0022-538X/91/084204-07$02.00/0
Copyright© 1991, AmericanSocietyforMicrobiology
Intragenic
Suppression of
a
Deletion Mutation of the
Nonstructural
Gene of
an
Influenza A Virus
JOHNJ.
TREANOR,'*
ROSALYNBUJA,1
AND BRIAN R. MURPHY2InfectiousDisease Unit, Universityof Rochester, 601 ElmwoodAvenue, Box689, Rochester, New York14642,'and Laboratory ofInfectious Disease, NationalInstitutesof Health, Bethesda, Maryland208922
Received 6February1991/Accepted 23 April 1991
Theinfluenza A/Alaska/77 (H3N2) virus mutant 143-1 is temperature sensitive (ts) due to a spontaneous
in-frame36-nucleotide deletionin thenonstructural(NS)genesegment,which leadstoa12-amino-aciddeletion
in the NS1 protein. In addition, it has a small-plaque phenotype on MDCK cell monolayers. However,
phenotypicallyrevertant(i.e.,ts+)viruseswereisolatedreadily following replication of the143-1 virusbothin vitroandin vivo. In ordertodetermine thegenetic mechanism bywhichescapefromthetsphenotype occurred,
weperformed segregational analysisandfoundthatanintrasegmentalsuppressormutationcaused the loss of
thetsphenotype.Nucleotidesequenceanalysisrevealed thepresenceofanintragenicmutation in each of the ts+ phenotypic revertantviruses, involving a substitutionof valine for alanine at amino acid 23 of the NS1 protein.Thismutation resulted inacquisition ofthets+phenotypeand also in thelarge-plaque phenotypeon
MDCKcells,characteristic ofthewild-typeA/Alaska/77parentvirus.Thisaminoacidsubstitutionispredicted togeneratean areaofalphahelixinthe secondarystructureoftheamino-terminal portionofthe NS1protein of the revertant viruses whichmaycompensatefor loss ofanalpha-helicalregion duetothe deletion of amino
acids66 to 77 in theNS1 proteinof the 143-1 virus. The genome of influenza A virus consists of eight seg-ments of single-stranded negative-sense RNA encoding at least10 viral polypeptides (11). The NSgene-segmentis the smallest of thesegene segments and encodes two polypep-tides found in infected cells but not in purified virions, referredtoasNS1 and NS2. Fewtemperature-sensitive (ts) orconditionally lethal mutations of the NSgenesegmentare
available, andrelatively littleisknownregardingtheroles of theNS1 and NS2proteins ininfluenza virus replication.
Recently, an influenza A virus with an unusual NS gene segmentwasisolated duringreassortmentbetween the cold-adapted (ca) influenza A/Ann Arbor/6/60 virus and the wild-type(wt) influenzaA/Alaska/77 (H3N2) virus (13). The reassortantvirus, denoted CR43-3, derived the hemaggluti-nin (HA) andneuraminidase (NA) gene segmentsfrom the influenzaA/Alaska/77 virus and the PB1, PB2, PA, nucle-oprotein (NP), and matrix protein (M) gene segments from the ca influenza A/Ann Arbor/60 virus, but the NS gene segment did not appear to be derived from either parent virus. Nucleotide sequence analysis subsequently
demon-strated that the NSgene segmentof the CR43-3 virus was
derived from that ofthe A/AlaskaI77 virus by an in-frame
36-nucleotide deletion, which ledtoapredicted deletion of
aminoacids 66to77oftheNS1protein (2).TheCR43-3virus
was ts, witha shutofftemperature for plaque formation of 37°C, andmanifested ahostrangephenotype notseenwith
eitherparentvirus, characterized by restrictedreplication in Madin-Darby canine kidney (MDCK) cells compared with replicationinprimary chick kidney (PCK) cells(13).
Inordertoevaluatespecifically thecontribution of the NS deletiontothephenotypes of the CR43-3 virus,abackcross
mating of this virus with the A/Alaska/77 wt virus was
performed (23). Reassortantvirus 143-1was isolated, which
derived the NSgenesegmentbearing the deletion mutation fromtheCR43-3virusand all othergenesegmentsfromthe
*Corresponding author.
A/Alaska/77wtvirus. The143-1 virus wasts, withashutoff temperature for plaque formation of37°C, but had only a
partial-host-range phenotype, manifestedby a reduction in
the size of the plaques formed on MDCK cells. These
phenotypes could also be transferred with the NS gene
segment from the 143-1 virus to the wt influenza A/Be-thesda/85 virus(23).
The significant degree oftemperature sensitivity associ-ated with the deletion in the NSgene segmentof the 143-1 virus suggested that this gene segment could be used as a
genetically stable component ofa live attenuated influenza virus vaccine. Therefore, further studies to evaluate the 143-1 virus inanimals with a 37°C body temperaturewere
performed. However,the143-1virusreplicated efficientlyin hamsters andchimpanzees, suggestingthat thetsphenotype of this virus was host dependent (23). In addition, viruses which had recovered the abilityto grow in MDCK cells at 40°C (i.e., ts+ viruses) were isolated from these animals,
suggestingthat the defect inreplication imposed by the NS deletion could be compensated for, most likely by one or
moresecond-site mutations(23). Analysis ofsuch mutations
mayprovide insightinto the structuralrTequirementsfor NS
gene function orinto possible interactions with other gene
products. Therefore,in thecurrentstudy,wedeterminedthe
nucleotide sequence of the NS gene segment of the 143-1 virusandanalyzedthe mechanisms bywhich the ts
pheno-type ofthe 143-1 virus was lost duringin vivo and in vitro
passage.The results of these studies suggestthat the tsand plaque-size phenotypes of the 143-1 virus can be directly attributed to the deletion in the NS gene and that loss of thesephenotypes can occurby intragenic suppression bya
pointmutation.
MATERIALS AND METHODS
Viruses and cells. The isolation, biological cloning, and characterizationof thewtinfluenzaA/Alaska/77 (H3N2)and A/Bethesda/85 (H3N2) viruses and of the 143-1 virus con-4204
on November 10, 2019 by guest
http://jvi.asm.org/
SUPPRESSION OF INFLUENZA VIRUS DELETION MUTATION 4205
tainingthe NS gene segment bearing the deletion mutation have been described previously (21, 23). The ts virus
C1332-8was isolated from the nasopharynx of seronegative chimpanzee 1332 on the eighth day following intratracheal inoculation with the 143-1 virus. The ts+ virus C1331-5 was isolated from the nasopharynx of seronegative chimpanzee 1331 onthefifth day following intratracheal inoculation with the 143-1 virus (23). The ts+ virus H2-7 was isolated from nasal turbinates of a cyclophosphamide-treated hamster on the seventh day following intranasal inoculation with the 143-1 virus (23). All viruses were isolated on MDCK cells (23). Each of these viruses was biologically cloned by
plaque-to-plaque passage in MDCK cells at 33°C and
ex-panded by a single passage in 9-day-old embryonated hen eggs.
The 143-1 virus was also subjected to serial passage in MDCK cellsatgraduallyincreasingtemperatures. The 143-1 virus was first biologically cloned by plaque-to-plaque pas-sageand growninMDCKcellsat33°C. This seed pool was thenseriallypassaged in aMDCK monolayer culturewith a
fluid overlay at38, 39, and 40°C. Cell culture supernatants
from wells inoculated with the highest dilution of virus that
induced cytopathic effect at the highest temperature were used for the nextpassage until a virus which grew equally well at40°C as at 33°C was isolated after four passages. This
virus, denoted P4-40 (passage 4 at40°C), was then
biologi-callyclonedby plaque-to-plaque passagein MDCK cells at
40°C and studied further. As a control, a parallel series of
passages ofthe 143-1 virusat 33°Cwerealso performed.
Efficiency of plaque formation. The level of temperature
sensitivity ofthewild-type viruses, the 143-1 virus, and the revertantviruses wasdeterminedby plaque assay on MDCK cells as described previously (19). The shutoff temperature
forplaqueformation wasdefinedasthelowest temperature at which a greater than 100-fold reduction in plaque titer relative to that at 33°C occurred. Viruses with a shutoff temperatureof39°Corless wereconsidered ts.
Genetic reassortment. A backcross of each of the ts+ isolates of the 143-1 virus with the A/Bethesda/85 wt virus was performed by previously described methods (19, 23). Briefly, confluent MDCKmonolayers werecoinfected with theA/Bethesda/85 virus andeitherthe 143-1virusor one of
the ts+ isolatesat a
multiplicity
of infection ofapproximately
5 for each virus. After a 24-h incubation at 33°C, the cell culture supernatant was plaqued on MDCK cells at
33°C.
Individualreassortantplaqueswerepicked,and theparental origin of the viral gene segments was determined as de-scribed below. Reassortant viruses
deriving
the NS gene segment from the 143-1 parent virus or the ts+ derivative were further plaque passaged and then tested for thetem-perature-sensitive phenotype as described above. Reassor-tantviruses exhibitinga shutoff temperature of39°Corless wereconsidered temperature sensitive.
Genotype analysis. Individual virus
plaques
wereinocu-lated directly into 9-day-old embryonated hen eggs. Viral
RNAwas
purified
from allantoicfluid,
and the genotypeofeachreassortantviruswasdeterminedbycomparisonof the
migrationof the viral RNA gene segments with those of the parent virus RNA gene segments on 2.6%
polyacrylamide
gels containing 4.5 or 6 M urea after silverstaining
of thegels, asdescribed
previously
(26).Nucleotidesequenceanalysis. The nucleotide sequences of the NS gene segments of the A/Alaska/77 wt
virus,
the ts 143-1virus, and the ts+ isolates were determinedby
directdideoxynucleotide
sequencing
of cDNAcopies
of the NSgene segmentgenerated by polymerasechain reaction
(PCR)
(10). Briefly, approximately 1 pug of purifiedvirus RNA and the synthetic DNA primer 5'-AGCAAAAGCAGG were heatedto 95°C for 1 min, annealedon ice, and added to a first-strand cDNA synthesis reaction mix containing avian
myeloblastosis virus reverse transcriptase (Life Sciences,
St.Petersburg,Fla.)inPCR buffer(50mMKCl,50mMTris
[pH8.4],25 mM
MgCl2,
100jig
of bovineserumalbumin perml) with 1.25mM each deoxynucleoside triphosphate.
Fol-lowing a 1-h incubation at 42°C, the products were added
directlyto a PCR reaction mix alsocontaining the reverse
primer 5'-AGTAGAAACAAGGand 2 UofTaqpolymerase (Cetus
Corp., Norwalk, Ohio).
The double-stranded DNAproduct correspondingtothe NS gene segmentwaspurified by polyacrylamide gel electrophoresis and sequenced
di-rectlywith Sequenase version2.0(US Biochemical,
Cleve-land, Ohio)
following
themanufacturer's instructionsexcept for the addition ofdimethyl
sulfoxide attheannealing
step(29). Oligonucleotide primers for PCR and nucleotide
se-quencing reactions were obtained from Genetic
Designs,
Houston, Tex. The nucleotide sequence of both strands of the cDNAs was determined. Nucleotide sequence analysisand
peptide secondary-structure predictions
weredone withthe programsof the
University
of Wisconsingenetics
group(9)ona VAX computer.
RESULTS
Characterization ofrevertantviruses. Ina
previous study,
phenotypicallyrevertantviruseswereisolated from animals
experimentally
infected with the 143-1 virus(23).
Inordertodetermine the mechanism
by
which these virusesescaped
from the
temperature-sensitive
phenotypespecified by
theNS1 deletion mutation, three of the viruses isolated from animals were selected for further
study
and their shutoff temperaturewas determined afterbiological cloning.
These resultsareshownin Table 1. VirusC1332-8,
isolated after 8days
ofreplication
inachimpanzee,
manifestedsignificantly
restricted
replication
at39°C.
In contrast, virusC1331-5,
isolated after 5days of
replication
inachimpanzee,
and virusH2-7, isolatedafter 7
days
ofreplication
inanimmunosup-pressed
hamster, wereable toformplaques
asefficiently
at40°C
as at33°C,
aphenotype clearly
different from thatofthe 143-1virus administeredtothese animals. We alsosubjected
the 143-1virustoselective pressure
by
serial passageof this virus in MDCK cells atprogressively increasing
tempera-tures,asdescribed in Materials and Methods. Theability
ofthe virusesisolatedateach passageleveltoform
plaques
at elevated temperatures is also shown in Table 1. Amarkedchange
in shutoff temperature was observed betweenpas-sage levels 2 and
3,
atwhichpoint
the virus recovered theability
toreplicate efficiently
at40°C.
Asacontrol,
the143-1virus was also
passaged
in aparallel
series oftitrations in MDCK cells at33°C.
The virus recovered after the fourth such passage showednochange
in its leveloftemperaturesensitivity.
The 143-1 virus also manifested a
partial-host-range
phe-notype, characterized
by
asmall-plaque phenotype
onMDCK cells at the
permissive
temperature of33°C.
Theplaque
morphology
on MDCK cells at33°C
of the virusesanalyzed
in thisstudy
is shown inFig.
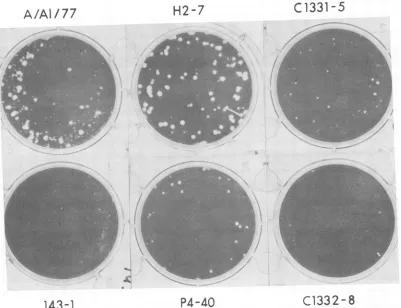
1. The 143-1 virusexhibiteda
small-plaque
morphology
on MDCKcells,
while the A/Alaska/77 wt virus formedlarge,
clearplaques
onthese cells. The
H2-7,
C1331-5,
and P4-40 viruses also formedlarge,
clearplaques
onMDCKcells,
similartothose of the A/Alaska/77virus,
while the ts virus C1332-8mani-VOL.65, 1991
on November 10, 2019 by guest
http://jvi.asm.org/
J. VIROL. 4206 TREANOR ET AL.
TABLE 1. Efficiency ofplaque formationatpermissiveandnonpermissive temperaturesoftheA/Alaska/77and143-1viruses and of virusesisolatedafter in vivo and in vitroreplicationof the 143-1 virus
Passage history of 143-1
.
.
*.__,- .:^ +;- 1ntin in;rtedReductionIn
plaque
titeraiogto)
3 atinuwmutempvstiterat33°C Shutoffplaque(eC)tempformation'for
38°C 39°C 400C
A/Alaska/77 NAb 0.00 0.00 0.63 -41
143-1 NA >3.00 >4.00 >4.00 .38
C1332-8 8days inchimpanzee1332 0.93 >3.00 >4.00 .39
C1331-5 5days inchimpanzee1331 0.00 0.00 0.78 -41
H2-7 7days in
hamstere2
0.09 0.20 1.00 -41P1-38 MDCK cellsat
382C
(P1)C >2.00 >3.00 >3.00 .38P2-39 39°C passageofP1 1.16 >3.00 >3.00 39
P3-39
390C
passageofP2 0.35 0.61 1.70 -41P4-40 40°Cpassageof P3 0.50 0.20 0.27 -41
P4-33 4passagesat330C 2.30 >3.00 >3.00 '38
aShutoff temperatureisdefinedasthelowesttemperatureatwhichagreaterthan100-fold reductioninplaque titer occursrelativetotheplaquetiter at330C.
bNA,notapplicable.
c
P1,
passage1.fested a
small-plaque
phenotype on these cells, similar to thatofthe 143-1 virus.Geneticanalysis. Inordertodeterminethegenetic mech-anismbywhichthe
phenotypic
revertantsescapedfromthets
phenotype,
wegenerated
reassortantsbetween the 143-1virus or the ts+
phenotypic
revertant viruses and the A/Be-thesda/85(H3N2)
wt virus. TheA/Bethesda/85
virus waschosenasthewtvirusparent because itreplicated
efficiently
at 40°C and each of its gene segments could be readily
A/Al/77
H2-,
distinguished
from those of the revertant viruses bypoly-acrylamide
gel
electrophoresis
(datanotshown).
Thegeno-type of the reassortant progeny viruses was then deter-mined, and reassortant viruses which
derived
the NSgene segment from the143-1virusoroneoftherevertant viruses wereplaquepurified
andtestedfor the tsphenotype.
Reas-sortant virusesexhibiting
a shutoff temperature of 39°C orless were
considered
ts. Underthesecircumstances,
reas-sortant viruseswhich derivetheNSgenesegmentfromthe
7
C1331-5
143-1
P4-40
C1332-8
FIG. 1. Plaquemorphologyof theA/Alaska/77,143-1,H2-7,P4-40, C1331-5,andC1332-8viruses.Biologicallyclonedvirusesweregrown
onMDCK cells underanagaroseoverlayfor 72 hat33°C.Cells werefixed withFormalinandstainedwith
crystal
violet.Vim,.c
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.71.559.97.231.2] [image:3.612.122.523.391.699.2]SUPPRESSION OF INFLUENZA VIRUS DELETION MUTATION 4207 TABLE 2. Results of reassortment between the 143-1 virus
and theA/Bethesda/85 virus
Origina ofgenesegment:
Reassortant Phenotype
1 2 3 4 5 6 7 8
8
]
A A B A A A ts9 p A
[JB
E A ts14 A A A A B A L A ts
18 A A A A A ts
23 E A
L
[SjA
E A A ts27 A A A A A A A ts
33 A A A E A A A ts
aA, gene segmentderived fromthe143-1 virus;B,genesegment derived fromtheA/Bethesda/85 virus.
143-1 virus should be ts. If an extrasegmental suppressor mutation were present in the ts+ revertant viruses, then reassortant viruseswhich derive the NS gene segment from arevertant virus butin which the gene segment bearing the suppressormutation is replaced by the corresponding gene segment of the A/Bethesda/85 virus should also be ts.
Alternatively, if the mechanism of reversion were intraseg-mentalsuppression, ortruereversion (unlikely in the case of alargedeletion mutation), then each reassortant virus
deriv-ingthe NS gene segment fromats+ revertant virus should be ts+ irrespective of the A/Bethesda/85 genes present in the reassortant.
The results of thisanalysisare shown in Tables2through 5. Asexpected,viruseswhichreceivedthe NS gene segment from the 143-1 virus and a variety of other gene segments
fromthe A/Bethesda/85 viruswere ts (Table 2). Each reas-sortantvirus which received the NS gene segment from the
A/Bethesda/85 viruswasts+ (datanotshown). This confirms the ability of the NS gene segment of the 143-1 virus to transfer the ts phenotype to theA/Bethesda/85 virus(23).
In contrast, the NS gene segments ofthe C1331-5, H2-7,
andp4-40 viruseswereunabletotransfer thetsphenotypeto theA/Bethesda/85 virus(Tables 3 through 5). Ineach case, reassortantviruses which derived the NS gene segment from oneof therevertantvirusestogether withone ormoregene segments of the A/Bethesda/85 virus retained the abilityto
replicateefficientlyat39°C. Each of theA/Bethesda/85virus genes (exceptthe NSgene) was present inone or moreof
these reassortants. Inaddition,theNS gene segment present in thets+revertant viruseswasidenticaltothatof the 143-1 virus bypolyacrylamide gelelectrophoresis, indicatingthat true reversion had not occurred. These results suggest that an intrasegmental suppressormutation was responsible for the loss of thetsphenotype bythets+ phenotypicrevertant viruses C1331-5, H2-7, andP4-40.
Nucleotide sequence analysis. To determine the sequence of the intrasegmental suppressor
mutation,
we next deter-mined the nucleotide sequences of the NS gene segmentsof the A/Alaska/77, 143-1, C1331-5, H2-7, and P4-40 viruses. Because multiple attempts tosequence theRNAof the NS gene segmentof the 143-1 virus directly were unsuccessful(due to the relatively poor growth of this virus and conse-quent difficulties in template preparation), we
directly
se-quenced PCR-generated cDNA copies of these gene seg-ments. The nucleotide sequence information generated by [image:4.612.312.555.95.332.2]this approach should accurately represent the sequence of the virion RNA, since products with randomPCR-induced
TABLE 3. Results ofreassortmentbetween the C1331-5virus andtheA/Bethesda/85 virus
Origina of gene segment:
Reassortant Phenotype
1 2 3 4 5 6 7 8
3 M M [FB B A * B C
ts+
S M M C B C E C C ts+
8 C C C CB C ts+
22 C C CC ri C C ts+
30 C C C C C C ts+
51 C C C[! B B 1 C ts+
54 C C
E[Lj
B B C C ts+55
p C C CBI
B C C ts57 C C [ C C B E C ts+
63 C C C C C B * c ts+
65
Iq
B C C CC ts67
C
[
H
B Cc
C ts+70 C C C B
[
* C C ts+74 C B B
C
]
C
ts+79 C [ B B C B C C ts+
81 C C B B p! B c ts+
106 C [E1C C C l C ts+
a C,genesegmentderived from theC1331-5 virus;B, genesegmentderived from theA/Bethesda/85virus;*,parental originofgenesegment could notbe
determinedunequivocally.
errors at any specific site would be expected to constitute
minor subpopulations of the bulk product. Each mutation was confirmed by sequencing of templates generated by at least twodifferent PCRamplifications of virion RNA.
The nucleotide sequence of the NS gene segment of the 143-1 virus was identical to that of the A/Alaska/77 virus except for the 36-nucleotide deletion, confirmingthat addi-tional mutations in the NS gene had not been selected in the generation of reassortant 143-1. The nucleotide sequence of the NSgene segment of the tsC1332-8 virus was identical to thatof the 143-1 virus. However, the nucleotide sequences of the NS gene segments of the ts+C1331-5, H2-7,and P4-40
TABLE 4. Resultsofreassortmentbetweenthe H2-7 virusand theA/Bethesda/85virus
Origina ofgenesegment:
Reassortant Phenotype
1 2 3 4 5 6 7 8
1 E!
P
E9
P Hts+
5
H[
H H H H H ts+12 H H B H H B H ts+
16 B B 1 H H B H ts
17 B * H B
[]
H B H ts+19 B H H B H [ * H ts+
20 B B B B H H H ts+
41
[BI BI
B [ H ts+48 H IB H B B H H ts
49 H H H
BJ
H H H ts+a H,genesegmentderived from theH2-7virus; B,gene segmentderived from theA/Bethesda/85virus;*,parentaloriginofgene segmentcouldnotbe determined unequivocally.
VOL.65, 1991
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.313.555.547.700.2]TABLE 5. Resultsofreassortment between the P4-40 virus and theA/Bethesda/85 virus
Origina ofgene segment:
Reassortant Phenotype
1 2 3 4 5 6 7 8
1 B P * * P ts+
3 B P * * p p ts+
18 P P B P ts+
28 P P P P B P P P ts+
34 P P P IBI P[ M P ts+
37 P P P P [ *[L] P ts+
40 p [! P P P P P P ts+
49 P P E[ B P P P
ts+
51 P P P JB B[] P ts+
54 P P E P
IBB
B P P ts+57
P
P
B B Pp
ts+59 [ jS P P P P i!P ts+
a
P,
gene segmentderivedfrom theP4-40virus; B,gene segmentderived fromtheA/Bethesda/85 virus;*, parental origin of genesegment couldnotbe determinedunequivocally.viruses each contained a U to C transition at nucleotide 94 (mRNA sense),leading to a predicted change from valine to alanineatamino acid 23of NS1. Because this change occurs within the NS2 intron, no alteration in NS2 would be predicted to occur. The mechanism of escape of these viruses from the ts phenotype was therefore attributable to
intragenic suppression. The C1331-5 virus also had a
non-codingnucleotide change from U to C at nucleotide 110. The sequences of the NS genes of the C1331-5, H2-7, and P4-40 viruses were otherwise identical to that of the 143-1 virus.
Inorder to determine when in the courseofserial in vitro passageof the P4-40 virus the U to C changeatnucleotide 94 occurred, we determined thenucleotide sequence of the NS gene segment from nucleotides 50 to 150 of viruses from each passage level. As shown in Fig. 2, the change from valine to alanine occurred between passage 2 and passage 3
of this virus, coincidentwith a significant change in shutoff temperaturefor plaqueformation from 39 to more than 40°C. This change was seen more clearly after further passage and
biologicalcloning of this virus.
The potential structural consequences of the amino acid
change present in the revertant viruses, as predicted by computer analysis of secondary structure (6), are shown in
Fig. 3. The 12-amino-acid deletion in the NS1 protein of the 143-1 virusdisrupts a region of alpha helix predicted to occur
approximately between amino acids 51 and 84 of the wild-type NS1 protein. The change from valine to alanine at amino acid 23 in the revertant viruses is predicted to result in
generation ofa new region of alpha helix between approxi-mately amino acids 18 and 27 of the NS1 protein. The effects of these changes on the three-dimensional structure of the NS1 protein are unknown.
DISCUSSION
In aprevious study, the 143-1 virus was shown to have twophenotypeswhich could be transferred with the NS gene segment to awtinfluenza A virus, a shutoff temperature for
plaque formationonMDCK cells of 37°C (ts phenotype) and a small-plaque phenotype on MDCK cells (23). In the current study, we confirmed the ability of the NS gene
P1 P2 P3 P4
G A T C G A T C G A T C G A T C
-T c--_
G ~~~~~~~~~...~ G
FIG. 2. Dideoxysequencingof theplusstrand ofaPCR copyof the NSgenesegment of the 143-1 virus after serial passageat380C (P1), 390C(P2and P3), and40'C (P4). A substitution of C for Tat
nucleotide 94 canbe seen to occur between thesecond and third
passage.
segment of the 143-1 virus to transfer these
phenotypes
to the wi' influenza A/Bethesda/85 virus and showed that the nucleotide sequence of the NS gene segment of the 143-1 viruswasidenticaltothatof theA/Alaska/77virusexceptfor the 36-nucleotide deletion. Thus, thetwophenotypes
of the 143-1 virus can bedirectly
attributed to the 12-amino-acid deletion in the NS1protein
of this virus.When the 143-1 virus was administered to hamsters and
chimpanzees
in theprevious study
(23) orpassaged
in vitroat elevated temperatures in this
study,
viruses whichcon-currently
lost the twophenotypes
of the 143-1 virus wereisolated. Loss of the i's and
small-plaque phenotypes
oc-curred
concurrently
ineachof the ts'sphenotypic
revertant viruses. Theseobservationssuggestthat theproducts
of the NSgene segment, either alone orincombination with those of other viralgenes,play
arole in the hostrangeof influenza A viruses(4,
24, 27).The
genetic
mechanismby
which thephenotypically
re-vertant viruses lost the i's
phenotype
was shown to be thedevelopment
ofanintragenic
suppressor mutation.Nucleo-tide
sequencing
revealed that therewas asingle-amino-acid
difference between the NS gene segmentof the 143-1 virus and the
corresponding
genes of the revertantviruses,
achange
from valine to alanine at amino acid 23 of the NS1protein. Intragenic suppression
ofpoint
mutations in tsmutantsof influenza A viruses has been
reported
previously
(18, 26), as hasintragenic
suppression
of i's and other mutations of animalviruses,
including
vesicular stomatitisvirus,
poliovirus,
foot-and-mouth diseasevirus,
vacciniavirus,
and Sindbis virus (12, 14,17).
Intragenic
suppression
of a deletion mutation of an influenza virus has not been
reported
previously.
Themechanism
by
which this mutation affects thestability
of the NS1
protein
is unclear. The deletion of the 12 amino acids of NS1 in virus 143-1disrupts
aregion
ofalpha
helix inthe NS1
protein
that extendsapproximately
from aminoacids 51 to 84 of the
wild-type
sequence.Recently,
an additional i's mutant of the influenza A/Udorn/72 virus wasdescribed in which this
alpha-helical region
wasdisrupted by
a
change
fromlysine
toasparagine
at amino acid 62 of theNS1
protein
(8). These observationssuggest thatthisalpha-helical
region
of NS1plays
animportant
role in thefunctionorstructural
stability
of the NS1protein.
Inthets's
revertant virusesanalyzed in thisstudy,
asingle-amino-acid
changeto alanine atposition
23 was able to restore this lostfunction. This mutation occurs within the amino-terminal half of theNS1
protein,
which has beensuggested
to be the activeportion
of the molecule (20). However, the presence of avalineat amino acid23 oftheNS1
protein
is notconserved within human influenza A viruses (3),suggesting
that theon November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.344.535.75.154.2] [image:5.612.63.303.95.274.2]SUPPRESSION OF INFLUENZA VIRUS DELETION MUTATION 4209
*E
C
A
w~~~W
FIG. 3. Chou-Fasmanpeptide structure predictions of the amino-terminal 150 amino acids of the NS1 protein. (A) Structure of the NS1 proteinoftheA/Alaska/77virus; (B) structure of the 143-1 virus around the deletion (the structure of the NS1 protein of this virus would otherwise be identical to thatof the A/Alaska/77 virus); (C) 1331-5 virus sequence at the site of the intragenic suppressor mutation (the structureoftheNS1protein of this virus would otherwise be identical to that of the 143-1 virus). Helices are shown with a sine wave, beta sheetsareshown with a sharp sawtooth wave, turnsare shown with1800 turns,and coils are shown with a dull sawtooth wave.
structure of this particular region may tolerate some vari-ability. While the change to alanine is a conservative one, computeranalysis predicts that this substitution would result in conversion of a region ofbeta-sheet structure to one of
alphahelix, approximately from aminoacids 18 to 27 of NS1. Itistherefore possiblethatthisnewregion ofalpha helix is
able to substitute for the loss ofthe original alpha-helical structure. However, more definitive analysis of the
func-tional significance ofthese mutations awaits the develop-mentofadirect invitro assayofNSprotein function.
Theobservationthat asinglepointmutation may result in the loss ofaphenotype specified by a deletion mutation is
relevant tothe development of livevirus vaccines. Animal viruses bearing a number of types of missense mutations,
including ts, ca, monoclonal antibody-resistant (mar), and protease activation (pa) mutations have been evaluated as
potential live attenuated vaccines.Inthe case of the ts, mar,
andpa mutants, reversion to the wild-type phenotype fol-lowing replication invitroorinvivo has beenobserved (5,
16,28), andthispossibilityrepresents amajorconcern inthe use of missense mutations in the construction of such
vaccines. Thishas been particularly true in the development
of ts influenza A viruses as live virus vaccines, where
phenotypic reversion ofa tsinfluenzaAviruscontainingtwo
independent ts lesions was observed following prolonged
replication in a seronegative child (25, 26). To date,
rever-sion of influenza virus vaccines containing the attenuating
genes ofthe ca influenza A/Ann Arbor/6/60 virus has not
been
observed,
possibly because this virus possesses fourindependent attenuating mutations (22).
Viable animal virusesbearing largedeletions represent an attractivealternative to missense mutations in the
construc-tion oflive attenuated vaccines, since the attenuation
phe-notype
imposed
by such mutations should be more stable (15). Inaddition,the recentdevelopmentoftechniquesfor invitro mutagenesis of influenza A viruses provides a means
forthereadyconstruction of suchmutations(7).Theresults of the current study, however, suggest that even this
ap-proachwill noteliminate the need for concern aboutpossible reversion, since both intragenic and potentially extragenic
suppressormutations may developand alter the phenotypes specified by deletion mutations. Asimilarneedfor concern has been suggested by the observation that second-site mutations in the simian virus 40 VP1 gene can suppress a large deletion in the agnogene ofthis virus following repli-cation in vitro (1). Therefore, it may be necessary to con-structinfluenza virusesbearingdeletionmutationsin several gene segments to ensure adequate genetic stability. Studies toaddress the feasibility of this approach areinprogress.
ACKNOWLEDGMENTS
Wethank R. Chanockfor critical review and helpful discussion and R. Rose and M.Perkins for adviceregarding the PCRtechnique.
REFERENCES
1. Barken, A., R. C. Welch, and J. E. Mertz. 1987. Missense mutations inthe VP1 geneof simian virus40thatcompensate for defects causedby deletions inthe viralagnogene.J. Virol. 61:3190-3198.
2. Buonagurio, D. A., M. Krystal, P.Palese, D. C. DeBorde, and H. F. Maassab. 1984. Analysisofaninfluenza Avirusmutant
withadeletion intheNSsegment.J.Virol. 49:418-425. 3. Buonagurio, D. A., S. Nakada, J. D. Parvin, M. Krystal, P.
Palese, and W. M. Fitch. 1986. Evolution ofhuman influenza A viruses over 50 years:rapid, uniformrateofchange in NSgene. Science232:980-982.
4. Brown, E. G. 1990. Increased virulence ofa mouse-adapted variant of influenzaA/FM/47 virus is controlled by mutations in genome segments4, 5, 7, and8. J.Virol. 64:4523-4533. 5. Chanock,R.M.,andB.R.Murphy.1980. Useof
temperature-sensitive and cold-adapted mutant viruses in immunoprophy-laxis ofacuterespiratorytractdisease. Rev.Infect.Dis. 2:421-432.
6. Chou, P. Y., and G. D. Fasman. 1978. Prediction of the secondarystructureofproteinsfromtheiramino acid sequence. Adv. Enzymol.47:45-148.
7. Enami, M., W. Luytjes, M. Krystal, and P. Palese. 1990. Introductionofsite-specific mutations intothe genomeof
influ-enzavirus. Proc. Natl. Acad. Sci. USA87:3802-3805. 8. Hatada, E., M. Hasegawa, K. Shimizu, M. Hatanaka, andR.
VOL.65,1991
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.135.468.71.255.2]Fukuda.1990.Analysis of influenza A virus temperature-sensi-tivemutants withmutations in RNAsegment8. J.Gen. Virol. 71:1283-1292.
9. Jameson, B. A.,andH.Wolf.1988. Theantigenic index:anovel
algorithm for predictingantigenic determinants. Comput. Appl. Biosci. 4:181-186.
10. Kawasaki, E. L., and A. M. Wang. 1989. Detection ofgene
expression, p. 89-98. In H. A. Erlich (ed.), PCR technology: principles and applications for DNA amplification, 1st ed. Stockton Press, New York.
11. Lamb, R. A. 1989.Genes andproteins of the influenza viruses,
p. 1-87. In R. M. Krug(ed.), The influenza viruses. Plenum
Publishing Corp., New York.
12. Li,J.-P., and D. Baltimore. 1990. Anintragenic revertant ofa
poliovirus 2Cmutanthasanuncoating defect. J. Virol.
64:1102-1107.
13. Maassab, H. F., and D. C. DeBorde. 1983. Characterization of
aninfluenza A hostrangemutant. Virology 130:342-350. 14. McFadden, B., K. Essani, andS. Dales. 1980. Anew
endonu-clease restriction site which is at the locus ofa
temperature-sensitivemutation in vacciniavirus isassociated with trueand pseudo-reversion. Virology 101:277-280.
15. Meignier, B., R. Longnecker, and B. Roizman. 1988. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: construction and evaluation in rodents. J. Infect. Dis. 158:602-614.
16. Mochizuki, Y., M. Tashiro, and M. Homma. 1988. Pneumo-pathogenicity in mice ofa Sendai virus mutant, TSrev-58, is accompanied by in vitro activation with trypsin. J. Virol. 62:3040-3042.
17. Morita, K., R. Vanderoef, and J. Lenard. 1987. Phenotypic
revertantsoftemperature-sensitive Mproteinmutantsof vesic-ular stomatitis virus: sequenceanalysis andfunctional
charac-terization. J. Virol. 61:256-263.
18. Mucke, K., andC. Scholtissek. 1987. Extragenic andintragenic suppression ofatransportmutation in thehemagglutiningeneof an influenza A virus as revealed by backcross and sequence
determination. Virology 158:112-117.
19. Murphy, B. R., L. J.Markoff,N. T. Hosier,J.G. Massicot,and
R.M. Chanock.1982.Production and level of genetic stability of
an influenza A virus temperature-sensitive mutant containing
twogeneswithts mutations.Infect. Immun. 37:235-242. 20. Norton, G. P., T. Tanaka, K.Tobita, S. Nakada, D.A.
Buon-agurio, D. Greenspan, M. Krystal, andP. Palese. 1987. Infec-tious influenza A and B virus variants with long carboxyl
terminaldeletions in the NS1 polypeptides. Virology 156:204-213.
21. Sears, S. D., M. L.Clements, R. F. Betts, H. F.Maassab,B. R. Murphy, and M. H. Snyder. 1988. Comparison of live,
attenu-atedHlNiand H3N2cold-adapted and avian-human influenza areassortant viruses and inactivated virus vaccine in adults. J. Infect. Dis. 158:1209-1219.
22. Snyder, M. H., R. F. Betts, D. DeBorde, E. L. Tierney, M. L. Clements, D.Herrington, S. D.Sears,R.Dolin,H. F.Maassab, and B. R. Murphy. 1988. Four viral genes independently
con-tribute toattenuation of liveinfluenza A/AnnArbor/6/60 (H2N2) cold-adaptedreassortantvirusvaccines.J.Virol.62:488-495. 23. Snyder, M. H., W. T. London, H. F. Maassab, R. M. Chanock,
and B.R. Murphy. 1990. A 36nucleotidedeletionmutation in thecoding regionof the NS1 geneofaninfluenzaAvirusRNA segment 8specifiesatemperature-dependenthostrange pheno-type.Virus Res. 15:69-84.
24. Tian, S.-F., A. J. Buckler-White, W. T. London, L. J. Reck, R. M. Chanock, and B. R. Murphy. 1985. Nucleoprotein and membrane protein genes are associated with restriction of replication of influenza A/Mallard/NY/78 virus and its reassor-tantsinsquirrelmonkey respiratorytract.J.Virol.53:771-775. 25. Tolpin, M. D., J. L. Clements, M. M. Levine, R. E. Black, A. J. Saah, W. C. Anthony, L. Cisneros, R. M. Chanock, and B. R. Murphy. 1982. Evaluation of a phenotypic revertant of the A/Alaska/77-ts-lA2 reassortant virus in hamsters and in
sero-negative adult volunteers: further evidence that the tempera-ture-sensitivephenotypeisresponsible for attenuation of ts-lA2 reassortantviruses. Infect. Immun.36:645-650.
26. Tolpin,M.D., J. G. Massicot, M.G.Mullinix,H. W.Kim, R. H. Parrott, R. M. Chanock, and B. R. Murphy. 1981. Genetic factors associated with loss of thetemperature-sensitive pheno-type ofthe influenza A/Alaska/77-ts-lA2 recombinant during growth in vivo. Virology 112:505-517.
27. Treanor, J., M. Snyder, W. London, and B. R. Murphy. 1989. TheB alleleoftheNSgeneof avian influenzaviruses, butnot
the Aallele, attenuates ahumaninfluenzaAvirusforsquirrel monkeys. Virology 171:1-9.
28. Tuffereau, C., H. LeBlois, J. Benejean, F. L. Coulon, and A. Flamand. 1989.Arginine orlysine in position333ofERAand CVS glycoprotein is necessary for rabies virulence in adult mice. Virology 172:206-212.
29. Winship, P. R. 1989.Animprovedmethodfor directly sequenc-ing PCR amplified material using dimethyl sulfoxide. Nucleic AcidsRes. 17:1266.