Copyright© 1991, AmericanSociety forMicrobiology
Potential Role for
Herpes
Simplex Virus ICP8
DNA
Replication
Protein in Stimulation of
Late
Gene Expression
MIN GAOAND DAVID M. KNIPE*
Department ofMicrobiology and Molecular Genetics, Harvard Medical School, 200 LongwoodAvenue,
Boston,
Massachusetts 02115Received 16 November 1990/Accepted 20 February 1991
Wehave identifiedatrans-dominantmutantform of the herpessimplexvirus(HSV) DNA-binding protein ICP8 which inhibitsviral replication.Whenexpressed bytheV2.6cellline,themutantgeneproductinhibited
wild-type HSVproduction by 50-to150-fold when the multiplicity of infectionwasless than 5.Production of
HSVtypes1and 2 butnotproductionofpseudorabies viruswasinhibited inV2.6 cells. The inhibitoryeffect wasnotduesolelytothehigh levelsofexpression, because the levels of expressionwerecomparabletothose in the permissive wild-type ICP8-expressing S-2 cell line. Experiments designedto define the block in viral production in V2.6cellsdemonstrated (i) that viralaand I8geneexpressionwascomparableinthedifferent
celllines, (ii) that viralDNAreplicationproceeded butwasreduced toapproximately 20% of the control cell level, and (iii) that late gene expression was similar to that in cells in which viral DNA replication was
completely blocked. Geneticexperiments indicatedthat themutantgeneproductinhibitsnormalfunctionsof
ICP8.Thus,ICP8mayplay distinct roles in replication of viral DNA and in stimulation of lategeneexpression.
Thedualroles ofICP8 in these twoprocessescouldprovideamechanismforcontrolling thetransition from
viral DNAsynthesistolategeneexpression duringthe viral growthcycle.
The majorDNA-binding protein of herpes simplexvirus (HSV), ICP8, is expressed as a p or delayed early gene productduring productive infection.ICP8 isoneof theseven
virus-encoded proteins thatare requiredfor the replication of the HSVtype 1 (HSV-1) genome (2, 4, 31, 44, 45). The functions and activities performed by ICP8 have not been completely established. Some known properties of ICP8 include(i)theabilitytolocalizetothe cell nucleus indepen-dent of other viral proteins, (ii) the ability to bind DNA nonspecifically in vitro and in vivo, (iii) the ability to down-regulate the expression ofICP4 under certain condi-tions(12), and (iv)theabilityto promoteassemblyof DNA replication structuresinthe infectedcellnucleus.
To study the functional domains ofICP8, we have
con-structed a variety of ICP8 gene mutations (9a, 10). Pheno-typic analysis ofICP8 gene mutants indicated that several regions of ICP8arerequired for its nuclearlocalization (9b). However, analysis of ICP8-pyruvate kinase fusion proteins showed that the carboxyl-terminal 28 residues of ICP8 comprisetheonly portion of ICP8thatcanfunctionaloneas anuclearlocalization signal.
ICP8 binds to single-stranded DNA (ssDNA) or
double-stranded DNAinvitro (1, 26,34, 39),withatleastafivefold
preference for ssDNA (28, 40), and can be isolated in DNA-protein complexes from infected cells (27, 29). The
ICP8 sequences required for ssDNA binding have been
mapped between residues 564 and 849, on the basis ofthe
studies of a variety of ICP8 mutant viruses, of in vitro transcription-translation products of ICP8, and of partial proteasedigestion of purified ICP8 (10, 30, 43).
Theintranuclear location of ICP8 is determined,atleast in part, by the status of viral DNA replication (5, 35). In infectedcells, before viral DNA synthesis ICP8 localizesto
nuclear framework-associated structures called prereplica-tive sites (5, 35, 36). As viral DNAreplicationoccurs, ICP8
* Correspondingauthor.
migrates to replication compartments (5, 35), where it is boundtoprogenyandreplicating viral DNA (27, 29).Viruses expressinganaltered ICP8moleculefailtoassemble prerep-licative sites.Therefore, ICP8isrequired fortheassembly of prereplicativesites(5).
To further studythefunctional domainsofICP8, wehave introduced various mutated ICP8 gene sequences into the viral genome (9a, 10). However, we were unable to intro-duce certain mutations from ICP8 gene plasmids into the viralgenomebyrecombination. One of thesemutantshasa small deletion between the sequences encoding the DNA-binding region and the nuclear localization signal ofICP8. Thisreportdescribes that thismutantICP8proteinexhibits
atrans-dominantphenotypeandcansignificantlyinhibitthe productionofwild-type(wt)HSV-1.Analysisof this mutant phenotypesuggests thatwt ICP8 playsa role in the
stimu-lation of lategeneexpression.
MATERIALS ANDMETHODS
Plasmids. Theplasmid pSV8and thenucleotidenumbering systemfor the ICP8genewere describedpreviously (9, 20).
The plasmid pSV8 was constructed by inserting the ICP8 codingsequences(mapunits0.374 to0.409) downstreamof the simian virus 40 early promoter in plasrrlid RL18:PK12
(24).Theplasmid pSV8.3wasderivedfrompSV8 bydeleting
the polylinker region and the SacI-BglII fragment ofthe pyruvate kinase gene. The plasmid pSVdlO5 was con-structedby deletion ofaBgllIfragmentofpSV8.3 after the conversion ofPvuII (nucleotide 3853) andNaeI (nucleotide 4111)sitestoBgllIsites.Thus, pSVdlO5lacks codons 1083 to 1168 of the ICP8 coding sequence but encodes four additional aminoacids,Gly-Arg-Ser-Ser, in theBglII linker sequence. The plasmid pSG28 was providedby M. Levine (University ofMichigan).
Cellsand viruses. Vero cellsweregrownand maintainedas
described previously (25). Thegrowth medium for the
neo-mycin-resistant cell lines S-2 (10) and V2.6 (see below)
2666
on November 10, 2019 by guest
http://jvi.asm.org/
POTENTIAL ROLE FOR HSV ICP8 IN LATE GENE EXPRESSION included 200 ,g of the antibiotic G418permlduring the first
passageof thecells after thawingor500,ug of G418permlof
mediumeveryfive passages.
TheHSV-1wtstrain KOS1.1waspropagated and assayed as described previously (25, 27). The mutant viruses d301 andnlOwere grownandpropagated in the ICP8-expressing S-2 cellline (10). Pseudorabies virus (PRV)wasprovided by
E. Sinn and R. Roeder (RockefellerUniversity).
For infectionsatamultiplicity ofinfection (MOI) of 2 PFU
percell for biochemical analysis, cell numberswere
deter-mined by cellcountsofaculture flaskofeach cell line being
used priorto theinfection.
Isolation of d105ICP8-expressing cell line. Verocellswere
transformed with the plasmids pSVdlO5 and pSVneo (Fig. 1) (42)asdescribed previously (6, 10).After growth in medium containing the antibiotic G418 (a neomycin analog), drug-resistant colonies were isolated, grown into cultures, and screenedbyindirect immunofluorescence (5) using the lOE-3 anti-ICP8monoclonalantibody (38) for the abilitytoexpress
the mutant form of ICP8 upon nlO infection. The lOE-3 antibody recognizes diO5 ICP8 butnotnlO ICP8 (9b).
ssDNA cellulose chromatography. ssDNA cellulose chro-matography of infected cell extracts was performed as
describedpreviously (10, 26).
Analysis of viral proteins and viral DNA replication. Cell monolayercultureswereinfected with KOS1.1ord301virus
and then labeled with [35S]methionine and harvested as
indicatedbelow. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of infected cell lysates was
performed asdescribed previously (25). After
electrophore-sis, the gels were fixed, dried, and exposed to Kodak SB5 film, orthe proteins weretransferred by electrophoresis to nitrocellulose filters for Western immunoblot analysis. Im-mune complexes were detected on blots by a procedure
involving acolor reaction for alkaline phosphatase activity
conductedas specified by the manufacturer (Promega
Bio-tec, Madison, Wis.). The rabbit polyclonal serumPP5 (46) and themousemonoclonalantibody lOE-3 (38)wereusedto detect HSV-1 DNApolymerase andICP8, respectively.
Analysis of viral DNA amplificationduring the courseof infection was performed as described previously (37). The
probes used were the plasmids pBR3441 (30) and pSG28
(13). To quantify the data, the slots were cut out and radioactivitywas measuredby liquid scintillation counting.
Northern (RNA) blotanalysis.Total cytoplasmic RNA for Northern blots was isolated and analyzed as described
previously (37). The plasmid used for the probewas
pEcoRI-BamHI-I-I (gC gene probe [8]). Band intensities on the autoradiogram were determined with an Ultrascan laser
densitometer and an on-line integrator (LKB Instruments,
Inc., Rockville, Md.).
RESULTS
The mutant d105 exhibits a trans-dominant phenotype.
WhenweattemptedtointroducevariousmutantICP8genes into the viral genome by recombination, we observed that
viruses containing certain mutant alleles could not be iso-lated. One of these was the diO5 mutation (Fig. 1). The mutantdlO5 ICP8genehas adeletion of 86 codons between
the sequence encoding the DNA-binding domain and the sequenceencoding the nuclear localization signal. One pos-sible reason for the failure of introduction of the diO5
mutation into the viral genome was that the diO5 gene product exhibited a trans-dominant mutant phenotype. To test this, we cotransfectedvarious amounts ofmutant dlO5
lz:::~~ ZBEl
0.41 0.40 0.39
KOS [
1196aa
DNA-bindingregion
Nuclear localizationsignal I
Nuclearfunction
d105 I U
FIG. 1. Functional domains ofICP8. Analysis ofa variety of ICP8 mutants indicated that the DNA-binding region of ICP8 is located within residues 564to 1081(10)and that theC-terminal28 residues of ICP8 can functionas anuclearlocalization signal (9b). MutantdlOl ICP8 which ismissing residues 17 to 563localizes to the nucleus and binds to ssDNA but fails to promoteviral DNA replication (10). This indicates that the N-terminal half of ICP8 has anuclearfunction other than DNAbinding. The d105mutantICP8 hasadeletionof residues 1082to1169 andexhibitsatrans-dominant negative phenotype.
plasmidwith 1 ,ugof infectious HSV-1DNAinto Vero cells; we observed that plaque numbers were dramatically
de-creased compared with those obtained when the wt ICP8
plasmid was cotransfected (Table 1). Even when the molar ratio of the amounts of ICP8 gene sequences in diO5 plasmid to those in the infectious viralgenome was only 2:1 (0.1 ,ug ofdlO5 plasmid), significant inhibition ofplaque formation was observed. Thus, the diO5 mutant showed a trans-dominantphenotype.Incontrast, many other ICP8 mutants did not causeaninhibition of viralgrowth (datanotshown). The diO5 gene product was able to localize into the
nucleuswhen it wasexpressedin Verocells transfectedwith
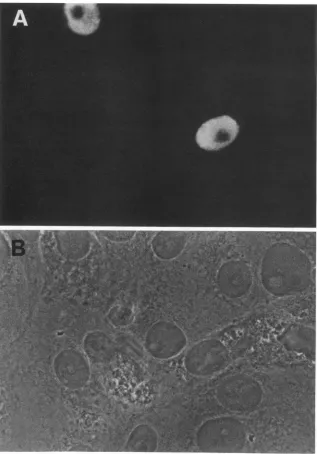
thepSVdlO5 plasmid (Fig. 2).Tostudy the phenotype ofthe
diO5 gene product in more detail, we isolated a cell line
named V2.6 which expressed dlO5 ICP8 upon HSV
infec-tion. TheV2.6cell linewas apoorhost forwtvirusgrowth, despite the presence ofwt ICP8 expressed from the viral genome. Theplating efficiency ofwtvirusonV2.6cellswas 100- to1,000-foldlower than that on Verocellsor onthewt
[image:2.612.318.556.72.218.2]ICP8-expressingS-2 cellline,and the sizeoftheplaqueswas
TABLE 1. Inhibition of HSV-1plaque formationby trans-dominant mutantICP8'
Type of ICP8DNA No. of plaques
andamt(,g) Expt 1 Expt2 Expt 3
wt
0.1 85 81
0.5 86 83 107
1.0 64 73 192
Mutant
0.1 25 12
0.5 0 2 0
1.0 0 2 0
aInfectiousKOS1.1DNA(1 ,ug)wascotransfectedwithvariousamounts
of eitherwtICP8plasmid(pSV8.3)ord105mutantplasmid(pSVdlO5)bythe calciumphosphateprecipitation procedure (9, 14).Plaqueswerecounted 3to 4days after transfection.
VOL.65, 1991 2667
ni
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.316.557.573.686.2]FIG. 2. Nuclear localization ofmutant dlO5 ICP8 in transfected cells. Vero cellswere transfected withpSVdlO5 and processed for immunofluorescenceby usingthe lOE-3antibody. (A) Immunofluorescence micrograph, (B) corresponding phase-contrastmicrograph.
extremely small (datanot shown). Single-cycle growth
ex-periments showed thattheyield ofwtvirusonthe V2.6 cell linewas 50- to150-fold lower than that on Vero cellsoron thewtICP8-expressing S-2 cell line when theMOI was less than 5 (Table 2). At an MOI of 0.1, the V2.6 cell line
exhibited ayield of HSV-1 KOS1.1 reduced approximately 150-foldfromthat on Vero cells or on the wt
ICP8-express-ingS-2 cellline. Theinhibition of growth on V2.6cellswas overcome at an MOI of 10. Thus, although the
diO5
gene product could inhibit viral replication in the presence of wtICP8atlowMOIs,thetrans-dominant effect appeared to be
due to competitive inhibition because the inhibition was
decreasedat ahigh MOI. Becausesignificantinhibition was
obtainedat an MOI of 2.5 andmostcells were infected, we
chose an MOIof 2 to 2.5 PFU per cellfor thebiochemical studies described below.
To examine the specificity of resistance of the V2.6cell
linetoHSV-1, wealso tested theability of the V2.6 cell line to restrict the growth of HSV-2 and ofa closely related herpesvirus, PRV. An inhibitory effect of similarmagnitude was observed for HSV-2 on the V2.6 cell line (data not
shown). However,nodecrease of virusyieldswasobserved when PRV was plated on the V2.6 cell line (Table 3). The induction of themutantform of ICP8 in the V2.6celllineby
PRV was evidenced by Western blot analysis by using an antibody specific for ICP8 (datanot shown). These results indicated that the inhibitory effect ofmutantdlO5wasHSV
specific.
Todetermine whether this inhibitory effectwasexertedon ICP8, the mutant diO5 plasmid was cotransfected with increasing amounts of wt ICP8 plasmid. When this was done, the inhibition of the production of wt virus was
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.153.470.68.522.2]POTENTIAL ROLE FOR HSV ICP8 IN LATE GENE EXPRESSION TABLE 2. Growth of wt HSV-1 on different cell lines
Yield(PFU percell)atthefollowing MOII
Cell line Expt
0.1 1.0 2.5 5.0 10.0
Vero 1 260 (153b) 380 (91) 420(3.8)
Neor 2 950 (158) 660(66) 410 (8.2)
S-2 1 260(153) 310 (74) 230 (2.1)
2 560 (92) 450(45) 420 (8.4)
V2.6 1 1.7 4.2 110
2 6.1 10 50
aVero cells, the wtICP8-expressing S-2cell line,andtheV2.6cell lineexpressingd105ICP8were infected with KOSwtvirusat theindicatedMOI and were
incubatedat 37°C.Cellswereharvested 24h afterinfection, andtiters of theprogenyviruseswere determinedonVerocells.
bNumbers inparentheses represent thefoldgreater yield on these cells relative to the yield on V2.6 cells.
partially relieved (Table 4). These results demonstrated that theinhibitory effect caused by the d105geneproductwas a
trans-dominant effect mediatedon wtICP8.
Themutantd105 ICP8 binds tossDNA. For many
DNA-binding proteins, such as yeastGCN4 (22), trans-dominant mutant forms retain their DNA-binding ability but lose transactivation activity ortheir ability to interact with
cer-tainotherproteins (reviewedinreference 18). Therefore,to determine whether theinhibitory gene product retained the
ability to bind DNA, we infected V2.6 cells with a
DNA-binding-negative ICP8 mutant d301 (10). Extract from in-fectedcellswaspassedover assDNAcellulose column,and ICP8 waseluted stepwise with buffer containing increasing
salt concentrations. Polypeptides in the various fractions
were analyzed by PAGE (data not shown) or Western
blotting (Fig. 3). In V2.6 cells infected with d301,a120-kDa
form of ICP8 was detected (Fig. 3, lanes 12 and 13), in agreement with thepredicted size of the diO5gene product
(9). Mutant d301 ICP8 bound poorly to ssDNA cellulose (Fig. 3, lanes 12 through 15) and was found in the
flow-through fractions (lanes 7 and 8). In contrast, d105 ICP8 bound to ssDNA (Fig. 3, lanes 12 through 15), and the majority ofitwaseluted with 0.5MNaCl (lane 13). In other
comparisons, dlO5 ICP8 boundtossDNA withanefficiency
similartothatofwtICP8 (datanot shown).
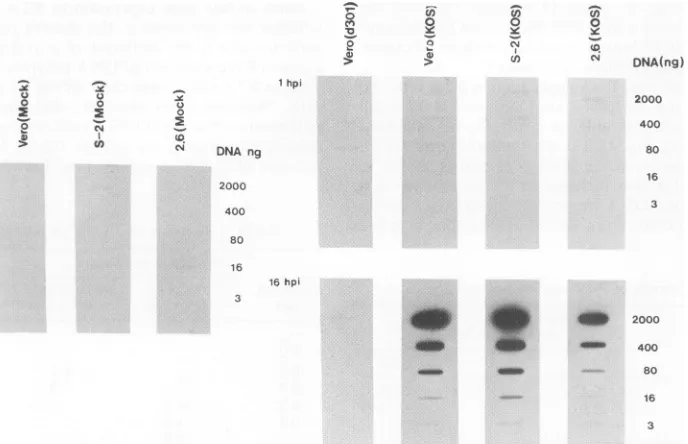
Partialinhibition ofviral DNAreplicationinV2.6 cells.To determinewhetherthe decreased viral yield inthe V2.6 cell linewasduetoablockinviralDNA synthesis,weexamined
viral DNAreplication (Fig. 4). The d301mutantvirus, which contains a large deletion in the ICP8gene and isunable to replicate its DNA (10), was included in the experimentas a
negative control. Total DNAwasisolated from mock-orwt virus-infected Vero cells, from the wt ICP8-expressing S-2
TABLE 3. Growthof PRVondifferentcelllines
Yield(PFUpercell)at
Cellline the followingMOI'
0.1 2.0
Vero 180(3.6b) 340(1.8)
S-2 100(2.0) 330(1.8)
V2.6 50 188
aVerocells, thewtICP8-expressingS-2 cellline, andthe V2.6 cell line expressing dlO5ICP8wereinfected with PRVatanMOI of 0.1or2 andwere
incubatedat37°C. Cellswereharvested 24 h afterinfection,andthe titers of theprogenyvirusesweredeterminedonrabbitkidneycells.
bNumbers in parenthesesrepresentfoldgreateryieldonthese cellsrelative totheyieldonV2.6 cells.
cellline, and from the inhibitory V2.6 cell line at 1 or 16 h postinfection. Fivefold serial dilutions of the DNA were bound to anitrocellulose filter, which was then hybridized with32P-labeled HSV-1 VP16 gene DNA as aprobe (32). As expected, mutant d301-infected Vero cells showed no am-plification of viral DNA during the course of infection,
[image:4.612.315.557.573.685.2]consistent withprevious results (10). In contrast, wt virus-infected Vero, S-2, and V2.6 cells showed substantial am-plification of viral DNA during the infection. Toquantitate theseresults, the amount of radioactivity hybridized to each slot was measuredby scintillationcounting, and the relative amounts of HSV-1 DNA in eachsample were determined. In thisparticular experiment, the amount of viral DNA synthe-sized in the V2.6 cells was about 33% of the level in Vero cells and 19% of that in the S-2 cells. In several separate experiments, viral DNA replication in V2.6 cells was re-ducedto anaverageof17% of the Vero cell level or S-2 cell level. Similar results were obtained when the EcoRI joint fragment (pSG28) was used as aprobe, indicating that the fulllength of the viral genome wasprobablyamplified.Thus, substantial viral DNAreplication occurred in the V2.6 cells. Block in late gene expression in V2.6 cells. To determine whether the inhibition of the growth ofwt HSV-1 in V2.6 cellswasduetothe inhibition of aorX geneexpression, we measured the amounts of DNApolymeraseexpressedby wt virus in V2.6 cells, Verocells, or thewtICP8-expressingS-2 cells. Western blots revealed that the quantity of DNA polymerase (Fig.5A) in V2.6 cellswasonlyslightly reduced relative tothat in Verocells orthe wtICP8-expressing S-2 cell line (Fig. 5A, compare lane 9 and lane 7 or 8 at 6 h
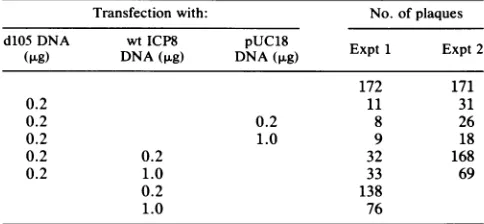
TABLE 4. Relief ofdlO5 ICP8 inhibition bywtICP8
Transfectionwith: No.ofplaques
d105DNA wtICP8 pUC18
(,ug) DNA
(p.g)
DNA(,ug) Expt 1 Expt 2172 171
0.2 11 31
0.2 0.2 8 26
0.2 1.0 9 18
0.2 0.2 32 168
0.2 1.0 33 69
0.2 138
1.0 76
aInfectious KOS1.1 DNA(0.2,ug)wascotransfected with variousamounts
of wtICP8 plasmid(pSV8.3), d105mutantplasmid(pSVdlO5),andpUC18
plasmid bythecalciumphosphateprecipitation procedure (9, 14).Plaques
werecounted 3days after transfection.
VOL.65,1991 2669
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.59.298.601.670.2]1 2 3 4 5 6 7 8 9 10 111213 1415
"agm
a_:". . . -8(WT)-8(2.6)
*~~~~~~~-8(d301)
FIG. 3. DNA binding of the dlO5 mutant gene product. V2.6 cells were infected with the DNA-binding-negative ICP8 mutant d301at anMOI of 10 PFU per cell and labeled with[35S]methionine from6to 8 h postinfection. The various protein fractions resolved on a ssDNA cellulose column were subjected to SDS-PAGE and electroblottedonto a nitrocellulose filter. The filter was then probed with a monoclonal antibody specific for ICP8 (1OE-3). Lanes: 1, purifiedwtICP8 as a size marker; 2, total cellularlysate; 3, pellet from high-saltDNase extraction;4,pellet after dialysis; 5, extract put onssDNA column; 6 to 11, flowthrough and wash; 12, 0.3 M NaClelute; 13, 0.5 M NaCl elute; 14, 1.0 M NaCl elute; 15, 4.0 M NaCl elute. The positions of wt ICP8, the dlO5 form of ICP8 expressed fromV2.6 cells,and d301ICP8areindicatedonthe right.
postinfection,orlane 12 andlane 10or11 at12h postinfec-tion).
Wealso examined the amountsof ICP8 expression in the differentcell types. Western blots(Fig. SB) revealed that the
levelsofwtICP8 expression were equivalentin Vero
(Fig.
5B, lanes 1 and 2) and V2.6 cells (Fig. 5B, lanes 5 and6).
Thisblot also shows that theamountofdiOS ICP8expressed
inV2.6 cells(Fig. SB, lanes 5 and 6)wasonly slightlygreater than theamountofwtICP8expressed in the permissive S-2 cells(Fig. SB, lanes 3 and4). Other Western blots demon-strated no apparent reduction of ICP4 expression. These results demonstrated that the majorpartof the inhibition of the growthofwtHSV-1 in the V2.6 cell linewas notdueto theinhibition ofotor ,3 geneexpression.
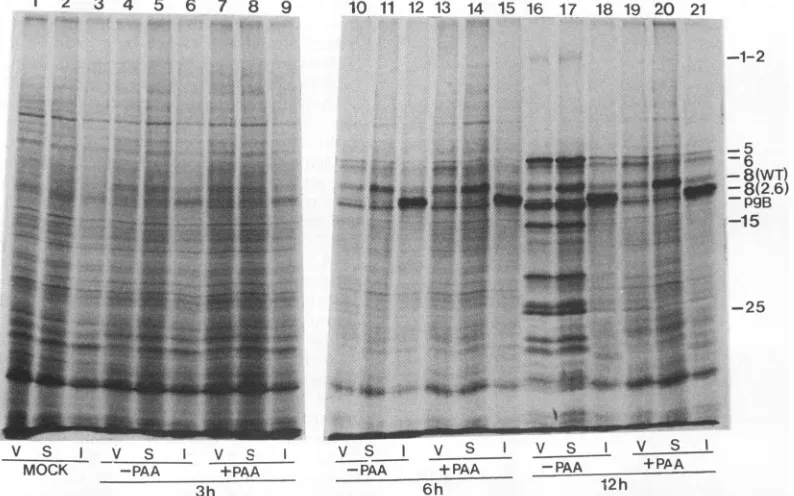
To further examine viralgeneexpression in V2.6 cells,we infected Verocells,wt ICP8-expressing S-2 cells, and V2.6 cells withwt HSV-1 in the absence orpresenceofsodium phosphonoacetate (PAA), a specific inhibitor of HSV-1 DNAsynthesis, andwepulse-labeled the cells with
[35S]me-thionine at3, 6, or12 h postinfection. The labeledproteins
were separated by SDS-PAGE and visualized by autoradi-ography(Fig. 6). The mutantform of ICP8 expressed from V2.6 cells, likewtICP8 expressed from S-2 cells, was not constitutively expressed butwas induceduponvirus infec-tion. The patterns of viral protein synthesis at 3 or 6 h postinfectionweresimilar in the 3 different celllines, except that significant amounts of the mutant form of ICP8 ex-pressed from the V2.6 cell line and ofwt ICP8 expressed from the S-2 cell line were observed. The amount of the mutant form of ICP8 produced by the V2.6 cell line was greater than that ofwt ICP8 in Vero cells (Fig. 6, lanes 21 and 19) but was comparable to that produced by the wt ICP8-expressing S-2 cell line (Fig. 6, lanes 21 and 20)at12 h postinfection. Although slightly greater amounts of d105 ICP8 than ofS-2ICP8 wereexpressedat6 hpostinfection in this experiment (Fig. SB and 6, lanes 14 and15), thiswas not observed in other experiments (data not shown). Because S-2 cells were aspermissivefor HSV as Vero cells(Table2), theinhibition of wt virusgrowthontheV2.6 cell line was not
o 0
2 N
A)
0N
1hpi
(3 0
(04
N
DNAng
2000 400 80 16
16hpi
cn 0 0 0
2 2 o o
A CA
-_a,
_r 4_ _
_~~~~~4%l
DNA(ng)
2000 400 80 16 3
2000 400 80 16 3
FIG. 4. HSV-1DNAreplication indifferentcell lines. Verocells, the wtICP8-expressing S-2cellline, and theinhibitoryV2.6cell line weremockinfectedorinfected with the ICP8 mutantd301or wtHSV-1. Total cellular DNA was preparedimmediatelyafter viraladsorption (1 h) or near the end of the infection cycle (16 h). Equal amounts of each DNA were subjected to fivefold serial dilutions, and the DNAs were boundto anitrocellulose filter,which wasprobedwith32P-labeledDNAspecific for the VP16 gene. Anautoradiographof the blot is shown. hpi, Hours postinfection.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.78.283.70.240.2] [image:5.612.140.484.447.669.2]POTENTIAL ROLE FOR HSV ICP8 IN LATE GENE EXPRESSION A 1 2 3 4 5 6 7 89 10 11 12
-pol
v S I V S I V S I v s I
MOCK 3h 6h 12h
B1
2 3 4 5 6 7 8 9 10 11 12_
-P.
_ .::II
_010mi,
_mm
-ICP8(wt)-ICP8(V2.6)
FIG. 5. Western blot analysis of the amounts of HSV DNA polymeraseandICP8 expressed in different cell lines. (A) Vero cells (V),the wtICP8-expressingcell line (S), and the inhibitory cell line (I)weremock infected or infected with wt HSV-1 at an MOI of 2 PFU per cell and harvestedat 3, 6, or 12 h postinfection. Proteins in thecell extracts were separated by SDS-PAGE and electroblotted ontonitrocellulose. The filter was thenprobed with the PP5 poly-clonal antisera against HSV-1 DNA polymerase (provided by D. Coen). Theposition of DNA polymerase (pol) is indicated to the right of the filter. (B)Westernblot forICP8,obtained by using the 1OE-3monoclonal antibodyspecific for ICP8. Pairs of panels contain threefolddilutions of extractobtainedfrom cells at 6 h postinfection. Lanes1and2, Vero cells; lanes 3 and 4, S-2 cells; lanes 5 and 6, V2.6cells; lanes 7 and 8, Vero cells with 400 ,ug of PAA per ml; lanes 9 and 10, S-2 cells with400,g ofPAAper ml; lanes11 and12, V2.6cellswith400 ,ugof PAA perml.
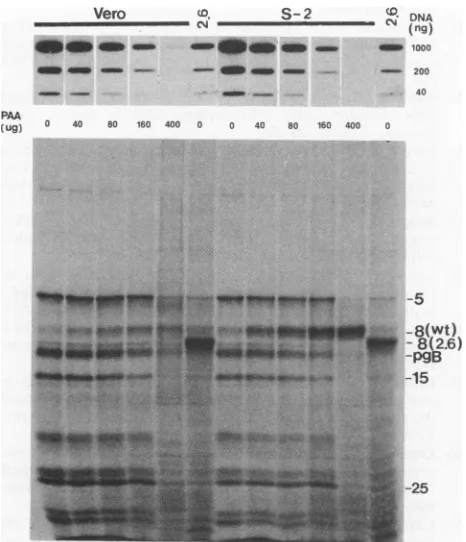
late gene expression. To determine whether this was the case, we used various concentrationsofPAA to attemptto
reduce viral DNA synthesis in Vero cells or in the wt
ICP8-expressing S-2cell line tolevels similar to that of the V2.6 cell line inthe absence of PAA. Thus, if the level of DNAsynthesiswasthesole determinant of the levels of late geneexpression,late geneexpressionwould then be similar
in the different cell lines. Cells were infected with wt virus and harvestedat16 hpostinfection for analysisof viral DNA and protein synthesis (Fig. 7). With increasing concentra-tions ofPAA, decreasingamounts of viralDNA
amplifica-tion and late gene expression were observed in Vero cellsor inS-2 cells. At a PAA concentration of 400 ,g per ml, viral DNA synthesis was completely inhibited, and no -y2 viral polypeptide synthesis (e.g., ICP15) was observed. At PAA concentrations between80 and 160 ,ug per ml, the amounts of viral DNA synthesized in Vero cells or in S-2 cells were similar to that synthesized in V2.6 cells infected without PAA. However, under these conditions, synthesis of viral
polypeptidesof
-yl
(ICP5 andICP25)and-y2(ICP15) classesin theV2.6 cells wasmarkedly reduced compared with that inVero cells or in S-2 cells. Quantitation of ICP5 synthesis by densitometry indicated that in V2.6 cells the rate of synthesisofICP5wasapproximately eightfoldlower than in Vero cells or S-2 cells.Therefore,weconclude that thelack oflate gene expression in the inhibitory V2.6 cell line was not due entirely to the decreased amount of viral DNA synthesis.
Tofurther define the level atwhich late geneexpression was decreased, we performed Northern blot analysis to measure the steady-state level of
y2
gC mRNA (Fig. 8) in different cells. Infection of Vero cells or V2.6 cells was carried out in the absence or in the presence of 100 ,ug of PAA per ml. Total cytoplasmic RNA was isolated from infected cells at 16 h postinfection. Equal amounts of total cellular RNA were probed on a Northern blot with a32P-labeledDNA fragmentfor the gC gene. NogC mRNA was detected in mock-infected V2.6 cells (Fig. 8). As ex-pected, partial inhibition of viral DNA synthesis by PAA
reduced the amounts ofgC mRNA in Vero cells orin S-2 cells. At a PAAconcentration of 100 ,g perml,the levelof
gCmRNA in V2.6cells was at leastsixfoldless than in the other cells. Therefore, the block in late geneexpression in V2.6 cells was due, atleast inpart, to a decreaseinmRNA accumulation.
simply
duetooverexpressionof the mutantform ofICP8. Inaddition, the amount of wt ICP8 produced by wt virus
infection on the V2.6 cell line was only slightly reduced
compared withthat on Vero cells(Fig. 6, lanes 19 and21).
This result was consistent with those inFig. 5, suggesting
that the inhibition ofwt virus growthon the V2.6 cell line was not aconsequenceof the mutant form of ICP8
attenu-atingtheexpression ofa. and L genes.
Incontrast, thesynthesisof viral polypeptides of-yl(ICP5
andICP25) andy2 (ICP1-2andICP15) classeswasmarkedly reducedin V2.6cells(Fig. 6,
lane
18)comparedwiththat in Verocells (lane 16) or the wt ICP8-expressing S-2cell line(lane 17).Infact,the
pattern
of viral polypeptide synthesisat 12 hpostinfection wasalmost identical to that observedintheabsence of viral DNAsynthesis(Fig. 6,compare lane 18 andlane19, 20,or21).Thus,viral lateproteinsynthesiswas reduced in V2.6 cells.
Separation of defects in viralDNAsynthesis andlategene expression. It was conceivable that the decreased viralDNA
replicationin V2.6cellswasthesolecauseof thedecreased
DISCUSSION
Wehaveexpressed atrans-dominantmutantformof the HSVICP8DNA-bindingproteinandobserved that it confers
significantresistance to HSV infection. In cells expressing
the mutantgeneproduct, viral DNAsynthesisand lategene
expression are decreased. Our results indicate that the decreased late geneexpression isnotdue to decreased DNA
replication. This suggests a new role for ICP8 in viral
replication, i.e., stimulation oflate geneexpression.
Inhibition of viralreplication by a mutant viral gene prod-uct. Viralreplication was inhibited
significantly
bythe mu-tantgeneproducteither in cotransfected cells or inacell lineexpressing the mutant gene product. Orberg and Schaffer
(33)previously isolatedacellline, U35, bytransformation of
largeamountsofwtICP8 gene DNA which showed reduced
permissiveness for HSV. The U35 cell line expresses high
levels of ICP8 upon viral infection, and it is believed that
overexpression ofwt ICP8 is the cause of decreasedviral
replication. Thiscell line also containedasignificantnumber
VOL.65, 1991 2671
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.67.295.70.350.2]1 2 3 4 5 6 7 8 9
ft
..,I.Wkw,-,.1
:.. .. *..f.
... i.
*.'
-10 11 12 13 14 15 16 17 18 19 20 21
...A "*1
-1-2
-5 -6 -8(WT) -8(2.6) -PgB -15
-25
v s I v s v- S I
MOCK -PAA +PAA
3h
-s~~~~~~~~~~-VS_ I V VS I V S I
-PAA +PAA -PAA +PAA
6h 12 h
FIG. 6. Polypeptide profile ofwtHSV-1-infected Vero (V), S-2 (S), and V2.6 cells(I). Cellmonolayer cultures wereinfected withwt
KOS1.1at anMOI of2 PFU percell in the absenceorpresenceof400 ,ugofPAA perml.At3, 6,or12 hpostinfection, the cellswerelabeled with[35S]methioninefor30min and thenharvested. Equalfractions ofeachcelllysateweresubjectedtoSDS-PAGE andautoradiography. Showntotherightof the gelarethepositions of several HSV-1proteins.
ofrearranged and deleted ICP8 genes (33). Thus, defective
geneproducts could be expressed in these cells. Twolines of evidence argue that the
diO5
defective gene product, notoverexpression of ICP8, isresponsible forthe inhibition of viral replication observed inthis work. First, parallel
trans-fections withequalamountsofdlO5andwtICP8 gene DNAs
ledtogreatlyreduced numbersofplaquesin the presenceof d105 DNA. Second, although S-2 cells and V2.6 cells ex-press approximately equal amounts of the two forms of ICP8, infectionof V2.6 cellsgives 150-fold less virus at a low
MOI. Therefore, thereis some uniquefeature of the defec-tive gene product which causes an inhibition of viral
repli-cation.
Because cotransfection of wt ICP8 gene DNA with the
diO5
DNArelievedtheinhibitoryeffect of thediO5
gene, we concluded that the dlO5gene product exhibitedadominantinhibitory phenotype by acting as a competitiveinhibitorof wt ICP8 function. The
diO5
protein retains thessDNA-binding properties of wt ICP8, and it seems likely that it
could compete with wt ICP8 forDNA-binding sites. Infec-tion of V2.6 cells at a high MOI also overcame the inhibitory
effect. These results indicate that the mutant gene product
has not acquired a new activity but instead acts as a
competitive inhibitorof the normal functions of wt ICP8. Two HSV-1 trans-dominant mutant gene products have been reported (7, 41). It was hypothesized that these mutant gene products interfered with wt viral functions either by
formation
of nonfunctional dimers (41) or by tying up cellfactorsininactivecomplexes (7). The reduction intiter of wt
virus stock observed on these different inhibitory cell lines varied from mutant to mutant, but in most cases it was in the range of 10- to 40-fold. The V2.6 cell line showed a much greater inhibitory effect on the production of wt virus
becausethereduction in theyield ofwtvirusproducedin a
single lytic infectious cyclewasas muchas 150-fold. Apotential role for ICP8instimulation of lategene expres-sion.Sevenviral geneproducts,
including
ICP8,arerequired forHSV DNA replication (2). Because HSV DNAreplica-tionisabsolutely requiredfortruelate(-y2) geneexpression during HSV infection (19, 21, 23), all DNA replication proteins areindirectly required forlate geneexpression. It hasbeen difficulttodemonstrateadirect role for any of these seven genes in late gene expression because these two events are tightly coupled. ICP8 mutant viruses cannot activate late gene expression, apparently because these mutants could notpromoteviral DNA synthesis (10). V2.6 cellsofferauniquesituationtoexamineapossible regulatory
roleof ICP8 in late geneexpression.
InV2.6cells,wtvirussynthesizedamountsof viral DNA
sufficienttosupportlate gene
expression,
butvirtuallyno-y2geneexpression was observed. Thiswasin contrast tothe results in which significant amounts of -y2 genes were ex-pressed when viralDNA replication was partially inhibited
byPAAto alevelsimilartothatonV2.6cells. Theseresults
suggestedthat late gene
expression
could bepartially sepa-ratedfromDNAreplicationand thatICP8 hasadistinct role in stimulationof late geneexpression.The ability of a ssDNA-binding protein to promote late genetranscriptionwould not beuniquetoICP8. The adeno-virusDNA-bindingprotein,the gene 32protein of bacterio-phage T4,and the SSBproteinof Escherichia coliexhibited
similar roles in activation oftranscription (3, 11, 15). The adenovirus DNA-binding protein can enhance the expres-sion ofa reporter genecontrolled by several different pro-moters within transfected cells (3). The adenovirus major
late promoter showed a greater response to the
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.119.516.75.323.2]POTENTIAL ROLE FOR HSV ICP8 IN LATE GENE EXPRESSION
Vero D _ S-2 C DNA
CM C4
~~~~~(ng)
_m_
-
-
_m
_ 1000_ - _ - -
---- 200 PAA
(ug)
r%l
(Dr
I8
0 100 0 100 0 0
40 80 160 400 0 0 40 80 160 400 0
:..
*mJrn Itn,
-gC mRNA
[image:8.612.66.299.73.344.2]18S
--25FIG. 7. The block in late gene expression in V2.6 cells is independent of decreased viral DNA replication. Vero, S-2, and
V2.6cells wereinfected withwtHSV-1atanMOI of 2 PFUpercell
with theindicated concentrations of PAA andwereharvestedat16 hpostinfection for the analysis of viral DNA and protein synthesis asdescribed in the legendstoFig. 4 and 5.
binding protein than to the ElA transactivator protein, suggestingthattheDNA-binding protein playsacentralrole
inactivation of the latepromoter. Several T4 bacteriophage gene32mutantssynthesizedhighlevels of viralDNA(30to 55% ofwtviruslevel)butweredefective for thesynthesisof late gene 23 protein, the major structuralprotein of the T4 capsid (11). The amounts of mRNA were also greatly
re-duced for at least 3 late genes in these gene 32 mutants. Theseresultsdemonstratedadirect requirement forgene32 proteininthe activation of lategeneexpression. The E. coli SSB protein activates promoters transcribed by the bacte-riophageN4virionRNApolymerase andis alsorequiredto initiatetranscriptionfromspecificpromotersiteson double-strandedDNA(15).Inaddition,the DNAreplication protein gp45ofbacteriophage T4 hasalso been demonstratedtobe directly required for late gene expression(16). These three T4-encoded DNApolymerase accessory proteins stimulate theopeningof T4late promoters, and the activation of late promoters in vivo by these three proteins is effected by moving replicationforksactingasmobileenhancers(16, 17). Thus, several DNAreplication proteinsappeartobe
neces-sary forstimulation of late viralgene expression.
Our current working model is that at late times after infection, wt ICP8 binds to newly synthesized double-strandedprogenyDNA andholds it in aformonwhichlate transcriptionisoptimal.This could be duetoICP8bindingto ssDNAregions onprogeny DNA andkeepingthe promoter regions open for transcription. Alternatively, ICP8 might bind to specific DNA structures in late promoters, or the
specificity ofstimulation of late gene expression might be
FIG. 8. Accumulation of gC mRNA inwtHSV-1-infectedcells. Vero, S-2, and V2.6 cellswereinfected withwtvirusatanMOIof 2 PFU per cell with or without 100 ,ug of PAA per ml. Total cytoplasmic RNA was prepared at 16 h postinfection. Equal amountsof RNAweresubjectedtoNorthern blotanalysis by using 32P-labeled probe specific for gC mRNA. Showntothesidesofthe gelarethemigrationpositions of rRNA and gC mRNA.
achievedby the interactionsbetween ICP8 and other viralor
cellular proteinsneeded torecognizelategenepromoters.In addition to altering the structure of viral chromatin, the binding ofICP8 to progeny DNA may target the DNA to nuclear sites such as replication compartments where late transcriptioncan occur.Themutantform of ICP8expressed by the inhibitory cell lineV2.6 may have lost its ability to interact with other viral or cellular proteins, but it still retains itsabilitytocompete withwtICP8 for DNAbinding. Thus, the d105 protein would behave in a trans-dominant defectivemanner.
ACKNOWLEDGMENTS
Wethank D.Coenfor PP5antipolymeraseserumand E. Sinnand
R. Roeder forPRV.
This workwassupportedbyPublic Health Servicegrant CA26345 from theNational Cancer Institute.
REFERENCES
1. Bayliss,G.J.,H.S.Marsden,andJ.Hay.1975.Herpes simplex virus protein:DNA-bindingproteinsininfected cells and inthe virusstructure. Virology68:124-134.
PAA
(ug) 0
-5 -8(Wt) -8(2.6) -PgB -15
28S
-VOL.65, 1991 2673
[image:8.612.322.550.75.420.2]t:.
...
on November 10, 2019 by guest
http://jvi.asm.org/
2. Challberg, M. D. 1986. A method foridentifying the viral genes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA 83:9094-9098.
3. Chang, L.-S., and T.Shenk. 1990. The adenovirus DNA-binding proteinstimulates the rateoftranscription directedby adenovi-rus and adeno-associated virus promoters. J. Virol. 64:2103-2109.
4. Conley, A. J., D. M. Knipe, P. C. Jones, and B. Roizman. 1981. Molecular genetics ofherpes simplexvirus.VII. Characteriza-tion of atemperature-sensitive mutant produced by in vitro mutagenesis anddefective in DNAsynthesis andaccumulation of -ypolypeptides. J. Virol. 37:191-206.
5. de Bruyn Kops, A., and D. M. Knipe. 1988.Formation ofDNA replication structures in herpes virus-induced cells requires a viral DNA binding protein. Cell 55:857-868.
6. DeLuca,N. A., A. McCarthy, and P. A. Schaffer.1985.Isolation andcharacterizationof deletion mutantsof HSV-1 inthegene encoding theimmediate-early regulatory proteinICP4. J. Virol. 56:558-570.
7. Friedman, A. D., S. J. Triezenberg, and S. L. McKnight. 1988. Expression of atruncated viral trans-activator selectively im-pedes lytic infection by its cognate virus. Nature (London) 335:452-454.
8. Frink, R.J.,R.Eisenberg, G. Cohen,and E. K.Wagner.1983. Detailed analysis oftheportion of the herpessimplex virustype 1genomeencodingglycoproteinC. J. Virol. 45:634-647. 9. Gao, M., J. Bouchey, K.Curtin, and D. M. Knipe. 1988.Genetic
identification of a portion of the herpes simplex virus ICP8 proteinrequired forDNA-binding. Virology 163:319-329. 9a.Gao, M., and D. Knipe. Unpublisheddata.
9b.Gao,M., and D. Knipe.Submitted forpublication.
10. Gao, M., and D. M. Knipe. 1989.Genetic evidence formultiple nuclearfunctions ofthe herpessimplex virus virus ICP8 DNA-binding protein. J. Virol.63:5258-5267.
11. Gauss, P., K. B. Krassa, D. S. McPheeters, M. A. Nelson, and L. Gold. 1987. Zinc(II) and the single-stranded DNA binding protein of bacteriophage T4. Proc. Natl. Acad. Sci. USA 84:8515-8519.
12. Godowski, P. J., and D. M. Knipe. 1986.Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc. Natl. Acad. Sci. USA 83:256-260.
13. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C. Glorioso. 1981. Cloning of herpes simplex virus type 1
se-quences representingthe wholegenome. J. Virol.38:50-58. 14. Graham, F. L., and A. J. Van der Eb. 1973. A newtechnique for
the assayof infectivityofhumanadenovirus5 DNA.Virology 52:456-467.
15. Haynes, L. L., and L. B. Rothman-Denes. 1985. N4virionRNA polymerase sites oftranscription initiation. Cell41:597-605. 16. Herendeen, D. R., G. A. Kassavetis, J. Barry, B. M. Alberts,and
E. P.Geiduschek. 1989. Enhancement ofbacteriophageT4late transcription bycomponents ofthe T4 DNA replication appa-ratus.Science 245:952-958.
17. Herendeen, D. R., K. P. Williams, G. A. Kassavetis, and E. P. Geiduschek. 1990. An RNApolymerase-binding protein that is required for communication betweenan enhancer and a pro-moter.Science248:573-578.
18. Herskowitz, I. 1987. Functional inactivationof genes by domi-nantnegative mutations. Nature(London)329:219-222. 19. Holland, L. E., K. P. Anderson, C. Shipman, and E. K. Wagner.
1980. Viral DNA synthesis isrequired for efficientexpression of specific herpes simplex virus type 1 mRNA. Virology 101:10-24.
20. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C. Glorioso, and M. Levine. 1984. Transcriptional and genetic analysesof the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J. Virol. 49:947-959.
21. Homa, F. L., J. C. Glorioso, and M. Levine. 1988. Aspecific 15-bp TATA boxpromoterelement isrequired for expression of
aherpes simplex virus type 1 late gene. Genes Dev.2:40-53. 22. Hope, I. A., and K. Struhl. 1986. Functional dissection of a
eukaryotic transcriptional activator protein, GCN4 of yeast.
Cell46:885-894.
23. Johnson, P. A., C.MacLean, H. S. Marsden, R. G.Dalziel,and R.D.Evertt.1986.The product of geneUS11ofherpes simplex type 1 is expressed as a true late gene. J. Gen. Virol. 67:871-873.
24. Kalderon, D., B. L. Roberts, W. D.Richardson,and A. E. Smith. 1984. A short amino acid sequence able to specify nuclear location. Cell 39:499-509.
25. Knipe, D. M., M. P. Quinlan, and A. E. Spang. 1982. Charac-terization of two conformational forms of the major DNA-binding protein encoded by herpes simplex virus 1. J. Virol. 44:736-741.
26. Knipe, D. M., and A. E. Spang.1982. Definition ofaseries of stages in theassociation oftwoherpesvirus proteins with the cellnucleus. J. Virol. 43:314-324.
27. Lee, C. K., and D. M. Knipe. 1983. Thermolabile in vivo DNA-binding activity associated with a protein encoded by mutantsof herpessimplexvirus type 1.J.Virol. 46:909-919. 28. Lee, C. K., and D. M. Knipe. 1985. Animmunoassayfor the
study of DNA-binding activities ofherpessimplex virus protein ICP8. J. Virol. 54:731-738.
29. Leinbach, S. S., and J. F. Casto. 1983. Identification and characterization ofdeoxyribonucleoprotein complexcontaining themajorDNA-binding protein of herpes simplexvirus type 1. Virology 131:274-286.
30. Leinbach, S. S., and L. S. Heath. 1988. A carboxyl-terminal peptide of the DNA-binding protein ICP8 ofherpes simplex virus contains a single-stranded DNA-binding site. Virology 166:10-16.
31. Littler, E., D. Purifoy, A. Minson, and K. L. Powell. 1983. Herpes simplex virus nonstructural proteins. III. Function of themajorDNA-binding protein.J. Gen. Virol. 64:983-995. 32. McKnight, J. L. C., T. M. Kristie, S. Silver, P. E. Pellett, P.
Mavromara-Nazos, G.Campadelli-Fiume,M.Arsenakis, and B. Roizman. 1986. Regulation of herpes simplex virus 1 gene expression: the effectofgenomic environmentsand its implica-tionsfor model systems. Cancer Cells 4:163-173.
33. Orberg,P. K., and P.A. Schaffer. 1987.Expressionofherpes simplex virus type 1 major DNA-binding protein, ICP8, in transformed cell lines:complementation of deletionmutantsand inhibition ofwild-type virus.J.Virol. 61:1136-1146.
34. Powell, K. L., and D. J. M.Purifoy. 1976.DNA-bindingproteins of cells infected by herpes simplex virus type 1 and type 2. Intervirology 7:225-239.
35. Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location ofa herpes simplex virus DNA-binding protein is determined by the status of viral DNAreplication. Cell36:857-868.
36. Quinlan,M.P.,and D. M.Knipe. 1983.Nuclear localizationof herpesvirus protein: potential role for the cellular framework. Mol. Cell. Biol. 3:315-324.
37. Rice, S.A., and K. M. Knipe. 1990. Genetic evidence fortwo
distincttransactivation functions of theherpes simplexvirus a
protein ICP27.J. Virol. 64:1704-1715.
38. Rose, D. S. C., K. Shriver, D. S. Latchman, and N. B. LaThangue. 1986. A filamentous distribution for the herpes simplex virus type 2-encoded majorDNA binding protein. J. Gen.Virol. 67:1315-1325.
39. Ruyechan, W. T., A. Chytil, and C. M. Fisher. 1986. In vitro characterization ofathermolabile herpes simplex virus DNA-bindingprotein.J. Virol. 59:31-36.
40. Ruyechan,W.T.,and A.C. Weir. 1984.Interaction with nucleic acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J. Virol. 52:727-733.
41. Shepard, A. A., P. Tolentino, and N. A. DeLuca. 1990.
trans-dominant inhibition of herpes simplex virus transcriptional regulatory protein ICP4 by heterodimer formation. J. Virol. 64:3916-3926.
42. Southern,P.J., and P.Berg. 1982. Transformation of
mamma-lian cells to antibiotic resistance with a bacterial gene under controloftheSV40earlyregion promoter. J. Mol. Appl. Genet. 1:327-341.
on November 10, 2019 by guest
http://jvi.asm.org/
POTENTIAL ROLE FOR HSV ICP8 IN LATE GENE EXPRESSION
43. Wang, Y., and J. Hall. 1990. Characterization of a major
DNA-bindingdomain in the herpes simplex virustype 1 DNA-binding protein(ICP8). J. Virol. 64:2082-2089.
44. Weller, S. K., K. J. Lee, D. J. Sabourin, andP. A. Schaffer.
1983. Genetic analysis of temperature-sensitive mutantswhich define the gene for the major herpes simplex virus type 1 DNA-bindingprotein. J. Virol. 45:354-366.
45. Wu, C. A., N. J. Nelson, D.J. McGeoch, and M. D. Challberg. 1988. Identification of herpes simplex virus type 1 genes re-quired for origin-dependent DNA synthesis. J. Virol.
62:435-443.
46. Yager, D. R.,A.I.Marcy, andD. M.Coen. 1990. Translational
regulation of herpes simplex virus DNA polymerase. J. Virol.
64:2217-2225.
VOL. 65, 1991 2675