Splicing-Dependent Subcellular Targeting of Borna Disease
Virus Nucleoprotein Isoforms
Shohei Kojima,
a,bRyo Sato,
cMako Yanai,
a,bYumiko Komatsu,
a,dMasayuki Horie,
a,eManabu Igarashi,
fKeizo Tomonaga
a,b,gaLaboratory of RNA Viruses, Department of Virus Research, Institute for Frontier Life and Medical Sciences (inFront), Kyoto University, Kyoto, Japan
bDepartment of Mammalian Regulatory Network, Graduate School of Biostudies, Kyoto University, Kyoto, Japan
cDepartment of Medicine, Faculty of Medicine, Kyoto University, Kyoto, Japan
dKeihanshin Consortium for Fostering the Next Generation of Global Leaders in Research (K-CONNEX), Kyoto University, Kyoto, Japan
eHakubi Center for Advanced Research, Kyoto University, Kyoto, Japan
fDivision of Global Epidemiology, Research Center for Zoonosis Control, Hokkaido University, Sapporo, Japan
gDepartment of Molecular Virology, Graduate School of Medicine, Kyoto University, Kyoto, Japan
ABSTRACT
Targeting of viral proteins to specific subcellular compartments is a
fun-damental step for viruses to achieve successful replication in infected cells. Borna
disease virus 1 (BoDV-1), a nonsegmented, negative-strand RNA virus, uniquely
repli-cates and persists in the cell nucleus. Here, it is demonstrated that BoDV
nucleopro-tein (N) transcripts undergo mRNA splicing to generate truncated isoforms. In
com-bination with alternative usage of translation initiation sites, the N gene potentially
expresses at least six different isoforms, which exhibit diverse intracellular
localiza-tions, including the nucleoplasm, cytoplasm, and endoplasmic reticulum (ER), as well
as intranuclear viral replication sites. Interestingly, the ER-targeting signal peptide in
N is exposed by removing the intron by mRNA splicing. Furthermore, the spliced
isoforms inhibit viral polymerase activity. Consistently, recombinant BoDVs lacking
the N-splicing signals acquire the ability to replicate faster than wild-type virus in
cultured cells, suggesting that N isoforms created by mRNA splicing negatively
regu-late BoDV replication. These results provided not only the mechanism of how mRNA
splicing generates viral proteins that have distinct functions but also a novel
strat-egy for replication control of RNA viruses using isoforms with different subcellular
localizations.
IMPORTANCE
Borna disease virus (BoDV) is a highly neurotropic RNA virus that
be-longs to the orthobornavirus genus. A zoonotic orthobornavirus that is genetically
related to BoDV has recently been identified in squirrels, thus increasing the
impor-tance of understanding the replication and pathogenesis of orthobornaviruses. BoDV
replicates in the nucleus and uses alternative mRNA splicing to express viral
pro-teins. However, it is unknown whether the virus uses splicing to create protein
iso-forms with different functions. The present study demonstrated that the
nucleopro-tein transcript undergoes splicing and produces four new isoforms in coordination
with alternative usage of translation initiation codons. The spliced isoforms showed
a distinct intracellular localization, including in the endoplasmic reticulum, and
re-combinant viruses lacking the splicing signals replicated more efficiently than the
wild type. The results provided not only a new regulation of BoDV replication but
also insights into how RNA viruses produce protein isoforms from small genomes.
KEYWORDS
bornavirus,
Mononegavirales
, RNA splicing
T
he regulation of the subcellular localization of proteins is critical for them to
precisely exert their function and trafficking. Therefore, eukaryotic proteins contain
several signal sequences, such as nuclear localization signals (NLSs), nuclear export
CitationKojima S, Sato R, Yanai M, Komatsu Y,
Horie M, Igarashi M, Tomonaga K. 2019. Splicing-dependent subcellular targeting of Borna disease virus nucleoprotein isoforms. J Virol 93:e01621-18.https://doi.org/10.1128/JVI .01621-18.
EditorRebecca Ellis Dutch, University of
Kentucky College of Medicine
Copyright© 2019 American Society for
Microbiology.All Rights Reserved. Address correspondence to Keizo Tomonaga, [email protected].
Received14 September 2018
Accepted14 November 2018
Accepted manuscript posted online12
December 2018
Published
OF VIRAL GENE EXPRESSION
crossm
19 February 2019
on November 6, 2019 by guest
http://jvi.asm.org/
signal (NESs), Golgi retrieval signals, endoplasmic reticulum (ER) retention signals, and
mitochondrion-targeting signals (1–4). In addition to these signal sequences,
posttrans-lational modifications and protein-protein interactions also determine the intracellular
distribution of proteins.
Viruses must unerringly control the subcellular localization of their proteins to
accomplish replication in host cells. To generate multiple proteins with diverse
func-tions and subcellular localizafunc-tions from relatively short viral genomes, viruses utilize
several transcription and translation mechanisms, such as cotranscriptional editing,
mRNA splicing, internal ribosome entry sites (IRESs), ribosomal shunting, leaky
scan-ning, non-AUG initiation, frameshifting, and readthrough (5, 6). For instance,
phospho-protein (P) mRNAs of many
Mononegavirales
are translated from downstream initiation
codons with leaky scanning and ribosomal shunting mechanisms, thereby generating
N-terminally truncated protein isoforms with different functions (7). Because
organelle-targeting sequences often locate at the N terminus, these P isoforms can localize to
distinct subcellular components.
mRNA splicing often deletes sequences encoding organelle-targeting signals in viral
proteins, producing isoforms with different subcellular localizations. In human T-cell
leukemia virus type 1 (HTLV-1) infection, alternative splicing of the bZIP factor results
in different subcellular distributions of its isoforms (8). Furthermore, the L6 region of
bovine adenovirus 3 expresses several isoforms with distinct subcellular localizations in
infected cells, which may arise by internal initiation of translation and alternative
splicing (9). However, the detailed mechanism of how viruses control the subcellular
localization of viral proteins by mRNA splicing has not been elucidated.
Borna disease virus 1 (BoDV-1) is a nonsegmented, negative-strand RNA virus that
replicates and transcribes in the nucleus. The BoDV genome harbors at least six open
reading frames (ORFs), as follows: nucleoprotein (N), X, P, matrix protein (M),
glycopro-tein (G), and large proglycopro-tein (L) (10). BoDV exploits the regulatory mechanism of the
intracellular localization of viral proteins to establish intranuclear persistent infection.
The intracellular localization of BoDV ribonucleoproteins (RNPs) is regulated by viral
proteins containing NLSs and NESs as well as their interactions. N, P, X, and L harbor
NLSs, and N and P contain NESs (11–18). Previous studies have demonstrated that the
interaction of accessory protein X with P enhances the nuclear export of P (19). In
contrast, P directly binds to N, leading to the retention of N in the nucleus (13, 20). N
transcripts encode two isoforms, namely, full-length isoform N (p40) and N-terminally
truncated isoform N
=
(p38), which is translated from the second initiation codon
downstream of the NLS (17). While N mainly localizes to the nucleus, the solo
expres-sion of N
=
is found in the cytoplasm (12, 13, 17). N
=
can modify the nuclear distribution
of P via their interaction (13). Furthermore, it has been reported that the expression
ratio of N and N
=
in cells is important for the elaborate control of BoDV polymerase
activity (21, 22). Thus, BoDV strictly regulates the intracellular localization of viral
proteins by intrinsic subcellular localization signals and protein-protein interactions to
accomplish viral replication and establish persistent infection in the nucleus.
BoDV utilizes the host mRNA splicing machinery for gene expression (23, 24). The
ORF of BoDV G overlaps those of M and L in a
⫹
1/
⫺
2 frame (10). To express M, G, and
L, the overlapping protein-coding region produces three different spliced transcripts
using the host pre-mRNA splicing machinery. Transcripts lacking introns I and II serve
as mRNAs for G and M, respectively (23, 24). mRNAs that lack both introns are for L (23,
24). Furthermore, rare alternative splicing has been observed in the transcripts
ex-pressed from the M/G/L overlapping coding region by reverse transcription-PCR
(RT-PCR) and Northern blotting (25). These reports have demonstrated that alternative
splicing of BoDV mRNAs plays a critical role for viral gene expression. However, no
splicing event has been demonstrated in BoDV transcripts, except for the M/G/L
polycistronic mRNAs.
The present study comprehensively analyzed the splicing junctions of BoDV
tran-scripts using next-generation sequencing (NGS) and discovered that the N transcript
contains two short introns and undergoes splicing. Although full-length N mainly
on November 6, 2019 by guest
http://jvi.asm.org/
accumulates at intranuclear viral replication sites, called viral speckles of transcripts
(vSPOTs), a spliced isoform of N accumulates in the ER. Mutational and structural
analyses revealed that intracellular localizations of spliced isoforms of N are regulated
by a combined mechanism with alternative initiation sites and splicing of the N
transcript. Interestingly, recombinant BoDVs (rBoDVs) lacking splicing signals in N
showed the ability to propagate faster than wild-type rBoDV (rBoDV-WT), suggesting
that the spliced isoforms of N negatively regulate BoDV replication. These results may
provide not only a new strategy for BoDV replication in the nucleus but also new insight
into the mechanism of how RNA viruses produce protein isoforms with different
subcellular localizations from a single transcript.
RESULTS
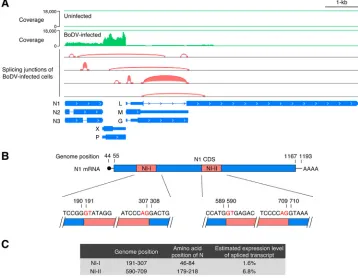
RNA sequencing defines the splicing landscape of BoDV transcripts.
To identify
BoDV splicing transcripts, transcriptome analysis of persistently BoDV (strain
He/80)-infected and unHe/80)-infected oligodendroglioma (OL) cells was performed by NGS (Fig. 1A).
Sequence reads were mapped to reference genomes of human and BoDV to avoid the
mismapping of host mRNAs to the viral genome. In addition to the previously reported
splicing patterns in the overlapping M/G/L coding region, two short spliced introns of
N, termed NI-I (nucleotides [nt] 191 to 307) and NI-II (nt 590 to 709), were detected
within the N sequence in the reads of BoDV-infected OL cells, suggesting that splicing
variants of BoDV N mRNA were produced in the infected cells (Fig. 1A). To verify the
splicing of N mRNA, RT-PCR was performed using RNAs from BoDV-infected cells. The
same splicing variants of N mRNA were observed by NGS (data not shown). Two newly
identified splicing variants in the N transcripts harbored canonical eukaryotic splicing
signals (GU-AG) and branchpoint sequences (Fig. 1B), strongly suggesting the
involve-ment of host splicing machinery. To estimate the expression levels of the N transcript
FIG 1Alternative splicing generates transcript variants of N. (A) IGV browser view showing the coverage and splicing junctions of mapped RNA reads from persistently BoDV-infected OL cells. (B) Map of the splicing donor and acceptor sequences of NI-I and NI-II. (C) Genome positions and amino acid positions corresponding to the N introns and estimated expression levels of spliced N transcripts. Expression levels of spliced N transcripts were estimated from the RNA-seq data of BoDV-infected OL cells using MISO (Mixture of Isoforms) software that quantitates the expression levels of alternatively spliced genes from RNA-seq data.on November 6, 2019 by guest
http://jvi.asm.org/
[image:3.585.41.399.75.353.2]variants, Mixture of Isoforms (MISO) software (26), which quantitates the expression
levels of alternatively spliced genes from RNA sequencing (RNA-seq) data, was used.
The levels of splicing of NI-I and NI-II introns were estimated to be 1.6% and 6.8%,
respectively (Fig. 1C), indicating that the expression levels of these splicing variants are
not high enough to be detected by Northern blotting (10, 27).
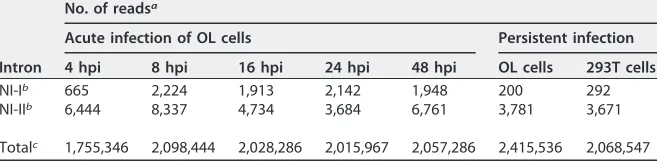
To elucidate if these splicing events are observed in the acute stage of infection,
amplicon deep sequencing of the N transcripts was performed. To obtain the N
transcript in the acute stage, OL cells were infected with BoDV, and total RNA was
extracted at 4, 8, 16, 24, and 48 h postinfection. As a control, total RNA of persistently
BoDV-infected OL and 293T cells was collected. Transcripts of N were amplified by
RT-PCR, and the sequences of the amplicon were analyzed by RNA sequencing. Splicing
variants of N mRNA were observed in both acute and persistent infection, indicating
that the spliced N transcripts are ubiquitously expressed in the course of BoDV infection
(Table 1).
Isoform N3 undergoes posttranslational modification.
After the splicing of NI-I
and NI-II, N transcripts retain the protein-coding sequence in the same reading frame
of N, suggesting that these variants produce truncated isoforms of N (Fig. 2A). To
further analyze the predicted N isoforms, we designated full-length N and the spliced
isoforms, which are translated from the first AUG codon of the N ORF, N1, N2, and N3,
and we designated those from the second AUG codon N1
=
, N2
=
, and N3
=
, respectively
(Fig. 2A). To investigate the expression pattern of N2 and N3 isoforms, expression
plasmids of N2 and N3 were generated. To prevent multiple splicing, silent mutations
were introduced at each splicing donor and acceptor site. While N2 was observed at the
predicted molecular weight (MW), N3 was observed at a lower position than the
expected size where the predicted molecular weight was 36 kDa, in both uninfected
and BoDV-infected cells (Fig. 2B). To discriminate the possibility that the transcripts
expressed from the plasmid encoding N3 undergo further unexpected splicing events,
N3 mRNA was synthesized
in vitro
and transfected into uninfected cells. As shown in
Fig. 2C, transfection of N3 mRNA also produced the shorter isoform, suggesting that
this isoform results from posttranslational modification of N3.
[image:4.585.42.374.86.168.2]To identify the modification of the shorter isoform, Flag-N3-Myc, which has 3
⫻
Flag
and Myc tags in the N and C termini, respectively, was expressed in uninfected cells.
While a 42-kDa band was detected by both anti-Flag and anti-Myc antibodies, the
30-kDa band was detected only by the anti-Myc antibody (Fig. 2D). Because the
molecular weight of the full-length product was approximately 40 kDa, the 42-kDa
band may represent the full-length Flag-N3-Myc product, and the 30-kDa form may
contain the C-terminal fragment of N3, designated N3C here (Fig. 2A). To evaluate if N3
=
can be processed into N3C, N3
=
was expressed from a plasmid. Although the expression
level of N3
=
was lower than that of N3, N3
=
also clearly expressed N3C (Fig. 2E),
indicating an intron NI-II-dependent production of N3C. The N3-encoding plasmid
potentially expresses N3
=
. To discriminate the influence of N3
=
on N3C expression from
the N3-encoding plasmid, we therefore made a plasmid encoding an N3 mutant,
named N3_M14A, which harbors mutations at the second ATG codon in the N3 ORF. As
TABLE 1Numbers of deep sequencing reads containing splice junctions
Intron
No. of readsa
Acute infection of OL cells Persistent infection
4 hpi 8 hpi 16 hpi 24 hpi 48 hpi OL cells 293T cells
NI-Ib 665 2,224 1,913 2,142 1,948 200 292
NI-IIb 6,444 8,337 4,734 3,684 6,761 3,781 3,671
Totalc 1,755,346 2,098,444 2,028,286 2,015,967 2,057,286 2,415,536 2,068,547
ahpi, hours postinfection.
bThe number of reads which harbored the splicing junction of the indicated intron. cThe number of reads mapped to the N transcript.
on November 6, 2019 by guest
http://jvi.asm.org/
shown in Fig. 2E, N3_M14A also produced N3C, indicating that both N3 and N3
=
can be
processed into N3C. Note that although the expression level of N3C seems to be high
in BoDV-infected cells compared to uninfected cells (Fig. 2B), it resulted from the
difference of primary antibodies used in these two panels. In addition, the signals of the
shorter isoforms appear to be weaker than those of the full-length proteins when
polyclonal anti-N antibodies are used for detection. This is probably due to the
difference in the numbers of epitopes that they harbor.
The N3C protein was sequenced to identify the N terminus of this product. To this
end, N3C with a Myc tag at the C terminus (N3C-Myc) was purified using an anti-Myc
tag antibody from transfected cells, and the sequence of purified N3C-Myc was then
analyzed by mass spectrometry (Fig. 2F). Peptides corresponding to amino acids (aa) 89
to 97 of N (AFVHGGVPR) were detected as the majority of the product, while no
N-terminal peptides (aa 1 to 81) were observed, indicating that N3C lacks the
N-terminal sequence and is started from aa 89 of N. Because the N terminus of N3C is
not a methionine, it is considered that N3 was cleaved into N3C by a host protease or
autoproteolysis.
N isoforms localize to distinct subcellular components.
To understand the
subcel-lular localization of N isoforms, we transfected N isoform expression plasmids in
persistently BoDV-infected HEK293T (293T) cells and visualized each isoform by
immu-nofluorescence analysis. We also transfected single-amino-acid substitution mutants of
N1, N2, and N3, named N1_M14A, N2_M14A, and N3_M14A, which do not express N1
=
,
N2
=
, and N3
=
, respectively, to exclude the possibility that the expression of N1
=
, N2
=
, and
FIG 2N isoforms expressed from splicing variants of N transcript. (A) Schematic representation of N isoforms. The amino acid positions corresponding to N1 are shown. Synonymous names are shown in parentheses. (B) Detection of N isoforms by Western blotting. Whole-cell lysates of uninfected and persistently BoDV-infected 293T cells transfected with plasmids encoding the indicated N isoforms were used. (C) Detection of N isoforms by Western blotting. Whole-cell lysates of uninfected 293T cells transfected with the mRNA encoding the indicated N isoforms were used. The asterisk represents nonspecific signals. (D) Detection of the N3 short form by Western blotting. The whole-cell lysate of uninfected 293T cells expressing Flag-N3-Myc was used. (E) Detection of N3C-Myc by Western blotting. Whole-cell lysates of uninfected 293T cells expressing the indicated N3 mutants were used. In the⫻10 sample, a 10-fold amount of the cell lysate was used for analysis. Coomassie brilliant blue (CBB) staining was used as a loading control. IB, immunoblotting; pAb, polyclonal antibody. (F) Detection of the N terminus of N3C. (Left) Purified N3C-Myc. N3C-Myc was purified from uninfected 293T cells expressing N3C-Myc using an anti-Myc tag antibody. Purified protein was stained with CBB. (Right) Coverage of N3C peptides and the N-terminal fragments of N3C detected by mass spectrometry. The arginine residue of which the peptide bond of the carboxy side is cleaved by trypsin during sample preparation is shown in blue.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:5.585.44.540.72.353.2]N3
=
affects the localization of the full-length isoforms. As described above, N1 and N1
=
mainly localize to the nucleus and accumulate at vSPOTs, the intranuclear replication
sites of BoDV. As shown in Fig. 3A, N2 was not incorporated into vSPOTs despite its
nuclear distribution. In contrast, N2
=
was localized in the cytoplasm. Furthermore,
transfection of N3 and N3
=
expression plasmids, both of which induce the N3C isoform
in cells (Fig. 2E), resulted in the distribution of the Myc tag signal only in the
cytoplasmic component. Interestingly, an immunofluorescence assay using an ER
marker revealed that the N3 isoform-accumulated component in the cytoplasm
corre-sponded to the ER. These observations revealed that N isoforms show different
subcellular localizations, indicating that N splicing may contribute to the distribution of
protein isoforms in BoDV-infected cells. We confirmed that the presence of the isoforms
FIG 3Subcellular localization and P- and chromatin-binding abilities of N isoforms. (A) Localization of splicing isoforms with a C-terminal Myc tag. Persistently BoDV-infected 293T cells were transfected with plasmids encoding the indicated constructs, and each N protein was detected using an anti-Myc tag antibody. vSPOTs were stained by an anti-P antibody, and the nucleus was counterstained with DAPI. Bars, 10m. (B) Immunoprecipitation analysis of N isoforms. (Top) Immunoprecipitation (IP) of N isoforms and coimmunoprecipitation (co-IP) of P. (Bottom) IP of P and co-IP of N isoforms. Uninfected 293T cells were transfected with plasmids encoding the indicated N and P constructs. IP and co-IP of N isoforms and P were detected by Western blotting. CBB staining was used as a loading control. (C) Chromatin-binding assay of N isoforms. Uninfected 293T cells were transfected with plasmids encoding the indicated N isoforms. (Left) Transfected cells were fractionated into cytoplasm, nucleoplasm, and soluble and insoluble chromatin fractions using micrococcal nuclease (MNase) digestion. (Right) Nuclei of transfected cells were fractionated into salt-extractable and insoluble fractions with 150 mM NaCl. Tubulin, HMGB1, HP1␣, and N isoforms were detected by Western blotting. CBB staining was used for detection of histones. For detection of tubulin, HMGB1, HP1␣, and histones, lysates of N1-Myc-transfected cells were used.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:6.585.43.473.75.475.2]initiated from the second AUG codon does not influence to the subcellular distribution
of their full-length isoforms.
We found that N2- and N3-derived isoforms did not localize to vSPOTs, probably due
to a lack of interaction with viral P protein (12). Thus, the interaction between the N
isoforms and P was investigated by immunoprecipitation (IP) analysis. As shown in Fig.
3B, P coimmunoprecipitated with N1 and N1
=
but not with N2, N2
=
, and N3C.
Consis-tently, N1 and N1
=
coimmunoprecipitated with P, but N2, N2
=
, and N3C did not. A
previous study demonstrated that aa 51 to 100 of N1 are important as a P-binding site
(20). Because N2, N2
=
, and N3C lack a part of the P-binding sequences (Fig. 2A), the loss
of P-binding ability may be critical for the vSPOT localization of these N isoforms.
We previously demonstrated that N1 binds to chromatin, allowing viral RNPs to
segregate into daughter cell nuclei with host chromosomes (28). Therefore, we
evalu-ated whether the nuclear isoforms of N conserve the chromatin-binding capacity. The
chromatin fraction was prepared by subcellular fractionation, and the chromatin was
digested with micrococcal nuclease (MNase), which yields soluble and insoluble
chro-matin. As described above, N1-Myc was detected in the soluble chromatin fraction
where HMGB1 was observed, while N1
=
-Myc was not. Interestingly, N2-Myc was not
observed in the soluble chromatin fraction but was detected with insoluble chromatin
(Fig. 3C). This suggested that the preferences for the binding region on chromatin may
be different between N1 and N2. To test the binding affinity for chromatin, we
extracted chromatin-binding proteins using salt extraction. As shown in Fig. 3C, both
N1 and N2 were extracted at a salt concentration of 150 mM, which is a relatively low
salt concentration, suggesting that the binding affinities of N1 and N2 are relatively
weak.
The N3 isoform translocates to the ER and is cleaved by host signal peptidase.
To identify the mechanism of N3 posttranslational modification that produces N3C,
which accumulates in the ER, alanine-scanning mutational analysis of N3 was
per-formed (Fig. 4A). Alanine replacement of aa 76 to 80 (AS76-80 mutant) completely
abolished the expression of N3C, indicating that the
76LVFLC
80region is essential for
N3C expression (Fig. 4B).
The region around aa 76 to 80 is highly hydrophobic, raising the possibility that
the sequence around
76LVFLC
80may contain the transmembrane domain (Fig. 4A).
TMHMM software (29), which predicts transmembrane helices in proteins,
demon-strated that the hydrophobic sequence from aa 68 to 91 exhibited a high probability of
being a transmembrane domain (Fig. 4C). Furthermore, SignalP (30), which predicts the
presence and location of signal peptide cleavage sites in proteins, revealed that the
sequence from aa 68 to 88 was a high-scoring signal peptide and that N3 is predicted
to be cleaved between A88 and A89 by host signal peptidase (SPase) (Fig. 4D). The
predicted cleavage site was at the same position as that of the N terminus of N3C (Fig.
2F), suggesting that N harbors the intrinsic ER-targeting sequence and translocates to
the ER, followed by the cleavage of the signal peptide by host SPase.
To test this hypothesis, the localization of N3 was investigated by
immunofluores-cence analysis. N3-Myc colocalized with calreticulin, an ER protein (Fig. 4E), in
unin-fected 293T cells. In contrast, the AS76-80 mutant did not localize to the ER, but it was
diffusely detected in the cytoplasm. These observations demonstrated that N3
trans-locates to the ER and that the sequence around
76LVFLC
80acts as the ER-targeting
signal peptide. To determine if N3 is cleaved into N3C by the host SPase, a competitive
SPase inhibition assay was performed. Preproinsulin (pPI) is a secretory preprotein that
translocates to the ER, where it is cleaved into proinsulin (PI). A mutant of pPI
(pPI-F25P), which harbors a single-amino-acid substitution following the signal peptide
cleavage site, specifically binds to SPase catalytic subunits and acts as a competitive
inhibitor of cellular SPase (31). As shown in Fig. 4F, N3C was expressed when N3 was
coexpressed with wild-type pPI, but the expression of N3C was completely abolished
when the host SPase was inhibited by pPI-F25P, demonstrating that N3C is cleaved by
host SPase. Taken together, these findings demonstrated that the intrinsic ER-targeting
on November 6, 2019 by guest
http://jvi.asm.org/
signal peptide around aa 76 to 80 is the molecular determinant for N3 to translocate to
the ER, where it is cleaved by the host SPase.
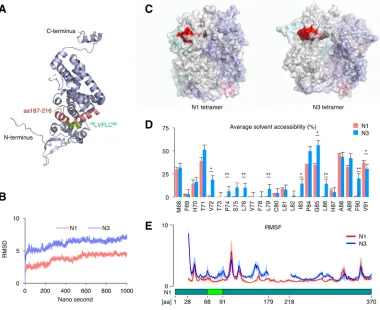
The N3 isoform signal peptide is predicted to be exposed to solvent.
Although
both N1 and N3 harbor signal peptides, only N3 translocates to the ER, suggesting that
NI-II splicing is a molecular switch for ER targeting. However, it is unclear how the
removal of the NI-II intron is the ER-targeting signal on N3. The crystal structure of N1
showed that the region between aa 76 and 80 is embedded inside the molecule and
that the region between aa 187 and 216 structurally covers
76LVFLC
80(Fig. 5A). Given
that the cover sequence within aa 187 to 216 is removed by NI-II splicing, it may
unmask the intrinsic ER-targeting signal peptide and make it available for interaction
FIG 4N3 translocates to the ER and is cleaved into a C-terminal fragment. (A) Protein sequences of N3 mutants used for alanine-scanning mutagenesis analysis. Underlined characters in the N3 sequence represent amino acids with hydrophobic residues. (B) Detection of alanine-scanning N3 mutants by Western blotting. Whole-cell lysates of uninfected 293T cells expressing each mutant were used. (C) Detection of transmembrane helix potential by TMHMM software, which predicts transmembrane helices in proteins. Full-length N3 (aa 1 to 330) was used for analysis. (D) Detection of signal peptide potential by SignalP, which predicts the presence and location of the signal peptide cleavage site in proteins. A partial N3 sequence (aa 68 to 127) was used for analysis. (E) Cellular localization of N3-Myc, AS76-80 –Myc, and N1-Myc in uninfected 293T cells. Each N protein was detected by an anti-Myc tag antibody. The ER was stained by an anticalreticulin antibody, and the nucleus was counterstained with DAPI. Bars, 10m. (F) N3 is cleaved into N3C by the host SPase. N3C expression was detected by Western blotting. Whole-cell lysates of uninfected 293T cells expressing the indicated constructs were used. pPI-Myc, preproinsulin with a C-terminal Myc tag; pPI-F25P-Myc, pPI-Myc with a single-amino-acid substitution, which functions as an SPase inhibitor. In panels B and F, CBB staining was used as a loading control.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.48.501.71.501.2]with the ER translocation machinery. To test this hypothesis, structural simulation of the
N1 tetramer and N3 tetramer was performed by molecular dynamics (MD) analysis
using the previously reported crystal structure of the N1 tetramer (32) to observe if the
ER-targeting signal peptide is exposed to the protein surface by NI-II splicing (Fig. 5B).
The predicted structure of the N3 tetramer was different from that of the N1 tetramer
(Fig. 5C). Although the solvent accessibility of atoms around
76LVFLC
80was quite low
in N1, it markedly increased in N3 (Fig. 5D). Furthermore, atomic fluctuation around
76
LVFLC
80and the N terminus was also increased in N3 (Fig. 5E). The crystal structure
of the N1 tetramer suggested that the region from aa 179 to 218 plays an important
role in the interaction with the N terminus of the neighboring monomer. The increased
atomic fluctuation of the N3 tetramer supports that the region from aa 179 to 218 is
crucial for forming the stable tetramer. These results suggested that NI-II splicing makes
the targeting signal accessible to the ER translocation machinery via a conformational
change and partial exposure to the protein surface.
Expression of N3C in the ER fraction of BoDV-infected cells.
To elucidate if N2
and N3C are expressed in BoDV-infected cells, rBoDVs, which harbor silent mutations at
splicing donor and acceptor sites within N, were generated (Fig. 6A). The successful
introduction of silent mutations was verified by RT-PCR and sequencing of the rBoDV
genome (data not shown). To detect the ER-specific expression of N3C, rBoDV-infected
OL, 293T, and Vero cells were fractionated into the cytoplasm, ER, and nucleus. As
shown in Fig. 6B, N3C was detected in the ER fraction of OL and 293T cells by Western
FIG 5The ER-targeting signal peptide is partially exposed to the protein surface in the predicted N3 structure. (A) Crystal structure of N1 (PDB accession number1N93). Green and red regions represent76LVFVC80and aa 187 to 216, respectively. (B) Root mean square deviation (RMSD) of the N1 tetramer and N3 tetramer. (C) Predicted structures of the N1 tetramer and N3 tetramer. The red region represents76LVFVC80. (D) Average solvent accessibility of each residue from aa 68 to 91 in N1 and N3. The data are presented as the means and standard errors of the means (SEM) of data from four independent simulations. Student’sttest was used for statistical analysis.*,P⬍0.05;**,P⬍0.01. (E) Root mean square fluctuation (RMSF) of each residue in N1 and N3. The data are presented as the means⫾95% confidential intervals (CIs) of data from four independent simulations. Solid lines show means, and shaded regions represent CIs.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:9.585.44.424.73.383.2]FIG 6Splicing of N isoforms negatively regulates BoDV infection. (A) Partial sequences of rBoDVs. Silent mutations that disrupt splicing signals in the N gene are shown in red. (B) Expression of N3C in BoDV-infected cells. Uninfected or persistently rBoDV-infected OL and 293T cells were fractionated into cytoplasm, ER, and nucleus fractions. The ER fraction was used for detection of N3C. The whole-cell lysate of uninfected 293T cells transfected with an N3-expressing plasmid was used for the positive control. N3C, tubulin, and calreticulin were detected by Western blotting. CBB staining was used as a loading control and for detection of histones. Asterisks represent an unidentified N product. (C) Expression levels of N isoforms during serum starvation. Persistently BoDV-infected OL cells were cultured in serum-free medium. (Top and bottom left) Distribution of cell cycles and cell numbers after serum starvation, respectively. For cell cycle detection, the cells were stained by propidium iodide (PI). (Bottom right) Expression levels of the N isoforms. N1, N1=, and N3C were detected by Western blotting. The whole-cell lysate was used for N1 and N1=, and the ER fraction was used for N3C detection. CBB staining was used as a loading control. Normal and starved represent cells cultured in serum-containing and serum-free media, respectively. The asterisk represents an unidentified N product. RFU, relative fluorescence units. (D) Expression of N2 and N3 inhibits BoDV polymerase activity in the minireplicon assay. Uninfected 293T cells were transfected with plasmids encoding the indicated proteins, and luciferase activity was measured at 48 h posttransfection. Gluc (Gaussialuciferase) activity, which was derived from the BoDV minireplicon, was normalized to Cluc (Cypridinaluciferase) activity, which was derived from a transfection control plasmid. The data are presented as the means and SEM of results from three independent experiments. One-way analysis of variance (ANOVA) and Tukey’spost hoctest were used for statistical analysis.*,P⬍0.05. RLU, relative light units. (E) Propagation of rBoDVs. OL cells were infected with rBoDVs at an MOI of 0.1. The levels of BoDV P mRNA and genome RNA at the indicated time points were measured by qRT-PCR. The data are presented as the means⫾SEM of results from three independent experiments. One-way ANOVA and Tukey’spost hoctest were used for statistical analysis.*,P⬍0.05;**,P⬍0.01; n.s., not significant.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:10.585.46.539.72.560.2]blotting, although the expression of N3C was not detected in Vero cells (data not
shown). These results confirmed that N3C is expressed in the ER during persistent
infection by BoDV.
On the other hand, the expression of N2 and N2
=
could not be evaluated in
BoDV-infected cells, suggesting that the expression of N2 and N2
=
is masked by the
signal of N1
=
. The expected molecular weights of N2 and N2
=
(approximately 34 to 36
kDa) were close to that of N1
=
(38 kDa), and the splicing efficiency of NI-I was quite low.
Further optimization of Western blotting is required to evaluate the expression of NI-I
splicing products.
The primary target of BoDV in natural infection is neurons. Neurons are
nonprolif-erating cells; therefore, we evaluated whether the expression levels of N isoforms
depend on cell proliferation. BoDV-infected OL cells cultured in serum-free medium
showed reduced proliferation and cell cycle arrest 72 and 96 h after serum depletion
(Fig. 6C), suggesting that quiescence was induced. While the expression of N1 and N1
=
did not show obvious changes by serum starvation, the expression of N3C appeared to
increase (Fig. 6C). This suggested that the expression of N3C may be regulated by cell
proliferation.
N2 and N3 negatively regulate BoDV infection.
The roles of N isoforms in BoDV
replication were investigated. A minireplicon system of BoDV, which synthesizes
re-combinant BoDV nucleocapsids containing a minigenome reporter RNA, was used
following transfection of expression plasmids encoding N, P, L, and the minigenome.
The minireplicon assay was performed in the presence or absence of plasmids
express-ing N1, N2, and N3. Consistent with previous results, viral polymerase activity was
strongly inhibited when itEBLN (an endogenous bornavirus-like nucleoprotein [EBLN] in
the thirteen-lined ground squirrel genome) (33) was cotransfected with the
minirepli-con minirepli-constructs. Although N1 did not inhibit the polymerase activity of the minirepliminirepli-con,
N2 and N3 significantly decreased the polymerase activity in the system (Fig. 6D). These
results suggested that N2 and N3 act as a negative inhibitor of viral transcription and
replication.
To understand the role of NI-I and NI-II splicing in viral replication, the
propa-gation of rBoDV lacking NI-I splicing, NI-II splicing, or both was investigated. OL cells
were infected with rBoDVs, and the levels of viral mRNA and genome RNA in
cells were monitored. As shown in Fig. 6E, levels of both viral mRNA and genome
RNA were significantly increased in cells infected with splicing-deficient rBoDVs
compared to wild-type rBoDV. These results suggested that the expression of N2
and N3 negatively regulates viral polymerase activity during BoDV infection.
Conservation of the splicing signals and hydrophobic region of N across
orthobornaviruses and endogenous bornavirus-like nucleoproteins.
To elucidate if
the splicing acceptor/donor sites are conserved in orthobornaviruses, the N genes of
orthobornaviruses were aligned. While the splicing signals were conserved across
mammalian 1 orthobornaviruses, other orthobornaviruses did not have the splicing
signals in the corresponding regions (Fig. 7A). The hydrophobic transmembrane region
was aligned in orthobornaviruses. All orthobornavirus N proteins contained many
hydrophobic residues in the regions corresponding to the predicted transmembrane
domain of N3 (Fig. 7B and D). These results suggested that the hydrophobic
trans-membrane potential is a conserved feature of orthobornaviruses.
To elucidate if such sequence features of orthobornavirus N are evolutionarily
conserved, the sequences of EBLNs, which are the ancient bornaviral sequences in host
genomes, were investigated. EBLNs are remnants of ancient viruses, and they provide
useful information for understanding the characteristics of ancient viruses. Although
most of the ORFs of integrated N sequences were disrupted by mutations, two EBLNs
in the human genome and bat genome retained almost intact ORFs (34, 35).
Interest-ingly, both EBLNs contained hydrophobic regions, suggesting that some antient
bor-naviruses might have already acquired the transmembrane potential of N (Fig. 7C).
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 7Comparison of splicing signals and hydrophobic regions across orthobornaviruses and endogenous bornavirus-like nucleoproteins. (A) Comparison of the splicing signals across orthobornaviruses. Nucleotide positions corresponding to the BoDV-1 genome are shown. Splicing signals are shown in boldface type. Asterisks represent conserved nucleotides. (B) Conservation of the signal peptide across orthobornaviruses. Amino acid positions corresponding to BoDV-1
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:12.585.41.538.64.686.2]DISCUSSION
The present study demonstrated that BoDV generates multiple N isoforms by
mechanisms such as downstream translation initiation and mRNA splicing (Fig. 1 and
2). The newly identified splicing in N had canonical eukaryotic splicing site sequences,
suggesting that BoDV uses host splicing machinery, as previously reported (23, 24). The
expression levels of the N spliced transcripts were relatively low at less than 7% of the
N transcripts in BoDV-infected cell lines by the estimation of NGS data. Previous data
suggested that BoDV harbors an exon splicing suppressor sequence and controls
alternative splicing of L transcripts (25, 36). Thus, it may be possible that BoDV also
controls the efficiency of N splicing to maintain the expression of N isoforms at low
levels. In this study, we could not detect N3C in Vero cells, while it was detected in OL
and 293T cells. Furthermore, the expression of N3C increased in nonproliferating cells
(Fig. 6). It would be of interest to determine how the expression level of N3C is
controlled in cells. It may also be necessary to estimate the expression levels of N
isoforms in BoDV-infected animal brains to understand the role of N splicing
in vivo
.
Many viruses replicating in the nucleus employ RNA splicing machinery to create
variant proteins from a single transcript, which generally play different roles in viral
infection. Because genome sizes and coding capacities are relatively limited in
nuclear-replicating RNA viruses, RNA splicing may be a conducive strategy to produce viral
protein diversity. For instance, influenza A virus expresses isoforms of NS and M
proteins using the host splicing machinery (37). It has been reported that the splicing
efficiency of the NS transcript coordinates the cellular antiviral response and the nuclear
export of viral RNPs (38). In addition, the splicing of the M transcript generates an ion
channel protein required for virus particle assembly, egress, and ingress, in addition to
the structural matrix protein (39). Furthermore, it has been reported that the mosquito
Culex tritaeniorhynchus rhabdovirus requires mRNA splicing to express mature L
protein, similarly to BoDV (40).
The utilization of alternative AUG start codons in the same transcript is another
mechanism to express multiple protein isoforms of different functions in many RNA
viruses. Ribosomal leaky scanning and ribosomal shunts are common mechanisms for
the P transcript of
Mononegavirales
. For example, the Sendai virus P transcript expresses
as many as five different isoforms using alternative AUG start codons, namely, P, C, C
=
,
Y1, and Y2 (41), suggesting that alternative usage of start codons may be a useful
mechanism for RNA viruses replicating in the cytoplasm. The present study showed that
mRNA splicing and alternative start codon usage cooperate for the expression of N
isoforms showing diverse subcellular localizations in BoDV-infected cells. This may be
the first report of the coordination between posttranscriptional and translational
regulations to create protein diversity in RNA virus infection.
The present study showed that the subcellular localizations of BoDV N isoforms are
elaborately controlled by their signal motifs within the sequence, such as the P-binding
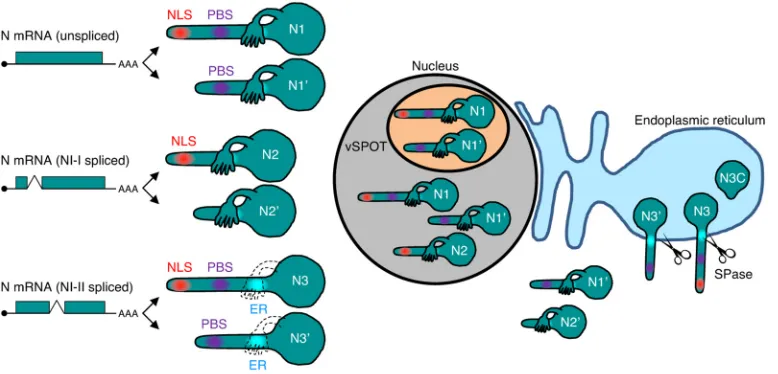
site, NLS, NES, and ER-targeting signal peptides (Fig. 3 and 8). The full-length N1 isoform
mainly localizes to the nucleus and accumulates in vSPOTs. N1
=
, which lacks an NLS,
localizes in the cytoplasm but translocates to the nucleus and accumulates in vSPOTs
via interaction with P. Although the spliced N2 isoform contains an NLS and localizes
to the nucleus, it does not accumulate at vSPOTs due to the lack of the P-binding site
(Fig. 8). N2
=
lacks both the NLS and P-binding site and, therefore, cannot enter the
nucleus. NI-II splicing generates N3 and N3
=
, which translocate to the ER, resulting in
FIG 7Legend (Continued)
N1 are shown. Underlined characters represent amino acids with hydrophobic residues. Asterisks represent conserved amino acids. (C) Conservation of the hydrophobic region in human and bat EBLN-1. Amino acid positions corresponding to BoDV-1 N1 are shown. Underlined characters represent amino acids with hydrophobic residues. Asterisks represent conserved amino acids. (D) Hydrophobicity of nucleoproteins across orthobornaviruses and an endogenous bornavirus-like element. Hydrophobicity was calculated by ProtScale. Full-length nucleoproteins of orthobornaviruses and endogenous bornavirus-like nucleoproteins were used for analysis. The regions corresponding to the hydrophobic transmembrane domain of N3 are shown in red. Accession numbers of protein sequences used for analysis are as follows:P0C796for BoDV-1,YP_009269413for variegated squirrel bornavirus 1 (VSBV-1),YP_009237642for aquatic bird bornavirus 1 (ABBV-1),YP_009268905for canary bornavirus 1 (CnBV-1),YP_009268893for parrot bornavirus 4 (PaBV-4),YP_009268899for PaBV-5,
YP_009055058for Loveridge’s garter snake virus 1 (LGSV-1), andNP_001186867forHomo sapiensEBLN-1 (hsEBLN-1).
on November 6, 2019 by guest
http://jvi.asm.org/
the development of a truncated form of N3C (Fig. 8). The isoforms produced from a
transcript showing different intracellular localizations comprise a unique mechanism for
BoDV.
Among the N isoforms, N3 and N3
=
may regulate processing and translocation in
infected cells. N3 and N3
=
translocate to the ER and are cleaved in the C-terminal
domain to generate N3C. The expression of N3 isoforms was detected in the ER fraction
of rBoDV-infected cells by Western blotting (Fig. 6). Transfection analysis revealed that
N3C was more abundant than N3, suggesting that a large portion of N3 in the ER is
cleaved by the host SPase. Although N3 and N3C contain an NLS at the N terminus,
most of the signals of these isoforms were detected at the ER and not in the nucleus.
Structural prediction of N3 revealed that the N terminus of N3 is more flexible than that
of N1, suggesting that the flexible N terminus of N3 isoforms contributes to escape
from the recognition of importin. Alternatively, the flexibility of the N terminus might
affect the oligomerization of N3, thereby preventing the nuclear localization of N3
isoforms.
Another interesting mechanism is the activation of the ER-targeting signal on N3
isoforms. ER targeting of N3 is accomplished by canonical ER translocation machinery.
The sequence from aa 68 to 91 contains several hydrophobic residues and is predicted
to form a helical structure (Fig. 4), suggesting that this region is a transmembrane
domain.
In silico
prediction and alanine substitution analyses demonstrated that this
region functions as a signal peptide that binds to the translocon in the ER membrane.
Although N1 also has this signal peptide, only N3 translocates in the ER. It is unknown
why N1 is not recognized by the ER translocation machinery. The prediction assay of
the N3 structure revealed that the signal peptides in N3 exhibit higher solvent
acces-sibility than N1 (Fig. 5), suggesting that the region from aa 179 to 218 (region deleted
by N-II splicing) masks the signal peptides in N1 (Fig. 8). Furthermore, N3 has a more
flexible structure than N1. This finding demonstrated that N3 may be highly susceptible
to conformational change following interaction with the translocation machinery in
cells.
The minireplicon assay and infection experiment using rBoDVs lacking N splicing
showed that N2 and N3 isoforms negatively regulate viral transcription and replication
(Fig. 6). N2 strongly inhibited the polymerase activity of the minireplicon, whereas N3
moderately inhibited the polymerase activity of the minireplicon. Because N2 cannot
FIG 8Schematic representation of N isoforms and their subcellular localization during BoDV infection. N1, N2, and N3 mRNAs are transcribed from the N gene. Each mRNA generates two isoforms by translation initiation from the first and second AUG codons. N1 translocates to the nucleus and accumulates in vSPOTs. The sole expression of N1=localizes to the cytoplasm, while N1=can enter the nucleus and vSPOTs via interaction with N1 and P in infected cells (Fig. 3A). N2 is transported into the nucleus, but N2 does not localize in vSPOTs. N2=localizes only in the cytoplasm. N3 and N3=
translocate to the ER and are cleaved into the N3C C-terminal fragment by the host SPase.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:14.585.42.431.73.260.2]interact with P and does not accumulate in vSPOTs, this isoform may act as a decoy for
host factors that are important for BoDV replication. Although the inhibition of
mini-replicon activity by N3 was not as strong as that by N2, rBoDV-ΔN3 propagated as
efficiently as rBoDV-ΔN2. Considering that N3C localized to the ER, it is likely that N3
plays a negative role in BoDV replication within the intracellular organelle. The finding
that the expression level of N3 increased during serum starvation suggested that N3C
may downregulate BoDV replication depending on the extent of cell proliferation.
BoDV N is a multifunctional protein that plays a central role in viral infection, such as
in viral RNP formation and nuclear transport of viral RNPs, as well as in inhibition of host
immune responses (13, 42–45). Although the present study could not demonstrate the
precise mechanisms of spliced N isoforms regulating BoDV replication, the distinct
cellular distributions of N may control and maintain a unique persistent infection by
BoDV in the nucleus. Further experiments, such as gene expression profiling and
identification of protein interactions, are required to understand the impact of N
isoforms on both viral and cellular functions.
The present data showed that the N proteins of other orthobornaviruses also have
many hydrophobic residues in the region corresponding to the transmembrane
do-main of N3 (Fig. 7), suggesting that these proteins also exhibit transmembrane
poten-tial in infected cells. Intriguingly, human EBLN-1, which originated from an ancestor of
orthobornavirus approximately 45 million years ago, also contains hydrophobic
resi-dues in the corresponding region, suggesting that some antient bornaviruses might
have already acquired transmembrane potential. According to analysis by TMHMM
(data not shown), the fact that nucleoproteins of other negative-strand RNA viruses
show little or no transmembrane potential suggested that the ER targeting of N might
be a strategy of orthobornaviruses crucial for their unique characteristics, such as
intranuclear replication and persistent infection.
In conclusion, the present study demonstrated that functions of BoDV N isoforms
are controlled by their subcellular localizations determined by splicing and downstream
AUG initiation. Such unique features of N, in addition to intranuclear persistent
infec-tion and noncytolytic replicainfec-tion, may distinguish BoDV from other negative-strand
RNA viruses. Further experiments on the mechanisms of N2 and N3 inhibition of BoDV
replication as well as the control of diverse subcellular localizations will aid in the
understanding of the unique characteristics of BoDV.
MATERIALS AND METHODS
Cell lines and viruses.OL (human oligodendroglioma) cells were cultured in high-glucose (4.5%) Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS). HEK293T (293T) (human embryonic kidney) and Vero (green monkey kidney) cells were cultured in low-glucose (1.0%) DMEM supplemented with 10% and 2% FBS, respectively. BoDV-infected OL cells, a cell line persistently infected with strain huP2br (46), was cultured under the same conditions as the parental cell line. BoDV-infected OL, 293T, and Vero cells, cell lines persistently infected with strain He/80/Fct (He/80/FR harboring a single-nucleotide substitution from C to T at genome position 4673) or rBoDV, were cultured under the same conditions as the parental cell lines.
Cell-free virus preparation. Cell-free BoDV was prepared as previously described (47). Briefly, BoDV-infected cells were treated with 0.05% trypsin and 0.48 mM EDTA and centrifuged. After centrif-ugation, the cells were washed once with phosphate-buffered saline (PBS) and suspended in Opti-MEM (ThermoFisher) or DMEM containing 2% FBS. The suspended cells were sonicated and centrifuged at 1,200⫻gfor 25 min at 4°C. The supernatant containing viral materials was stored at⫺80°C until use.
Virus infection.For amplicon sequencing, OL cells were infected with He/80 at a multiplicity of infection (MOI) of 1. After absorption for 1 h, the cells were washed with PBS. For propagation assays, OL cells were infected with either rBoDV-WT, rBoDV-ΔN2, rBoDV-ΔN3, or rBoDV-ΔN2N3 at an MOI of 0.1. After absorption for 1 h, the cells were washed with PBS and passaged at 1, 4, 7, and 10 days postinfection. After 10 days postinfection, the cells were passaged within the appropriate interval of a few days. Virus propagation was detected by immunofluorescence analysis of P.
Plasmid construction.To generate the eukaryotic expression plasmid of N1 (pcDNA3-N1), the PCR-amplified BoDV N1 coding DNA sequence (CDS) fragment was cloned into the plasmid pcDNA3 (Invitrogen). The N1 gene was amplified from cDNA from the cells infected with BoDV strain He/80/Fct. The expression plasmids of N isoforms were constructed from pcDNA3-N1 by PCR mutagenesis to delete the intron in focus and introduce the synonymous point mutations in another splicing signal. A Myc tag, a Flag tag, and a hemagglutinin (HA) tag were fused to the 5=end or the 3=end of the CDS by PCR mutagenesis. To generate the expression plasmid of P with an N-terminal Flag tag (pcDNA3-Flag-P), the
on November 6, 2019 by guest
http://jvi.asm.org/
PCR-amplified BoDV P CDS fragment from pCXN2-P (48) was cloned into the plasmid pcDNA3. A Flag tag was fused to the 5=end of the CDS by PCR mutagenesis. The RNA polymerase II (Pol II)-driven BoDV minigenome plasmid pFct-BDV was described previously (49). pFct-BDV_ΔN2, pFct-BDV_ΔN3, and pFct-BDV_ΔN2N3 were constructed from pFct-BDV by PCR mutagenesis to introduce the point mutations in the splicing signals in the N gene. To obtain the eukaryotic expression plasmids of pPI (pCAG-pPI-Myc) and pPI-F25P (pCAG-pPI-F25P-Myc), the PCR-amplified pPI gene was cloned into the plasmid pCAGG-S.MCS. A Myc tag was fused to the 3=end of the pPI CDS by PCR mutagenesis. pCAG-pPI-F25P-Myc was constructed from pCAG-pPI-Myc by PCR mutagenesis to introduce the point mutation.
BoDV reverse genetics.Reverse-genetics assays were carried out based on data from a previous study (49). Briefly, 293T cells were transfected with either the pFct-BDV, pFct-BDV_ΔN2, pFct-BDV_ΔN3, or pFct-BDV_ΔN2N3 plasmid and pCA-N, pCXN2-P, and pCAGGS-L using polyethylenimine max (linear; MW, 25,000) (Polysciences, Inc.). Three days after transfection, the cells were passaged and cocultivated with Vero cells. The Vero cells were passaged every 3 days, and the rescue efficacy of recombinant BoDV was evaluated by immunofluorescence analysis of P.
Minigenome assay.A minigenome assay was performed according to a protocol described previ-ously (50). Briefly, 293T cells were transfected with a Pol II-driven minigenome plasmid carrying the Gaussialuciferase gene; helper plasmids expressing the BoDV-1 N1, P, and L genes; and a control plasmid expressing theCypridinaluciferase gene using polyethylenimine max. Forty-eight hours after transfec-tion,Gaussialuciferase activity was measured with a BioLux luciferase assay kit (NEB) and normalized to the correspondingCypridinaluciferase activity.
Subcellular fractionation. 293T cells (7.5⫻106) were resuspended in 120l cold HMKE buffer (20 mM HEPES [pH 7.2], 5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 250 mM sucrose, 400g/ml digitonin, protease inhibitor), followed by pulsed vortexing and incubation on ice for 5 min. The lysates were centrifuged in Microfuge tubes at 1,000⫻g for 3 min at 4°C. The supernatant from this process represents the cytoplasmic fraction. The pellets were gently resuspended in NP-40 buffer (0.5% NP-40, 1⫻PBS, protease inhibitor), followed by pulsed vortexing and incubation on ice for 1 min. The lysates were centrifuged for 3 min at 1,000⫻gat 4°C. The supernatant and pellet from this spin represent the ER and nuclear fractions, respectively. For the fractionation of OL cells, the same protocol for 293T cells with one modification (HMKE buffer containing 800g/ml digitonin) was used.
Serum starvation.OL cells (1⫻104cells) were seeded into 10-cm dishes and incubated for 24 h in medium containing 5% FBS. The medium was removed, and the cells were washed four times with fresh serum-free medium. The cells were then cultured in serum-free medium for 24 to 96 h. Ninety-six hours after serum starvation, cells were collected, and the ER fraction was prepared using the same protocol as the one described above. For propidium iodide (PI) staining, cells were collected 24, 48, 72, and 96 h after serum depletion and stained with a Tali cell cycle kit. The fluorescence of PI and the cell number were measured using a Tali image cytometer.
Chromatin-binding assay (MNase digestion).Cells (2.5⫻106cells) were resuspended in 100l buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol [DTT], protease inhibitor) containing 0.1% Triton X-100. After incubation for 10 min on ice, nuclei were collected in a pellet by centrifugation (5 min at 1,200⫻g). Nuclei were washed three times with buffer A and lysed in 100l buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease inhibitor) for 30 min. The chromatin-enriched pellet was collected by centrifugation (5 min at 1,700⫻g), and the supernatant was removed to yield a nucleoplasmic fraction. The pellet was washed twice with buffer B and once with MNase buffer (20 mM Tris-HCl [pH 8.0], 5 mM NaCl, 2.5 mM CaCl2) and resuspended in 100l MNase buffer containing 20 U MNase. After incubation for 5 min at 37°C, the MNase reaction was stopped by the addition of an EDTA-EGTA solution to a final concentration of 5 mM, and the supernatant was then collected by centrifugation (5 min at 2,000⫻g) as a chromatin-binding fraction. The pellet was washed three times with MNase buffer and collected as an insoluble fraction. All steps were carried out at 4°C if the condition was not specified.
Chromatin-binding assay (salt extraction).Nuclei were prepared using the same protocol as the one for the chromatin-binding assay with MNase. Nuclei were washed three times with buffer A and lysed in buffer A containing 150 mM NaCl. Nuclei were incubated for 30 min and centrifuged for 1 h at 21,500⫻g. The supernatant and pellet from this process represent salt-extractable and insoluble fractions, respectively. All steps were carried out at 4°C.
Western blotting.The cell lysates were subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The membranes were blocked with Tris-buffered saline (TBS)– 0.1% Tween 20 with 5% (wt/vol) low-fat milk powder (TBSTM) and then reacted with anti-N (HB01 or HB01K), anti-P (HB03), anti-Myc (9E10 [Millipore] or 9B11 [Cell Signaling]), anti-Flag M2 (Sigma-Aldrich), antitubulin (B-5-1-2; Sigma-Aldrich), anticalreticulin (ab92516; Abcam), anti-HMGB1 (ab18256; Abcam), and HP1␣(catalog number 2616; Cell Signaling) antibodies, which were diluted appropriately in TBSTM, followed by a reaction with horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch). The protein bands were visualized with the ECL Prime Western blot detection reagents (GE Healthcare).
Immunofluorescence analysis. Persistently BoDV-infected 293T cells were transfected with the plasmids expressing the N isoforms and incubated for 24 h. The cells were fixed in 4% paraformaldehyde (PFA), permeabilized by incubation in PBS containing 0.25% Triton X-100, and then blocked with PBS containing 1% bovine serum albumin (BSA). Uninfected 293T cells were transfected with the plasmids expressing the N isoforms and the N3 mutant and incubated for 24 h. The cells were fixed in 100% methanol, because the anticalreticulin antibody was available only in the cells fixed in methanol. Methanol-fixed cells were blocked with PBS containing 1% BSA. Both PFA- and methanol-fixed cells were then incubated with the anti-BoDV P (HB03), anti-Myc (9E10), and anticalreticulin (ab92516) antibodies.
on November 6, 2019 by guest
http://jvi.asm.org/
This was followed by incubation with the appropriate Alexa Fluor-conjugated secondary antibodies (Invitrogen). The cells were counterstained with DAPI (4=,6-diamidino-2-phenylindole). An Eclipse Ti confocal laser scanning microscope (Nikon) was used for data collection.
Immunoprecipitation.293T cells were transfected with pCXN2-P (48) and pcDNA3 plasmids encod-ing Myc-tagged N isoforms for precipitation of N isoforms. 293T cells were transfected with pcDNA3-Flag-P and pcDNA3 plasmids encoding Myc-tagged N isoforms for precipitation of P. At 48 h posttrans-fection, the cells were collected and washed with PBS twice. The cells were lysed with lysis buffer (1⫻ PBS, 0.5% NP-40, protease inhibitor). After centrifugation (3,000⫻gfor 3 min), the supernatant was collected. Protein G Dynabeads (Invitrogen) were incubated with anti-Myc tag antibody My3 (MBL) or anti-Flag tag antibody FLA-1 (MBL) and incubated for 10 min with rotation. The beads were washed with lysis buffer once, and the supernatant was added. After incubation with gentle rocking for 2 h, the beads were washed with lysis buffer three times, and the immunoprecipitated proteins were eluted with the Myc tag peptide or the 3⫻Flag tag peptide. All steps were carried out at 4°C.
Purification of Myc-tagged N3C.293T cells were transfected with the pcDNA3-N3-Myc plasmid using polyethylenimine max. At 48 h posttransfection, the cells were collected and washed with PBS twice. The cells were lysed with lysis buffer (1⫻PBS, 0.5% NP-40, protease inhibitor). After centrifugation (3,000⫻gfor 3 min), the supernatant was collected. Protein G Dynabeads were incubated with anti-Myc tag antibody 9E10 and incubated with rotation for 10 min. The beads were washed with lysis buffer once, and the supernatant was added. After incubation with gentle rocking for 2 h, the beads were washed with lysis buffer three times, and the immunoprecipitated protein was eluted with the Myc tag peptide. All steps were carried out at 4°C.
Antigen-specific antibody purification. The peptide corresponding to aa 331 to 370 of N1 conjugated with 5 cysteines at the N terminus was synthesized. The C-terminus-specific polyclonal anti-N antibody (HB01K) was purified from the polyclonal anti-N antibody (HB01) with the peptide using a high-affinity antibody purification kit (GenScript).
qRT-PCR analysis. Total RNAs were extracted using the NucleoSpin RNA kit (Macherey-Nagel). Reverse transcription was performed using the Verso cDNA synthesis kit (ThermoFisher). RT-quantitative PCR (qRT-PCR) analyses were carried out using the Thunderbird SYBR qPCR mix and the Thunderbird probe qPCR mix (Toyobo) as previously described (50).
RNA-seq and data processing.The sequences of mRNAs from uninfected and He/80/Fct-infected OL cells were analyzed by NGS. Total RNA was extracted using TRIzol reagent (Invitrogen) and the Direct-zol RNA MiniPrep kit (Zymo Research). The qualities of RNA samples were checked with a high-sensitivity RNA kit (Agilent). Next, poly(A)⫹ RNA was extracted using the Dynabeads mRNA purification kit (ThermoFisher). An NGS library was prepared using TruSeq stranded total RNA with the Ribo-Zero Gold LT sample prep kit (Illumina) according to the manufacturer’s instructions, except for the Ribo-Zero process. NGS was performed on the MiSeq Illumina platform using MiSeq reagent kit v3 (150 cycles). The RNA sequencing reads were mapped to the human genome assembly GRCh38 and the BoDV genome using the Gencode v24 annotation and TopHat v2.0.13.
Amplicon sequencing and data processing.The amplicon sequences of the N transcripts from acutely He/80/Fct-infected OL, persistently huP2Br-infected OL, and persistently He/80/Fct-infected 293T cells were analyzed by NGS. Total RNA was extracted using TRIzol reagent, and the cDNA was synthesized using the oligo(dT) primer and the Verso cDNA synthesis kit. The cDNA of the N transcripts was amplified by PCR using the PrimeSTAR GXL DNA polymerase kit (TaKaRa). Amplicon libraries were prepared using the Kapa Hyper Prep kit. Deep sequencing was performed on the MiSeq Illumina platform using MiSeq reagent kit v3 (150 cycles). The RNA sequencing reads were mapped to the N transcript sequence using TopHat.
Molecular dynamics simulations.The initial coordinates of the N1 tetramer were taken from the cocrystal structure (PDB accession number1N93) (32). The missing loops at positions 315 to 322 in each monomer were added using Molecular Operating Environment (MOE) software (version 2018; Chemical Computing Group). The structural model of the N3 tetramer was constructedin silicousing the N1 tetramer structure. After deleting amino acid residues 179 to 218 of the N1 tetramer in each monomer, the Loop Modeler module of MOE was used to create a bond between amino acids 178 and 219 (N1 numbering) in N3. Protonation states of the ionizable residues were assigned at pH 7.0 using the PDB2PQR Web server (51). All missing hydrogen atoms were added with the LEaP module in AMBER 16 (Conflex USA) (52). The ff14SB force field was applied for N1 and N3 tetramers (53). The total charges of the proteins were neutralized by the addition of chloride counterions. The systems were then solvated in a truncated octahedral box of transferable intermolecular potential with 3 points (TIP3P) water molecules with a distance of at least 10 Å around the protein. All energy minimization and molecular dynamics (MD) simulations were performed using the pmemd.cuda program in AMBER 16, with a cutoff radius of 10 Å for the nonbonded interactions. The locations of hydrogen atoms, water molecules, and counterions were optimized to remove bad contacts. The energy of each system was then minimized without any constraints using the steepest-descent method for 500 steps, followed by the conjugate gradient method for 1,500 steps. After minimization of energy, the system was gradually heated from 0 K to 310 K over 300 ps with harmonic restraints (with a force constant of 1.0 kcal/mol · Å2). Two additional rounds of MD (50 ps each at 310 K) were performed with a decreasing restraint weight reduced from 0.5 to 0.1 kcal/mol · Å2. Next, 1.0s of an unrestrained production run was performed, and the production trajectories were collected every 10 ps. All MD simulations were performed using the NPT ensemble and the Berendsen algorithm to control temperature and pressure (54). The time step was 2 fs, and the SHAKE algorithm was used to constrain all bond lengths involving hydrogen atoms (55). Long-range electro-static interactions were treated using the particle mesh Ewald method (56).
on November 6, 2019 by guest
http://jvi.asm.org/
The stability of the trajectories was assessed by monitoring the root mean square deviations (RMSDs) of the backbone heavy atoms in comparison with the initial structure. After confirming that RMSDs in all systems reached equilibrium within 500 ns, the trajectories were extracted from the simulations from 500 to 1,000 ns in the analyses.
Data availability.The sequences reported in this article have been deposited in the DNA Data Bank of Japan (DDBJ) under Sequence Read Archive accession numbersDRA007567(total mRNA sequences of uninfected and BoDV-infected OL cells) andDRA007646(amplicon sequences of BoDV N mRNA).
ACKNOWLEDGMENTS
This study was supported in part by JSPS KAKENHI grants JP17H04083 (K.T.); MEXT
KAKENHI grants JP16H06429, JP16K21723, and JP16H06430 (K.T.); JSPS Core-to-Core
Program A; the Advanced Research Networks (K.T.); and AMED grants JP18am0301015
and JP18fm020814 (K.T.).
REFERENCES
1. Wasilewski M, Chojnacka K, Chacinska A. 2017. Protein trafficking at the crossroads to mitochondria. Biochim Biophys Acta Mol Cell Res 1864: 125–137.https://doi.org/10.1016/j.bbamcr.2016.10.019.
2. Ullman KS, Powers MA, Forbes DJ. 1997. Nuclear export receptors: from importin to exportin. Cell 90:967–970. https://doi.org/10.1016/S0092 -8674(00)80361-X.
3. von Heijne G. 1990. The signal peptide. J Membr Biol 115:195–201.
https://doi.org/10.1007/BF01868635.
4. Hauri H-P, Schweizer A. 1992. The endoplasmic reticulum-Golgi inter-mediate compartment. Curr Opin Cell Biol 4:600 – 608.https://doi.org/ 10.1016/0955-0674(92)90078-Q.
5. Paterson RG, Lamb RA. 1990. RNA editing by G-nucleotide insertion in mumps virus P-gene mRNA transcripts. J Virol 64:4137– 4145. 6. Firth AE, Brierley I. 2012. Non-canonical translation in RNA viruses. J Gen
Virol 93:1385–1409.https://doi.org/10.1099/vir.0.042499-0.
7. Chenik M, Chebli K, Blondel D. 1995. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J Virol 69:707–712. 8. Murata K, Hayashibara T, Sugahara K, Uemura A, Yamaguchi T, Harasawa H, Hasegawa H, Tsuruda K, Okazaki T, Koji T, Miyanishi T, Yamada Y, Kamihira S. 2006. A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J Virol 80:2495–2505.https://doi.org/10.1128/JVI.80.5.2495 -2505.2006.
9. Kulshreshtha V, Ayalew LE, Islam A, Tikoo SK. 2014. Conserved arginines of bovine adenovirus-3 33K protein are important for transportin-3 mediated transport and virus replication. PLoS One 9:e101216.https:// doi.org/10.1371/journal.pone.0101216.
10. Briese T, Schneemann A, Lewis AJ, Park YS, Kim S, Ludwig H, Lipkin WI. 1994. Genomic organization of Borna disease virus. Proc Natl Acad Sci U S A 91:4362– 4366.https://doi.org/10.1073/pnas.91.10.4362. 11. Schwemmle M, Jehle C, Shoemaker T, Lipkin WI. 1999. Characterization
of the major nuclear localization signal of the Borna disease virus phosphoprotein. J Gen Virol 80:97–100.https://doi.org/10.1099/0022 -1317-80-1-97.
12. Kobayashi T, Shoya Y, Koda T, Takashima I, Lai PK, Ikuta K, Kakinuma M, Kishi M. 1998. Nuclear targeting activity associated with the amino terminal region of the Borna disease virus nucleoprotein. Virology 243: 188 –197.https://doi.org/10.1006/viro.1998.9049.
13. Kobayashi T, Kamitani W, Zhang G, Watanabe M, Tomonaga K, Ikuta K. 2001. Borna disease virus nucleoprotein requires both nuclear localiza-tion and export activities for viral nucleocytoplasmic shuttling. J Virol 75:3404 –3412.https://doi.org/10.1128/JVI.75.7.3404-3412.2001. 14. Walker MP, Lipkin WI. 2002. Characterization of the nuclear localization
signal of the Borna disease virus polymerase. J Virol 76:8460 – 8467.
https://doi.org/10.1128/JVI.76.16.8460-8467.2002.
15. Yanai H, Kobayashi T, Hayashi Y, Watanabe Y, Ohtaki N, Zhang G, de la Torre JC, Ikuta K, Tomonaga K. 2006. A methionine-rich domain medi-ates CRM1-dependent nuclear export activity of Borna disease virus phosphoprotein. J Virol 80:1121–1129.https://doi.org/10.1128/JVI.80.3 .1121-1129.2006.
16. Wolff T, Unterstab G, Heins G, Richt JA, Kann M. 2002. Characterization of an unusual importin alpha binding motif in the Borna disease virus p10 protein that directs nuclear import. J Biol Chem 277:12151–12157.
https://doi.org/10.1074/jbc.M109103200.
17. Pyper JM, Gartner AE. 1997. Molecular basis for the differential subcel-lular localization of the 38- and 39-kilodalton structural proteins of Borna disease virus. J Virol 71:5133–5139.
18. Shoya Y, Kobayashi T, Koda T, Ikuta K, Kakinuma M, Kishi M. 1998. Two proline-rich nuclear localization signals in the amino- and carboxyl-terminal regions of the Borna disease virus phosphoprotein. J Virol 72:9755–9762.
19. Kobayashi T, Zhang G, Lee B-J, Baba S, Yamashita M, Kamitani W, Yanai H, Tomonaga K, Ikuta K. 2003. Modulation of Borna disease virus phos-phoprotein nuclear localization by the viral protein X encoded in the overlapping open reading frame. J Virol 77:8099 – 8107.https://doi.org/ 10.1128/JVI.77.14.8099-8107.2003.
20. Berg M, Ehrenborg C, Blomberg J, Pipkorn R, Berg AL. 1998. Two domains of the Borna disease virus p40 protein are required for inter-action with the p23 protein. J Gen Virol 79:2957–2963.https://doi.org/ 10.1099/0022-1317-79-12-2957.
21. Schneider U, Naegele M, Staeheli