comprehen-sivevirology,vol.3.PlenumPublishing Corp., New York.
13. Salzman,L.A., and L. A.Jon.1970.Characterization ofthe Kilham rat virus. J. Virol. 5:114-122.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
REPLICATIVE INTERMEDIATES OF RATVIRUS DNA
14. Salzman, L. A., and W. White.1973.In vivo conversion of the single-stranded DNA of the Kilhamratvirus to adouble-strandedform. J. Virol.11:299-305. 15. Salzman,L.A.,W. L.White, and T. Kakefuda 1971.
Linear, single-stranded deoxyribonucleic acid iso-lated from Kilhamratvirus.J.Virol. 7:830-835. 16. Siegl,G., andM.Gautschi.1973.The multiplicationof
parvovirus Lu HI inasynchronizedculturesystem.I.
Optimum conditions ofvirusreplication.Arch. Ges-amteVirusforsch. 40:105-118.
17. Straus, S. E., E. D. Sebring, andJ. A. Rose. 1976. Concatemers of alternating plus andminus strands are intermediates in AAV DNA synthesis. Proc.
Natl. Acad. Sci.U.S.A. 73:742-746.
18. Studier, F. W. 1965. Sedimentation studies of the size
andshape of DNA. J. Mol. Biol. 11:373-390. 19. Tapiero, H.,M. N. Monier, D. Shaool, and J. Harel.
1974.Distributionof repetitioussequencesinchick
nuclear DNA. Nucleic Acids Res.1:309-322. 20. Tattersall, P.,L. V.Crawford, and A. J. Shatkin. 1973.
Replication of the parvovirusMVM.H.Isolation and
characterization of intermediatesinthe replication of the viral deoxyribonucleic acid. J. Virol. 12:1446-1456.
21. Tenant,R.W., and R. E.Hand.1970.Requirement of cellularsynthesisfor Kilham ratvirus replication. Virology 42:1054-1063.
22. Vogt, V.M. 1973. Purification and further properties of single-strand-specificnuclease from Aspergillus ory-zae.Eur. J. Biochem. 33:192-200.
VOL. 20, 1976
95
on November 10, 2019 by guest
http://jvi.asm.org/