Epstein-Barr Virus Latent Membrane Protein 2A Regulates c-Jun Protein through Extracellular Signal-Regulated Kinase
Full text
Figure
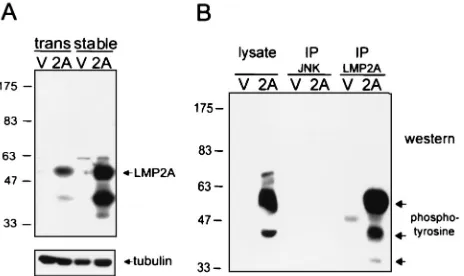
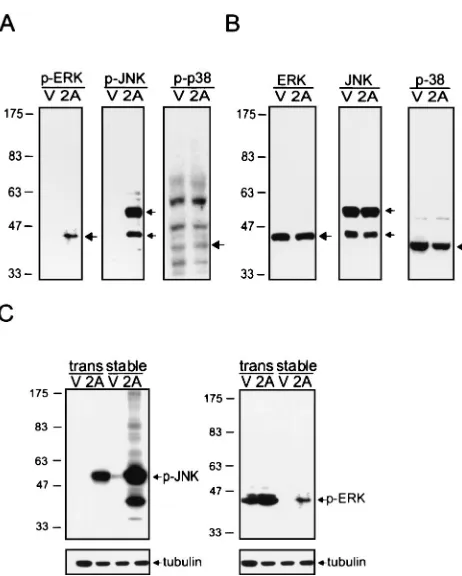
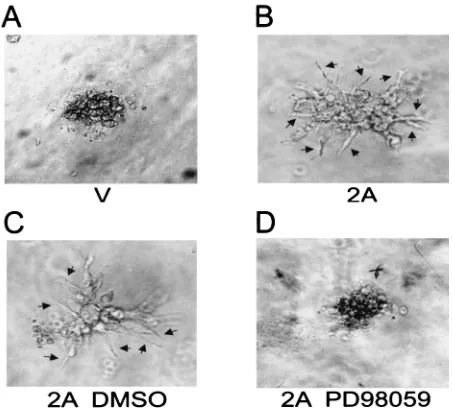
Related documents
Von den eingeschlossenen Patienten wurde 42 aufgrund einer isolierten TI behandelt, während 22 Patienten kombiniert an der Mitral- und Trikuspidalklappe behandelt
The groups of patients that underwent AT and ST showed significant clinical improvements in the motor symptoms of PD, as well as in functional capacity, indicating that pre-
Such as murine cytomegalo virus infection, avian influenza (H9N2), Chistosoma Mansoni Infection, PPR virus, Broad bean mosaic virus, HIV virus, Hepatitis C Virus,
The employment rate of graduates in the different disciplines is not uniform: In the last year of school about 90% of graduates of engineering and
A total of 878 patients were studied in the treatment-experienced trials of raltegravir: one phase IIb clinical study and 2 large identical phase III studies performed
In summary, to our knowledge, this was the first prospective intervention study to show that universal screening of respiratory specimens from individuals with presump- tive TB using
A study of artificial bee colony variants for radar waveform design Zhang and Zhang EURASIP Journal on Wireless Communications and Networking (2016) 2016 13 DOI 10 1186/s13638 015 0510
You are required to teach a comprehensive seven-day unit Before teaching the unit, describe contextual factors, identify learning goals based on your state or district
