0022-538X/87/041301-09$02.00/0
Copyright © 1987, American
Society
forMicrobiologyProcessing of the Semliki
Forest
Virus
Structural
Polyprotein: Role
of
the
Capsid Protease
PAULMELANCONtAND HENRIK GAROFF*
European Molecular Biology Laboratory,Programof Cell Biology, 6900Heidelberg 1, FederalRepublicofGermany Received 21July 1986/Accepted1 December 1986
Theprotease activities responsible forthe cotranslational processing of the Semliki Forest virus structural polyprotein wereinvestigated by usinganin vitrotranscription-translation system. Three cleavages released theindividual chains from thenascentpolyprotein in theordercapsid,p62, 6K (a nonstructural peptide), and El.We showeddirectly that theproteaseactivityresponsibleforthe release of thecapsid protein resides in the capsid itself: by progressive truncation of the cDNA used for the SP6 transcription,weshowedthataprecursor containingasfewas38 residues ofthep62 protein leftattheCterminus of thecapsidwasstillveryefficiently cleaved in vitro.Wefurther tested the possibility thatserine-219 of the capsid is involved in autoproteolysisby site-directed in vitro mutagenesis. A changein the sequenceGly-Asp-Ser(219)-Gly, atetrapeptide conserved
among several animal serine proteases, to Gly-Asp-Arg-Ser-Thr was shown to completely abolish in vitro cleavage. Thissupportsthenotion that the capsid isaserineprotease. The role ofthecapsidproteasein the processing of the 6Kjunctionswastheninvestigated bytranslationsofahybridpolyprotein in which the capsid
andmostof the p62 sequences are replaced by those of the secretory protein Iysozyme. The cleavages and concomitant appearance ofthe6K peptide occurred efficiently and were shown to require the presence of membranes. This demonstrates thatthecapsidproteaseis notrequiredfor thosecleavages andsuggests that amembrane-associated hostproteaseisresponsible for the cleavage.
Semliki Forest virus (SFV) is a small, enveloped RNA virus that belongs to the genus Alphavirus of the family Togaviridae. It consists of a nucleocapsid containing the
single-stranded virus RNA complexed with a basic capsid protein,surroundedbyalipid bilayercontainingtwointegral glycosylated membrane proteins (E2 and El) and a small peripheral protein E3 (for areview, see reference 18). The translation of thestructural proteins of SFV is initiatedata single siteon asubgenomic 26S RNA. Threecotranslational
cleavage events release the capsid (33 kilodaltons [kDa]), p62 (62 kDa; a precursor of the E3 and E2 proteins), a
nonstructural 6K peptide (60 amino acids [aal long), and finally the El protein (50 kDa).
The first cleavage event occurs between the tryptophan (aa 267) and serine (aa 268) of the polyprotein and releases thecapsidprotein from the polysome. The newly exposed N terminus of the p62 protein is then involved in initiating chain translocation across the endoplasmic reticulum (ER)
membrane. Chain translocation continues until it is arrested atthetransmembrane segmentof thep62protein but is then reinitiated to allow translocation of the El protein. The signal for this second translocationeventresides within the 6K peptide, the nonstructural peptide presentbetween the p62 and El proteins (36). It remains unknown whetherthe twocleavage events that are responsible for the release of thep62, 6K, and Elproteins occurbefore, during, or after the reinitiation oftranslocation. A final cleavage converts theP62glycoproteintoE2 andE3. This lastcleavage ismost probably a late Golgi event occurring en route to the cell surface.
The chymotrypsinlike protease responsible for the first cleavage (between C and p62) is probably virus encoded. The strongest evidence comes from the in vitro study of
*Correspondingauthor.
tPresent address:DepartmentofBiochemistry,Stanford
Univer-sitySchool ofMedicine, Stanford, CA94305.
Aliperti and Schlesinger (4). Using the fact that the transla-tion of26S RNA of SFV and the related Sindbis virus in cell-free systems gives riseto rapid and efficient release of the capsid protein (7, 9, 11, 12, 19, 21, 41), these authors have shown (i) that the appearance of the capsid protein
could be inhibited by the addition of aa analogs in the cell-free translation system and (ii) that almost complete cleavage of the accumulated precursor could be observed
after a chase with cold and normal aa. Theseresults dem-onstratethatde novosynthesis (from the viral RNA) of the
protease isrequired and that the proteasecan actintrans. Several observations suggest that the viral protease is located near the N terminus of the polyprotein. First, in
synchronized in vitro translations, the capsid protein is liberatedvery soonafter it issynthesized (8, 19). Second, the
analysis of the sequence of the cDNAs of temperature-sensitive(ts)mutantsdefectiveincleavage(and their
respec-tive revertants) has shown that the criticalaa substitutions
were located in the capsid protein (25). Moreover, the sequence surrounding serine-219 in SFV (serine-215 in Sindbis virus) shows homology with that around the serine present atthe active centerof several serine proteases (6). For these reasons, and the fact that one ofthe mutations discussed above mapsclose to serine-215, itwasproposed that the protease activity responsible for the capsid-p62 cleavageresides within thecapsidproteinand that itactsas a serineprotease(25).
Incontrast, thecleavagebetween the6K and Elproteins is most probably catalyzed by the host protease signal peptidase.We haverecentlylocated the translocationsignal forthe Elprotein to the last 26 residues of the6Kpeptide (36). This region contains a stretch of hydrophobic aa residues andhasadistribution of small and basicresiduesat thecleavage sitecharacteristic ofsignal peptidase cleavage sites. Much less is known about the cleavage between the p62 and 6k proteins. If that cleavage occurs in the cyto-plasm, the protease involved is most probably virus en-1301
on November 10, 2019 by guest
http://jvi.asm.org/
coded,
since no hostcytoplasmic
protease with theappro-priate
specificity
has yet been described. Thepossibility
therefore exists that the
putative capsid
proteaseplays
some role in thep62-6K
cleavage.
In thework
reported here,
weusedtheclonedcDNAfor the SFV 26S RNAtostudy
the involvementofcapsid
in theprocessing
ofthestructural
polypeptide. By truncating
fromthe 3' end the cDNA used as a
template
for in vitrotranscription-translation
experiments,
we showeddirectly
thattheprotease
activity
is restricted tothecapsid region.
Second,
we introducedby
in vitromutagenesis
two aachanges
at theputative
active serine-219 and showed thatthis
completely
abolishescapsid cleavage.
Thissupportsthe notion that thecapsid
is aserine
protease.Third,
using
ahybrid protein
in which thecapsid protein
and the luminalectodomain of
p62
was substituted with chicken oviductlysozyme
(a
secretoryprotein),
we demonstrated that thecapsid protein
was notresponsible
for thecleavages
oneither side of the6K
peptide.
MATERIALS AND METHODS
Materials. All
restriction endonucleases,
as well as calf intestinephosphatase,
T4 DNApolymerase,
Escherichia coliDNApolymerase
1(Klenow fragment),
and SP6 poly-merase, wereobtained fromBoehringer
GmbH, Mannheim,
Federal
Republic
ofGermany. Mung
bean nuclease(fast
protein liquid
chromatography grade)
waspurchased
fromPharmacia,
Freiburg,
FederalRepublic
ofGermany,
BAL31was
purchased
fromBethesda ResearchLaboratories, Inc.,
Gaithersburg, Md.,
RNasin waspurchased
fromPromega
Biotech, Madison,
Wis.,
andproteinase
K waspurchased
from E. Merck
AG,
Darmstadt,
FederalRepublic
ofGer-many. T4 DNA
ligase
was agenerousgift
ofE. Winkler.Theribonucleotides
(equine)
andspermidine
usedforRNAsyn-thesis werefrom
Sigma
ChemicalCo.,
St.Louis,
Mo. Thedeoxy-
anddideoxyribonucleotides
usedforDNAsequenc-ing
and thecapanalog 7mGpppG
werefrom Pharmacia. The DNA linkers(Sall
octamer)
andthe SP6 promoterprimer
were obtained from
Boehringer.
RibonucleaseA,
phenyl-methylsulfonyl fluoride, iodoacetamide, lysozyme,
creatinephosphate,
and
creatinephosphokinase
were fromSigma.
The rabbit
reticulocyte
lysate, L-[35S]methionine,
L-[35S]cysteine,
andlow-range
molecularweight
(MW)
mark-erswere
purchased
from AmershamCorp., Braunschweig,
Federal
Republic
ofGermany.
Themiddle-range
MWmark-ers and
En3Hance
were obtained from NewEngland
Nu-clear
Corp., Dreieich,
FederalRepublic
ofGermany.
Seph-adex G-75and
Sephacryl
S-300werefromPharmacia.Low-melting-point
agarose waspurchased
from BethesdaRe-search Laboratories. Salt-washed
dog
pancreasrough
microsomes
(50
A280units/ml)
wereprovided by
B.Dobberstein. Nikkolwas a
gift
fromD.Meyer.
General DNAmethods. For
rapid screening, plasmid
DNA wasprepared by
theboiling
methodofHolmes andQuigley(29),
as describedpreviously (43).
Largequantities
ofplas-mid were
prepared by
a modification of the alkali-sodiumdodecyl
sulfate method ofBirnboim
and Doly (5), asde-scribed
previously (36).
DNAfragments
wereisolated overlow-melting-point
agarose. Restriction endonucleases andDNA-modifying
enzymes were used as specified by themanufacturer. All molecular biological manipulations were
performed by
standardprotocols
(35). The preparation ofcompetentbacteria
(E.
coliHB101)
and theirtransformation were as describedby
Hanahan (26). The sequencing of double-strandedpGEM plasmids
by using the SP6 primer wasdoneessentially
asdescribedbyChen and Seeburg (10).Construction of the vectors. Late simian virus 40vectors for in vivoexpression were constructedasfollows.The SFV cDNA for these constructions was obtained from
plasmid
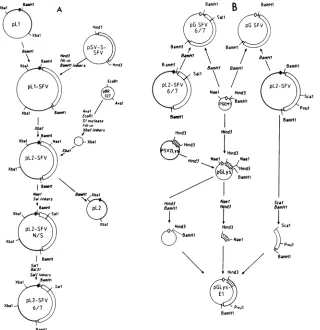
pSV-S-SFV(37) as aHindlIl fragment. The cohesive ends of the isolatedfragmentwerefilledin,andBamHIlinkerswere added, (Fig. 1A). Thisfragmentwasinserted at the BamHIsite ofvectorpLl (originally described as pAll-SV-L-2by
Gruss et al. [23]) to produce pLl-SFV. This vector was modifiedby substitutingapieceofpBR327for thelarge XbaI pBR322 fragment originallypresent,thereby removing
sev-eralNaeI sites and apoison sequencethat interferes in cis
with simian virus 40 replication (34). Plasmid pBR327 was cut with EcoRI and AvaI and treated with S1 nuclease to
destroy the EcoRI andAvaIsites (Fig. 1). Afterrepairwith
DNA polymerase, XbaI linkers were added, and the small
fragmentwaspropagatedas aplasmid.Thisplasmidwas cut
withXbaI, treated with phosphatase, andsubstituted for the pBR322 fragment between the XbaI sites of pLl-SFV to
produce pL2-SFV.
The modification of the capsid-encoding region was
ac-complished with the exonuclease BAL31. Aplasmidwith an
out-of-frame insertion ofalinkeratthe site ofmodification was first constructed (Fig. 1). Plasmid pL2-SFV was cut
extensively with NaeI untilmostof the DNA was linearized.
Theendswerefilledin, the linear fragmentwasisolatedover agarose, and a
Sall
linker (octamer)was added. Aplasmid containing the linker attheproperlocation, pL2-SFVN/S,was chosen for further work. SalI linker insertion was necessary to overcome thevery inefficientdigestion atthe
NaeI site ofinterest.
Plasmid pL2-SFV N/S was cut to completion with Sall and treatedbriefly withBAL31inabuffercontaining 0.5M
NaCl at 30°C. After inactivation of the nuclease, the ends wererepaired withDNApolymerase, and Sall linkerswere
added. Ampicillin-resistant transformants were first
screened
by
restriction forthe presence ofplasmids with aSall linker. Positiveplasmidswerethen screenedby
trans-fection of COS cells for the
synthesis
of full-length Elprecursor, as described below. Since the starting plasmid could not
synthesize
El protein as a result of the out-of-frame insertion of thelinker,
any plasmid in which the translation reading frame had been restored following thegentle
BAL31 treatmentmusthave sufferedamodification in thesequenceof interest.Approximately one-fourth of the plasmids tested containedalinker thatwasin frame with therestofthecodingsequence.Thesewerefurther analyzed by restrictionwithAvaItodeterminethe extentofthedeletion. PlasmidpL2-SFV
6/,
showingaminimalmodification, waschosen for further work. The 216-base-pair AvaI fragment
was subcloned at the AvaI site of plasmid pGEM 1 and
sequenced by using the SP6 promoterprimer.
Transcription vectors for in vitro expression. The SFV insertsofplasmids pL2-SFV and pL2-SFV 6/7were isolated as BamHIfragments andinserted at theBamHI site of the SP6 vectorpGEM 1(PromegaBiotech)(Fig. 1B).
The cDNA for chicken lysozyme was obtained from
plasmid pSV-2-Lys (32). The HindIII lysozyme insert was subcloned atthe HindIII site ofthepolylinker of pGEM 1. Thehybridgeneforaprotein containing almost the entire sequence oflysozyme fused to the C-terminal half of the SFVpolyprotein was constructedby a three-fragment
liga-tion. The lysozyme portion wasisolated as a HindIII-NaeI
fragment,and that for SFV was isolated as a ScaI-BamHI
fragment (Fig. 1B). The vector was reconstructed with a
HindIII-BamHI fragment containing almost the entire
pGEM 1 plasmid.
on November 10, 2019 by guest
http://jvi.asm.org/
BamHl Xba1 A
/ ~Xbal Hid
BapHl id SFV )
BamHl fSl-in
Xba Bl BamHn s - 3
EcoRl
pL1-SFV ,L
C ) / ~~~~~~~~~~Aval
Xbal aem1 AvalEcoR?
Stnuclease
Xba1 ~~~~~~Fill-mn XbaI Xba?Inkers
sBamHl
Xbal Nael cI~ Xbal
X7;XbaI
pL2-SFV
Xbal i J
BamHB
Naae Ba&/i Xbal Sal linkers No
BamHrl pL2
Xb 1 > L S 11 \ ( / \\ ~~~~~~~~Xbal
pL2-SFV
N/S
Xbal
SalI Ba131 Salinkers
Xbal BamHl
pL2-SFV
Xbal 6/7
BamHl
BamHr
Hind3 Hind3 IHind3
|PSVLy/Hind3
P/,nd3 Nael ,Nael
~/ Hind3 Nje1 \BBamHl
Hind3 Na.?
Bar/l 7d3
cI
Hind3 Hind3
BamHl 4
_S Nael
\ Hind3//
Peul
[image:3.612.152.472.66.396.2]BamHl
FIG. 1. Outline oftheplasmid constructions for in vivo (A) and in vitro (B)expression studies. Thin lines indicate pBR-derived DNA
sequences,thick lines indicatesequencesfrom simian virus40,andboxed regionsrepresentSFVcDNAorchickenoviductlysozymecDNA
(hatched areas). The largestboxesrepresentcodingregionswithin the cDNA sequences.All circlesare drawnto scale(forcomparison, pSV-S-SFV is 7,603 base pairs long). For detailsseeMaterials and Methods.
Transfection of COS cells.Plasmid DNA for the screening of in-frame pL2-SFV N/S mutants was prepared by the
boiling method. The DNA (2 pig) wastransfected on COS cells grown on cover slides (5 mmby 5 mm) by using the
DEAE-dextranmethod described by Cutleretal. (13). The
appearanceof the SFV El proteinwasdetected by indirect
immunofluorescence in Triton X-100-solubilized cells by a modification of themethod described by Timmetal. (43). As
anegative control, COS cellsweretransfected withplasmid
pL2orunmodifiedpL2SFV N/S. In thosecases, no or very
few cells were positive for El. In about one-fourth of the transfections with the modified PL2-SFV N/S DNAs, 5 to 10% ofthe cells in the monolayer showed bright reticular staining. Thepolyclonal rabbit anti-El antibodywas a gen-erous gift of D. Louvard and G. Warren. The
rhodamine-conjugated anti-rabbit secondary antibody was a generous
gift of Thomas Kreis.
Transcription and translation. CappedmRNAwas
synthe-sized in vitro from the pGEM vectors derivatives by using thebacteriophage SP6 polymerase, asdescribedpreviously (30, 37).LinearizedplasmidDNA(0.5 pug )wastranscribed at40°Cwith3to4U of SP6polymerase inafinal volume of 10 pd containing 0.5 mM each ATP, UTP,andCTP,0.1 mM GTP, 0.25 mM 7mGpppG, 10 U of RNase inhibitor, 2 mM spermidine, 100 ,ug of bovineserumalbuminperml,40 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES)-KOH (pH 7.4), 6mM magnesiumacetate, and 10
mMdithiothreitol. Aftera 10-min incubation, anadditional
100pmol of GTPwasadded, and the reactionwascontinued
for a further 30 min. A 25-,Il rabbit reticulocyte cell-free translation assay mixture contained 2 pul of transcription
mixtureand 50%commerciallysate andwasadjustedtothe following final concentrations: 100mM potassium acetate, 1.2 mMmagnesiumacetate,0.4 mMspermidine, 40 ,uM each 19 aa minus methionine (or cysteine), and 60 ,Ci of L-[35S]methionine(orL-[35S]cysteinewhenindicated). Someof the translations were carried out with dog pancreas salt-washed membranes present at aconcentration of 2.0 A280
units/ml. The membranes were treated onice with 25 mM
ethylene glycol-bis(,-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA) (pH 7.5) and with 0.005% Nikoll (Nikko, Tokyo, Japan) priorto addition. Translations were
carriedoutat30°C for60min.
Protease assay. Proteinase K digestion of the translation products was carried out at0°C for 30or60 min at afinal concentration of 0.25mgofproteaseperml in thepresence
orabsenceof 1% Triton X-100. Proteolysis wasstopped by the additionoffreshly dissolvedphenylmethylsulfonyl fluo-ride(20mg/mlinisopropanol)toafinalconcentration of1 to 2 mg/ml. After incubation for 5 min at 0°C, 2 volumes of sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample bufferwasadded and thesampleswereimmediately heatedto95°Cfor 5 min.
Electrophoresis. Samples from confluent monolayers of
Sca? BamHl
IScal
BPvul
BamHl
on November 10, 2019 by guest
http://jvi.asm.org/
BHK-21 cells infected with SFV
(wild
type orts3)
wereprepared
as describedby
Green et al.(22).
The infectedmonolayers
were labeled5.5 hpostinfection
with 50,uCi
ofL-[35S]methionine
for4 h(ts3)
orwithL-[35S]cysteine
for10min
(wild type).
Some of the infected cells werepretreated
with4 ,gof
tunicamycin
per mlfor2.5 hand labeledfor4hin the presence of
tunicamycin.
Cells werelysed
with hotsodium
dodecyl
sulfatesample
buffer andpassed
severaltimes
through
a Hamiltonsyringe
to break downchromo-somal DNA. All
protein samples
were heatedto95°C
for5 minin thepresenceof dithiothreitol and thenalkylated
withiodoacetamide.
Electrophoresis
of cell-free translationprod-uctsof8 and10%
polyacrylamide
slabgels
andsubsequent
processing
forfluorography
wereessentially
as describedpreviously (13). Electrophoresis
on22%polyacrylamide
slabgels
containing
6 Murea was done as describedby
Swankand Munkres
(42).
Middle-range
MW markers werephos-phorylase
b(92,500),
albumin(69,000),
ovalbumin(46,000),
carbonic
anhydrase (30,000),
andlactoglobulin
A(18,370).
Low-range
MWmarkerswere carbonicanhydrase (30,000),
trypsin
inhibitor(21,500), cytochrome
c(12,500), aprotinin
(6,500),
andinsulin B chain(3,400).
RESULTS
Cleavage
of thecapsid protein
in a cell-free transcription-translation system. Theprocessing
ofthepolyprotein
was studiedby
using
a cell-free translation systemprogrammed
with RNA transcribed in vitro. The construction of the
plasmids
used astemplate
for RNAsynthesis
is outlinedinFig.
1 and is described in more detail in Materials andMethods.
Templates
for RNAencoding
thecomplete
poly-protein
wereprepared by
linearization ofthepGSFV
plas-mids withanenzyme,
PvuII,
thatcuts verynearthe 3' end of the4-kilobase-long
SFV cDNA in the vector sequence.Translation-competent
RNA wasproduced by transcription
with thephage
SP6
polymerase
in the presence ofthecapanalog 7mGpppG.
Under suchconditions,
the RNApro-duced has the size
expected
foratranscript
ofa4-kilobase cDNA(data
notshown).
A rabbitreticulocyte lysate
pro-grammed
with thewild-type
RNA(labeled
SFV in lanes 2and
5)
produced large quantities
ofaprotein
ofapparent MW33,000
(Fig.
2).
Thisprotein
has almostthesamemigration
inpolyacrylamide gel
electrophoresis
asdidcapsid produced
in BHK cells infected with the SFV ts3 virus(lane
4). (Note
that the ts-3 virus infection was carriedout at
semipermis-siveconditions[37°C]
toensuretheaccumulation of boththepolyprotein
precursor[MW, 130,000]
and the structuralprotein products.)
Theslight
differenceinmigration
ratemaybe duetothe mutationin
ts3,
which is knowntoresideinthecapsid
protein,
orto someposttranslational
covalent modi-fication of thesame which occursinvivo,
orboth(33).
Weconclude that the
capsid protein
is veryefficiently
synthe-sized and cleaved from the nascent chain in our in vitrosystem. This is in agreement with
previous
translationsperformed
with26S RNAisolatedfrominfectedcells (7, 9,11, 12, 19,
21, 41).
Someofourwild-type
translations weremore efficientand allowed the
synthesis
ofa distinct [image:4.612.347.526.364.624.2]high-MW
protein species (Fig.
2,
lane2)
inadditiontothecapsidprotein.
This 97-kDaproduct
must correspond to theC-terminal
portion
ofthepolyprotein (containing
thep62,6K,and El
sequences)
that is left aftercleavage
ofthe capsidprotein
(19, 33).
No materialcorresponding
tothefull-length
polyprotein
observed in ts3-infected cells(pl30)
(lane 4) accumulates in thewild-type
RNA translations (lane 2). Note thatlanes1and 6(see
alsoFig.
3,lanes2and4) showanalyses of the
polyprotein
with a mutatedcapsid
protein
thatwill be described below.
Protease activity resides within the capsid protein. The protease
activity
encodedby
thewild-type
RNA wasmapped
by progressively truncating
from the 3' end the cDNAtemplate
used for thesynthesis
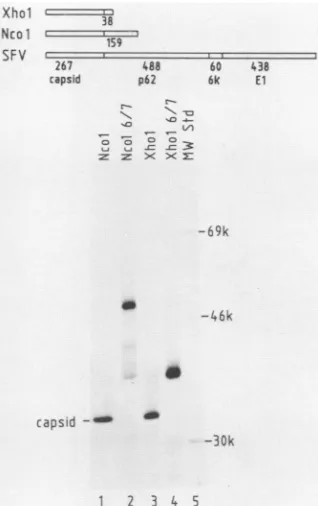
of the RNA. The RNA prepared from templates cleaved with the enzymes NcoI andXhoI encode proteins that nowcontain only 159 and 38 residues of p62 attached to thecapsid
protein,
respectively(Fig. 3,top).Ifthese
proteins
didnotcontainan active protease we would expect the accumulation of pre-cursorsof 50 and 37kDa, respectively.Thefluorogram
(Fig.
3, bottom)shows that the 33-kDacapsidband isproducedin thelysates programmedwith bothtruncated RNAs(lanes 1 and3). The C-terminal fragments released
by
thecleavage
are toosmalltobe observedon ourgels.Sincenodetectable amountof 50- and 37-kDa precursor moleculesaccumulates,
thecleavageof thecapsidappearsinbothcases toberather efficient. This demonstratesdirectly
that the proteaseactiv-ityresides within thecapsid protein, since it isvery
unlikely
that the 38-residuefragmentofp62has suchanactivity.
Changes at serine-219 destroy the proteaseactivity. As first notedby Boegeetal. (6),the sequencearoundserine-219 of thecapsidof SFV(serine-215in Sindbisvirus)has
homology
with a tetrapeptide sequence conserved around the activeresidue in several animal serine proteases. Inaddition,theaa substitutionresponsiblefor thecleavagephenotype ofsome
~> ~>
A
L/ 'iVri>
B r
p130--92k
-92k --b9k
-46k
- -30k
1 2 3
-69k
E1,
2[I
-46k
capsid
-30k
4 5 6
FIG. 2. Cell-free translations of RNA transcribed from full-length wild-typeandmutantSFVcDNA. Thein vitro transcription-translation ofplasmids pGSFV (labeledSFVinlanes2and5) and pGSFV6/7(labeledSFV6/7 in lanes1and6) linearized with PvuII
wascarriedout asdescribedinMaterialsandMethods. The trans-lation products from two different experiments were separated eitheron a10%gel(A)oron an8%gel (B). Lysateobtained from BHKcells infectedat 37°CwithSFV-ts3 was included inthe gel shown inlane4.Standard molecularweightmarkersarealsoshown (describedindetail in Materials andMethods).Thefigurerepresents
afluorogramof thegel.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
ts mustants ofSindbis virus (ts2 and ts5) has been shown to map atresidue218, near the putative active serine-215. We
wished to further test the hypothesis that the capsid is a serine protease by changing the sequence at the serine-219
residueanddetermining the cleavage phenotype in vitro with
complete and truncated precursors. Taking advantage of a
NaeIsite that overlapsthecodon forglycine-220,thecDNA
was mutagenized in vitro by using the enzyme BAL 31 and
linker addition,asdescribed in Materials and Methods. The nature of the changes introduced in one of the mutated
cDNAsequences (SFV6/7)wasobtained by DNA
sequenc-ing (data not shown). The deduced wild-type and mutant
protein sequences are shown in Fig. 4: a new arginine
residue was introduced on the N side of serine-219, and
glycine-220 was changed to threonine. Transcription-translation experiments with the full-length and truncated
mutatedtemplates were carried out as described above for
the wild-type one. The results with the full-length template
(Fig. 2, lanes 1 and 6) clearly show that no capsid can be
produced from the mutated cDNA. Instead, one observes
the accumulation of a high-MW band, absent from the
wild-type translations. Thisnew band comigrated with p130
produced inBHK cells infected with ts3 SFV (lane 4) and most probably corresponds to the full-length unprocessed
polyprotein.
(Note that many other medium-sized productsareseeninlanes 1 and 6. Theseprobablyrepresentvarious runoff products formed during translation ofthis very long mRNAinvitro.) Weconclude thatthe limitedchanges that we haveintroduced clearly destroy the protease activity. As
Xhol
38
Nco I z
159
SFV
267 488 60 438
capsid p62 6k El
X ' X
s0 m0
o o ( )< 2
-69k
-46k
capsid -0 do
-30k
1 2 3 4 5
FIG. 3. Analysis ofthe autoprotease activity ofthe capsid by truncation of the wild-type and mutant cDNA templates. Top, Diagramsof theexpectedtranslationproductsof the RNAprepared
fromthefull-lengthcDNA(SFV)and from cDNAs truncated with therestrictionenzymesNcoI and XhoI. The sizes of the individual
capsid p62andElproteinsand those of theexpected p62fragments
arealso indicated. Transcription-translation wascarriedoutasfor
Fig.2. Theproductswereseparatedon a10%gel.Thefluorogramof
thatgel ispresentedinthe lower part of thefigure.Thenatureofthe
templateis indicated above each lane.
w.t.
capsid mutant
C () p62 6k El
r6IyvAsPSer GlTy-(219)
c x p62 6k El
FIG. 4. DiagramoftheSFV polyprotein,indicating the sequence of theconserved and modified tetrapeptidethought to beinvolvedin theautoproteaseactivity of the capsid protein.
expected, translations withthetruncated mutated templates
(XhoI 6/7andNcoI 6/7) did not produce capsideither, but,
instead,theyproducedlargerproteins that have the mobili-tiesexpected fortheunprocessedtruncated precursors(Fig.
3, lanes 2and4). These resultsfurther demonstrate thatthe
cleavages observed withthe wild-type templates were
spe-cific and required the presence of a wild-type capsid se-quence. We believe that the mutation does not act by changing the structure of the cleavage site, since (i) the
changes were introduced 48 residues away fromthe siteof action ofthe protease and (ii) Aliperti and Schlesinger (4)
have shown that the introduction of aa analogs in the
substrate didnotpreventcleavageby the wild-typeprotease. Ourresultssupport thenotionthat theregion around
serine-219isimportant forproteolysisand that thecapsidis aserine
protease. Capsidprotease isnotinvolved incleavageofthe
p62-6Kjunction.
Thein vitro cleavageofthe6Kjunctionsandreleaseofthe
p62andElproteins has been observed onlyin the presence
ofmicrosomes (7, 19). Asdiscussed above, weexpect that the
6K-El
cleavage is catalyzed by signal peptidase atthe luminal surfaceof theERmembrane(36).This doesnotruleoutthepossibility that the other cleavagebetweenp62 and
El occursinthe cytoplasm, however.The latterprocessing
eventmayrequiremembranesfortheattachmentof the p62
protein andpropertopologyofthe 6Kpeptide relativetothe ER membrane. Toaddressthe possible involvement ofthe
capsid proteasein thecleavage ofthe 6Kjunctions,thegene
for a hybrid protein was constructed that was missingthe
capsid sequence but maintained the regions thought to be
important for correct topogenesis of the 6K peptide. The
construction ofa hybridgene with the cDNAs fora secre-toryprotein, chicken lysozyme, and the SFVpolyprotein is outlinedin
Fig.
1 anddescribed in moredetail inMaterialsand Methods. The resulting hybrid protein, Lys-El, is representedin Fig. 5. Allbutthe C-terminmal2 residuesof
thelysozymesequence arepresent,includingthe 18-residue cleavable signal sequence. This was substituted for the
capsid and most of the p62
protein.
Thehybrid
protein
retains the transmembrane segment and tail of the p62 protein in addition to the total 6K and El sequences. We expect that during translation ofthat
hybrid
protein
in the presenceofmembranes, translocationwillbeinitiatedby
thelysozyme
signal
sequence, then arrestedby
the transmem-brane region of the p62 protein, andfinally
reinitiated to allow the translocation of theElprotein, leaving
thetailofp62(30 aa)and
possibly
somepart of the 6Kpeptide
exposed
in thecytoplasm. Upon
translocation,
thesignal
sequenceof lysozyme, as well as the 6K-Eljunction,
should be effi-ciently cleavedby
signal
peptidase.
The release of the 6Kpeptide will be
dependent
onwhethercapsid
isinvolved in thep62-6Kcleavage.Incasecapsid
isnotinvolved,
the 6Kpeptide should be
generated
in addition to aon November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.345.532.70.146.2] [image:5.612.103.262.377.630.2]1306 MELANCON AND GAROFF
lysozyme
18 129
127
66
I
11s) (
267 433 1 24 1 31 60 412') 24 V2
[image:6.612.61.295.73.167.2]Capsid p62 6Kpeptide E1
FIG. 5. Diagram of the hybridprotein constructed tostudythe involvement of the capsid protease in the processing of the 6K peptide. The intact proteins encoded by the chicken lysozyme cDNA and the SFV26S cDNAareshownassingle lines above and below, respectively. The vertical bars represent proteasecleavage sites, and the wavy lines indicate the approximate positionof the lipidbilayer. The portions of thetwoproteins present in the hybrid areshown in the middle as boxes connectedbyavertical line. The size of theprotein domains bound by the bars and wavy lines are also given.
bound form of the lysozyme molecule (MW, 18,5000) and
finallyEl. On the otherhand, werecapsid requiredfor this cleavage event, the 6K peptide would remain as a C-terminal
extension ofthetransmembranelysozyme.Theapproximate MWofsuchahybrid is 26,000.
Theresults of the cell-free transcription-translation with thehybridgene areshowninFig. 6 and 7. To detect the 6K
peptide (Fig. 6), translations were performed with
L-:E: vr
!:
L30k -SM 21.5k-12.5k
[35S]cysteine (the 6K peptide has five cysteines and onlyone methionine) and the products were separated on a high-resolution gel containing urea. Under suchconditions, one can detect the appearance of a small band (lane 4) that
comigrates with the 6-kDa band produced in BHK cells infected with SFV (lane 2). This bandcanariseonly if both 6Kjunctionswerecleavedin vitro. Note that theprocessing of the 6K junctions occurs only in the presence of microsomes (compare lanes 3 and 4). Figure 7 shows the analysis ofa L-[35S]methionine translation on a 10% poly-acrylamide gel, onwhich the larger products canbebetter resolved. In the absence ofmicrosomes, amajor product of the size expected for the unprocessed Lys-El hybrid (65 kDa) accumulated (Fig. 7, lane 2). A minor abortive product migratingat50.5 kDawasalso present. When thetranslation wascarried out in the presence of microsomes (lane 3), less Lys-El precursor was seen and two new major bands appeared: one migratingat18.5kDa and anotherone migrat-ingat50 kDa (just below the abortive product of 50.5 kDa). The bandat18.5 kDamostprobably represents membrane-bound lysozyme which arises from (i) translocation of lysozyme with concomitantcleavage of the signal sequence, (ii) arrest of translocation at the transmembrane region of p62, and (iii) correct cleavage of the p62-6K junction. Note that no band with anapparent MW of26,000(corresponding
y ys-El
69k --y
4w _d_o
46k
-30k
-
6.5k-- - 6k peptide 18k
-3.3k- A r,
in vitro -M +M
1 2 3 4
FIG. 6. In vitro generation of the 6K peptide. Transcription-translation ofthelysozyme-El hybrid protein was carried out in the presence(lane 4) and absence (lane 3) of membranes, as described in Materials and Methods, with
L-[35S]cysteine
forlabeling. The prod-ucts were separated on a 22% gel containing 6 M urea. The fluorogramis presented. Lysate from BHK cells infected with SFV was also included (lane 2). The MW markers (low range) are described in Materials and Methods.MM M M
p p p TX
1 3 4 5 6
FIG. 7. Cell-free translation of thelysozyme-Elhybrid protein. Transcription-translation was carried out as described in Materials and Methods.Translations were done in the absence (lane 2) and presence(lanes3to6) of salt-washed microsomes (M). Some of the translationproducts were treated with exogenous proteinase K (P) for30(lanes4and6)or60(lane 5) min intheabsence(lanes4and 5)orpresence(lane 6) of Triton X-100 as described in Materials and Methods.
IS
.:
4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.390.481.337.649.2] [image:6.612.114.250.385.645.2]to a membrane-bound lysozyme with the 6K peptide at-tached) is observed. The above interpretation is supported by the protease challenge experiments shown in lanes 4 and 5. The membrane-bound lysozyme is resistant to the exogenous protease K and is shifted down by about 2 kDa. This is exactly the shift expected if translocation has oc-curred, the anchor of p62 spans the membrane, and the tail of p62 remains exposed to the cytoplasm (13, 20). As expected, the patternwas not affected by doubling the time of digestion from 30 to 60 min (compare lanes 4 and 5). No protection from the protease was observed when the diges-tion was carried out in the presence of Triton X-100 for 30
min(lane 6). The other major band, the one with an apparent MW of 50,000 (lane 2), most probably represents glycosyl-atedEl. It is protected from the added protease (lanes 4 and 5) and comigrates with glycosylatedElobtained from SFV-infected BHK cells which have been pulse-labeled with
L-[35S]methionine(data not shown) (36). The weak protected band migrating in front of glycosylated Elat approximately 48 kDa has been observed before in vitro translations and represents translocated unglycosylated El chains (36). We conclude that the cleavage of the 6K junctions does not require capsid but, as discussed below, does require a membrane-associated host protease. We further conclude that the transmembrane region of p62 can anchor the luminal domain of a foreign protein. Similar observations have been made with the anchors of other transmembrane proteins (24, 46).
DISCUSSION
We have established an in vitro transcription-translation system for studying the cotranslational processing of the SFV structural polyprotein. Since the RNA used for the translations is produced in vitro with the SP6 polymerase, the cDNA templates can be modified at will and the effect on the processing can be conveniently assessed in a rabbit reticulocyte cell-free system. The in vitro system was used first to study the protease responsible for the release of the capsid protein. We demonstrated directly that the activity resides within the structural protein capsid by using a combination of two approaches. (i) The cDNA used for transcription was truncated with restriction enzymes, and we showed that a capsid precursor containing only 38 additional residues from the N terminus of the p62 protein was very efficiently cleaved in vitro. (ii) The cDNA was mutagenized at the site of the putative active serine-219
residue, and this was shown to completely abolish the cleavage of the capsid protein. Since theprocessingrequires de novo synthesis (4) and since it is very unlikely that the 38-residue fragment of p62 has any activity, we conclude that the capsid is an autoprotease, responsible for its own release from the polysome.
The evidence to suggest that the SFVprotease isaserine protease has been recently reviewed by Hahn etal. (25). In the work described in this paper, we directed our mutationto the serine-219 thought to be part of the catalytic triad of the serine protease activity in the capsid and found that the mutation completely abolishedcleavage. Sincethemutation
is far removed fromthe siteofcleavage (48residues away), it is unlikely to act by changing the nature ofthe substrate. Our results therefore support the notionthat the capsid is a serine protease. Previous attempts to obtain in vitro direct evidence by using specific inhibitors such as tolylsulfonyl
phenylalanyl chloromethyl ketone have failed (4) and were not repeated inthis study. A definitive proofofthecatalytic
[image:7.612.322.555.68.149.2]a b c d
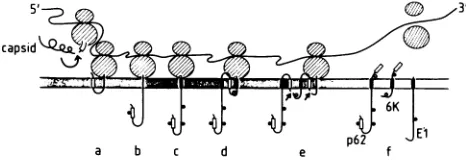
FIG. 8. Summary of the various cotranslational protease cleav-ages which are required to generate the SFV structural proteins. Details are given inthe text.
role ofserine-219 and the mechanism ofthe
capsid
protease in general requires the use of a series of mutations withwell-defined single-residue substitutions around each of the residues at theproposed active site.
The residues postulated to be involved in the serine protease activity of thecapsidmap to theC-terminalpartof
the molecule (25). This part, in contrast to the N-terminal one, ishighlyconserved (14, 39).
Interestingly,
this homol-ogy has recently been extended tothe
coatproteins
of picornaviruses (S. Fuller and P. Argos, unpublisheddata),
the crystal structure of some of which hasrecently
been solved (28,40).Onthebasis of thisfinding
(and
thefactthat the alphavirus nucleocapsid is arranged with the same(tri-angulationnumber,T = 3) icosahedral symmetryasthoseof
the picornaviruses; S. Fuller andJ.
Dubochet,
unpublished
data), it waspredicted that the C-terminalpartof thecapsid
polypeptide is folded into a eight-strand
P-barrel
structure typical of the shell domains found in all the available high-resolution structures oficosahedral viruses(1, 27,
28,
40). Theregionsthoughttobe involved in theserine
proteaseactivity ofthealphavirus
capsid
coincide on one side ofthepredicted 3-barrel. The positioning of the protease active site within a structural element of the virus
clearly
differsfrom the arrangement in picornaviruses. The
picornavirus
polyprotein is composed ofa structural and anonstructural
region. Almost all the cleavages,
giving
rise to both struc-tural and nonstrucstruc-turalproteins,
arecatalyzed
by
two proteases which are located in the nonstructural part of themolecule (fordiscussion, see reference
44).
Thecapsid of SFV appears tobeinvolved in
only
asingle
proteolytic cleavage event, which releases the C sequence from thatofp62.The fact thatweobserved correct process-ing at the 6K peptide in the Lys-Elhybrid
protein
demon-strates clearly that the
capsid
protease is not involved in these other cleavage events of the SFV structuralpolypro-tein. At present, we favor a model in which both 6K
cleavagesarecatalyzed
by
the host proteasesignal
peptidase
in the ER lumen (see also reference
39).
This model issupported bythe facts that
(i)
cleavages
occurin vitroonly
in the presence of microsomal membranes and
(ii)
bothcleavagesites arepreceded
by
asignal
peptide-like
sequence (45), i.e., a hydrophobic stretch of aaplus
aparticular
distribution around thecleavage
site. These features are present at the C-terminal ofp62
and 6Kin allspecies
of thegenus Alphavirus in which these sequences have been deter-mined (SFV [18], Sindbis virus
[39],
and Ross River virus [14]). Figure 8 summarizesschematically
our model for the way in which we think that the various structuralproteins
are formed from the
polyprotein.
Thedrawing
first illustrates theautoproteolysis
ofthecapsid
protein
occurring
in cison the nascent chain. Acis-acting
capsid
protease must beinvolved toaccountfor the first
capsid
protein.
Theexperi-ments ofAliperti and
Schlesinger
(4)
suggest thatcapsid
can fon November 10, 2019 by guest
http://jvi.asm.org/
alsoactintrans. The
newly exposed
Nterminusofp62acts as asignal
sequence andinitiatesthetranslocation ofthep62protein (stage
"a" in thedrawing).
Thesignal
sequence(represented by
an emptyrectangle)
is not cleaved butbecomes
glycosylated
andtransferredtothe lumen(stage b).Translocation of the
p62 protein
is arrested by the trans-membrane segment,represented by
asolidrectangle
(stagec).
Apossible
arrangement of thep62
tail and 6Kpeptide
allowing cleavage by signal peptidase
for both 6Kjunctions
isdepicted
instages d andeof thedrawing.
Thehydrophobicregion
at the C terminus ofp62(residues
395 to428,
openrectangle)
could act as asignal
sequence that initiates the translocation ofthe 6Kpeptide through
the ER membrane. The 6Kpeptide
isshown anchored in the membrane viaaninternal, 15-residue-long apolar region (residues
22 to36,
solidrectangle),
with the N terminus in the lumen. It has been shown that a 15-residuehydrophobic region
islong
enough
to serve as a membrane anchor(2, 3,
13,15,
16).Note that such stretches of
hydrophobic
residues are also present in the middle ofthe 6Kregion
of Sindbis virusand RossRiver virus(14,
39).
Thehydrophobic
Cterminus ofthe 6Kpeptide (residues
45to58,
openrectangle),
asmentionedabove,
acts asthesignal
fortranslocationof the Elprotein
(36). Upon cleavage by signal peptidase,
the tailofp62
mustflip
back on thecytoplasmic side,
since it is known to beavailable to antitail antibodies
microinjected
in thecyto-plasm (13;
L.Roman,
unpublished observations).
The same isthought
tohappen
with the El translocationsignal
(stagef).
This kind offlip-back
has beenreported
for thesignal
sequence ofpreprolactin
after it had been fused to the C terminus of thealpha-globin protein (38). Finally,
stage f shows the Elprotein fully
translocated andanchoredby
itstransmembrane segment
(solid rectangle).
We
hope
to use our in vitrotranscription-transla-tion/translocationsystemtotestthe model of 6K
processing
and
topology.
Thetemporal relationship
between thetwo6Kcleavages
and thetranslocationof the Elprotein
canalso bestudied with this system.
ACKNOWLEDGMENTS
WearegratefultoT. Kreis and G. Warrenforgenerousgiftsof antibodies. ThanksarealsoduetoB.Dobbersteinforprovidingsalt washedmicrosomes.Furthermore,we are mostgratefultoS. Fuller forthecommunication ofunpublishedresultsandfor criticalreading of the manuscript. We thank L. Roman and D. Cutlerfor useful discussions in theearlypartof this work.
P. MelanconwastherecipientofaNorthAtlanticTreaty Organ-izationpostdoctoral fellowship, obtainedthrough theNational
Sci-ence and Engineering Research Council of Canada, and of a short-termEuropean MolecularBiology Laboratory fellowship.
LITERATURE CITED
1. Abad-Zapatero, C.,S. S.Abdel-Meguid, J.E.Johnson,A.G. W. Leslie,I.Rayment,M.G.Rossmann,D.Suck,and T.Tsukihara. 1980. Structure ofsouthern bean mosaic virusat2.8 A resolu-tion.Nature(London)286:33-39.
2. Adams,G.A.,andJ.K.Rose. 1985.Structuralrequirement ofa membrane-spanningdomain forprotein anchoringand cell sur-facetransport. Cell41:1007-1015.
3. Adams,G.A., andJ.K. Rose. 1985. Incorporationofcharged amino acid into the membrane-spanning region blocks cell surfacetransport butnotmembraneanchoringofaviral glyco-protein. Mol. Cell. Biol. 5:1442-1448.
4. Aliperti, G., and M. J. Schlesinger. 1978. Evidence for an autoproteaseactivityofSindbis virus capsid protein. Virology 90:366-369.
5. Birnboin,H.C., andJ. Doly. 1979. Arapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic
AcidsRes.7:1513-1523.
6. Boege, U.,G. Wengler, G. Wengler, and B.Wittmann-Liebold. 1981.Primarystructureof thecoreproteinsof thealphaviruses SemlikiForestvirus and Sindbis virus.Virology 113:293-303. 7. Bonatti, S., R. Cancedda, and G. Blobel. 1979. Membrane
biogenesis: in vitrocleavage,coreglycosylation andintegration into microsomal membranes of Sindbis virusglycoproteins. J. Cell Biol. 80:219-224.
8. Bonatti, S., G. Migliaccio, G. Blobel, and P. Walter. 1984. Role of signal recognition particle in the membrane assembly of Sindbis virusglycoproteins. Eur. J. Biochem. 140:499-502. 9. Cancedda, R., R. Swanson, and M. J. Schlesinger. 1974. Viral
proteins formed in acell-free rabbit reticulocyte system pro-grammed with RNA from a temperature-sensitive mutant of Sindbis virus. J. Virol. 14:664-671.
10. Chen,E.Y.,and P. H.Seeburg. 1985. Supercoilsequencing: a fast and simple method for sequencing plasmid DNA. DNA Lab. Methods4:165-170.
11. Clegg, J.C.S.,andS.I. T.Kennedy. 1974. In vitrosynthesis of structuralproteins of Semliki Forest virus directedby isolated 26SRNAfrom infected cells. FEBSLett.42:327-330. 12. Clegg, J. C. S., and S. I. T. Kennedy. 1975. Translation of
Semliki Forest virus intracellular26S RNA: characterisation of theproducts synthesized in vitro.Eur.J.Biochem. 53:175-184. 13. Culter, D. F., P. Melancon, and H.Garoff. 1986. Mutants of the membrane-binding region of Semliki Forest virus E2 protein. II. Topology and membrane binding. J. Cell Biol. 102:902-910. 14. Dalgarno, L., C. M. Rice, andJ. H. Strauss. 1983. Ross River
virus 26S RNA: complete nucleotide sequence and deduced sequence of the encoded structural proteins. Virology 129: 170-187.
15. Davis,N.G.,J. D. Boeke, and P. Model.1985. Finestructureof a membrane anchordomain.J. Mol. Biol. 181:111-121. 16. Davis, N.G.,and P. Model. 1985. Anartificial anchor domain:
hydrophobicity suffices to stop transfer. Cell 41:607-614. 17. Garoff,H. 1985. Using recombinantDNAtechniquesto study
protein targeting in the eukaryotic cell. Annul. Rev.Cell Biol. 1:403-445.
18. Garoff, H., C. Kondor-Koch,and H.Riedel. 1982. Structure and assembly of alphaviruses. Curr. Top. Microbiol. Immunol. 99:1-50.
19. Garoff, H.,K. Simons,and B. Dobberstein. 1978.Assembly of the SemlikiForestvirus membraneglycoproteins in the mem-brane of the endoplasmic reticulum in vitro. J. Mol. Biol. 124:587-600.
20. Garoff, H.,and H.Soderlund.1978.Theamphiphilic membrane glycoproteins of Semliki Forestvirusareattached to the lipid bilayer by their COOH-terminal ends. J. Mol. Biol. 124: 535-549.
21. Glanville, N., J. Morser, P. Uomala, and L. Kaariainen. 1976. Simultaneous translation of structural and nonstructural pro-teins from SemlikiForest virusRNA in two eucaryotic systems in vitro. Eur. J.Biochem.64:167-175.
22. Green,J., G. Griffiths,D. Louvard, P. Quinn, and G. Warren. 1981. Passage of viral membrane proteins through the Golgi complex.J. Mol.Biol. 152:663-698.
23. Gruss, P.,N.Rosenthal,M.Konig, R. W. Ellis, T. Y. Shih, E. M. Scholnick,and G. Khoury. 1982. The expression of viral and cellularp21 rasgenes using SV40as avector, p. 13-19. In J. Gluzman(ed.), Eukaryotic viral vectors. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
24. Guan, J.-L.,andJ. K. Rose. 1984. Conversion of a secretory protein intoatransmembrane protein results in its transport to theGolgi complex but not to thecell surface. Cell 37:779-787. 25. Hahn,C. S., E.G.Strauss,andJ.H. Strauss. 1985. Sequence analysis ofthreeSindbis virus mutants temperature-sensitive in the capsid protein autoprotease. Proc. Natl. Acad. Sci. USA 82:4648-4652.
26. Hanahan,D.1983.Studiesontransformation of Escherichia coli withplasmids. J. Mol. Biol. 166:557-580.
27. Harrison,S.C.,A.J. Olson, C.E.Schutt, F. K. Winkler, and G. Bricogne. 1978. Tomatobushy stuntvirus at 2.9 A resolution. Nature(London) 27:368-373.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
28. Hogle, J. M., M. Chow, and D.J. Filman. 1985. Three-dimen-sional structure of poliovirus at 2.9 A resolution. Science 229:1358-1365.
29. Holmes, D. S., and M.Quigley. 1981. A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 114: 193-197.
30. Konarska, M. M., R. A. Padgett, and P. A. Sharp. 1984. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell 38:731-736.
31. Kondor-Koch, C., B. Burke, and H.Garoff. 1983. Expression of Semliki Forest virus proteins from cloned complementary DNA. I. Thefusion activity of the spike glycoprotein. J. Cell Biol. 97:644-651.
32. Krieg, P., R. Strachnan, E. Wailis, L. Tabe, and A. Colman. 1984. Efficient expression of cloned complementary DNAs for secretoryproteins afterinjection into Xenopus oocytes. J. Mol. Biol. 180:615-643.
33. Lachmi, B., N.Glanville, S. Keranen, and L. Kiairiainen. 1975. Tryptic peptide analysis of nonstructural and structural precur-sorproteins from Semliki Forest virus mutant-infected cells. J. Virol. 16:1615-1629.
34. Lusky, M., and M. Botchan. 1981.Inhibition of SV40 replication in simian cells by specific pBR322 DNA sequences. Nature (London) 293:79-81.
35. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982.Molecular cloning:alaboratory manual. ColdSpringHarborLaboratory, ColdSpring Harbor, N.Y.
36. Melancon, P.,and H.Garoff. 1986.Reinitiation oftranslocation in theSemliki Forest virus structural polyprotein: identification of thesignal for the El glycoprotein. EMBO J. 7:1543-1550. 37. Melton,D.A.,P. A. Krieg, M. R.Rebagliati, T. Maniatis,and
M. R. Green. 1984. Efficient in vitro synthesis ofbiologically active RNA and RNA hybridization probes from plasmids containingabacteriophage SP6 promoter. Nucleic Acids Res. 12:7035-7056.
38. Perara, E.,and V.R.Lingappa. 1985. A formeramino terminal signal sequence engineered to an internal location directs translocation ofboth flanking protein domains. J. Cell Biol. 101:2292-2301.
39. Rice, C. M., and J. H.Strauss.1981.Nucleotide sequence ofthe 26S mRNA of Sinbis virus and deduced sequence of the encoded virus structuralproteins. Proc. Natl. Acad. Sci. USA 78:2062-2066.
40. Rossmann, M. G., E. Arnold, J. W. Erickson, E. A. Frankenberger, J. P.Griffith, H.-J. Hecht, J. E. Johnson, G. Kamer,M.Luo,A.G.Mosser, R. R.Rueckert,B.Sherry,and G.Vriend. 1985. Structure ofahumancommoncold virus and functional relationship to other picornaviruses. Nature (Lon-don) 317:145-153.
41. Simmons,D.T.,andJ.H.Strauss.1974.Translation ofSindbis virus26S RNA and 49SRNAinlysates of rabbitreticulocytes. J. Mol. Biol. 86:397-409.
42. Swank, R. T., and K. D. Munkres. 1971. Molecular weight analysis of oligopeptides by electrophoresis inpolyacrylamide gel with sodiumdodecyl sulfate. Anal. Biochem. 39:462-477. 43. Timm, B., C. Kondor-Koch, H. Lehrach, H. Riedel, J.-E.
Edstrom, and H. Garoff. 1983. Expression of viral membrane proteins from cloned cDNA bymicroinjection into eukaryotic cell nuclei. Methods Enzymol.96:496-511.
44. Toyoda, H.,M.J.H.Nicklin,M.G. Murray, C. W. Anderson, J. J. Dunn, F. W. Studier, andE. Wimmer. 1986. A second virus-encodedproteinase involved inproteolytic processing of poliovirus polyprotein. Cell 45:761-770.
45. vonHeine, G. 1985. Structural andthermodynamic aspects of thetransfer of proteins into and across membranes.Curr. Top. Membr.Transp. 24:151-179.
46. Yost, C. S., J. Hedgpeth, and V. R. Lingappa. 1983. A stop transfersequence confers predictable transmembrane orienta-tionto apreviously secreted protein in cell-free systems. Cell 34:759-766.