Vol. 65, No. 1 JOURNALOFVIROLOGY, Jan. 1991, p.225-231
0022-538X/91/010225-07$02.00/0
CopyrightC) 1991, AmericanSociety forMicrobiology
Evolution of Human Immunodeficiency Virus
Type
1nef and
Long
Terminal
Repeat Sequences
over
4 Years In
Vivo and
In
Vitro
SYLVIE DELASSUS, REMI CHEYNIER, AND SIMON WAIN-HOBSON*
Laboratoire deRetrovirologie Moleculaire, Institut Pasteur,
28 Rue de Dr.
Roux,
75724 Paris Cedex15,
FranceReceived 2 August 1990/Accepted 19 October 1990
The evolutionofan851-bpsegmentof the human immunodeficiency virustype 1(HIV-1)genomeencoding
thenefopen reading frame and U3/R elements of the long terminal repeat has been followedover a 4-year
period in vivo andinvitro. The population of viralsequencesatanygiven timewasestablished by sequencing
clonedpolymerase chain reaction products. Thesamples studied werederivedfromthesamemanfor whom
adetailed analysis of thetatgene waspreviouslydescribed (A. Meyerhans, R. Cheynier, J. Albert, M.Seth,
S. Kwok, J.Sninsky, L. Morfeldt-Manson, B. Asjo, and S.Wain-Hobson, Cell 58:901-910, 1989).Once again
invitro culture resulted in the selection ofminorforms. Overa4-year period in vivo, there was noobvious
selection for,oroutgrowthof,anyparticularneforU3/Rsequence. Fewdefective nefproteinsequenceswere
observed, whicharguesagainst nef actingas a negative regulatoryfactor. Althoughnofunctionally defective
promoter/trans-activation-responsive elementswereidentified, the transactivation efficiencies varied between
0.2 and 2 times that of the control. Thesequenceencodingthemostefficient trans-activation-responsiveregion
didnotoutgrowothers. Theextremegeneticheterogeneity of thedifferentsamplesofthe locus,either in vivo
orinvitro, indicates that there isnosuchthingas asingle, distinct HIVsequence.It is suggested thatdifferent
HIV-1loci evolveindependently, recombination being responsible for their uncoupling.
Theplasticity ofthe humanimmunodeficiency virus(HIV)
genome hasbeenamplydescribed. Itisdueto amultitude of
phenomenaencompassing viral polymerase miscopying, du-plication, deletion, recombination,andhypermutation of the viral genome. These events, while rendering the task of
molecular biologists particularly arduous, are probably an
advantageto thevirusinits continual efforttoadapttolocal
environmentsorrespond toselectionpressures. Inorderto
describe such complexity inherent in all RNA viruses, the
conceptofaquasispecieshas beendeveloped(11,12, 34). In
brief, a quasispecies may be defined as a population of distinct but relatedviral genomes. The 10-kb sizeofthe HIV
type 1(HIV-1) provirus effectively prohibitsaccurate
anal-ysis of populations of complete sequences. A sequence
analysis of cloned DNAfragments, derived by polymerase chain
amplification
of the HIV-1 provirus, is perhaps ascloseascan be got to a
description
ofaHIV-1 quasispeciesatthe nucleotide sequence level (28).
In a
previous
longitudinalstudy
of the HIV-1 tat genesequencesin vivoandin
vitro,
itwasshown that the onedidnotreflect the other(28). There was no selection for
more-efficient tat genes with time despite the suggestion that
viruses isolated from asymptomatic seropositives, which
grow poorly, could be complemented by a cell line
perma-nently expressing tat(2). These isolates are referred to as
slow/low isolates (3). Bycontrast,isolates from patientswith
AIDS
replicate
well, thegrowth ofthe isolates beingunaf-fectedbypassageto atat-producing cellline. These viruses
are called
rapid/high
isolates (3). These observationssug-gested that perhaps there was selection, coincident with
disease,
of isolates with increased trans-activationeffi-ciencies. Asit happened no selection atthe level of the tat gene wasdescribed (28).
* Correspondingauthor.
225
Theviral longterminal repeat (LTR) carries the sequences
essential for transcription, reverse transcription, and
inte-gration. A particular feature is the
trans-activation-respon-sive (TAR) sequence in the Rregion, which is a target for
tat-dependent trans-activation of provirus expression (29,
31). The tat, as well as host proteins, binds TAR RNA,
resulting in efficient transcription and a positive feedback
loop (10, 38). Thus, it was possible that the differences
betweenviruses inearlyand latestagediseasemight bedue
to subtle differences in the TARregion.
HIV-1is endowed with at leastninegenes. The ninth and
most 3' gene, called nef, is a uniquefeatureofthe primate
lentiviruses,asopposedtothe otheranimal lentiviruses (30).
It wasoriginally thought to be anegative regulatory factor
(14, 18, 27, 36), perhaps interacting with the negative
regu-latory elements in the U3 region ofthe LTR (1), hence the
mnemonic nef(16). The role of nef
is,
however, now noclearer than when it wasfirst identified (19, 25). While it is
notessential toviral growth, its conservation inall primate lentiviruses arguesforan important role.
Sincethe U3 elementofthe LTRoverlapswith the 3' half
of the HIV-1
nef
openreading frame,
it was decided toamplify the entire nef open reading frame and LTR
se-quences. Apart from
addressing
the problem of possiblefunctional differences within the LTR and TARregions, it
wouldalsopermitthe mostextensive(approximately 10%of theprovirus)high-resolution studyof the evolution ofHIV-1
quasispecies coincident with disease
progression.
The dataprovided here show that, as in the tatgene
analysis,
therewas noselection of more efficient TAR elements.Inaddition
no completely defective LTR sequences were identified.
Thus, differences between the so-called slow/lowand
high/
fast viruses mustbe encoded elsewhere within the genome.
Little evolution of
nef
orLTRsequenceswasnotedfrom theasymptomaticto thedisease stage.
on November 10, 2019 by guest
http://jvi.asm.org/
226 DELASSUS ET AL.
I
env | IF
revtat
|nef I
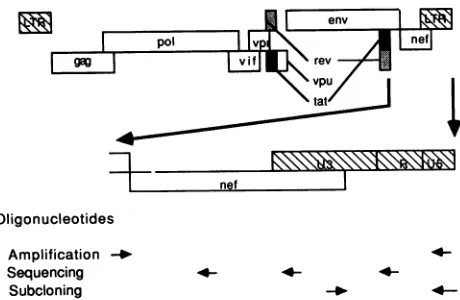
Oligonucleotides Amplification -_ Sequencing Subcloning
FIG. 1. HIV-1nef-U31R locus amplified.Theregionof the HIV-1
genomestudied is shownexpanded.The locations and orientations
of the oligonucleotides used for amplification, screening and
se-quencing of M13 recombinants, and subcloning of the promoter/ TARregionsaremarkedAmplification, Sequencing, and Subclon-ing,respectively.
MATERIALS AND METHODS
Bloodand culturesamples. Freshperipheral blood
mono-nuclearcells fromaHIV-1-infectedman wereanalyzedover
a4-year period. Samples Li, L2, L3, and L6were takenin
June 1985, March 1986, June 1986, and June 1989,
respec-tively. Viral isolates Vi, V2, V3, and V6 were derived by
coculture using seronegative donor
phytohemagglutinin-stimulated blasts. SamplesLi,L2, L3, Vi, V2, and V3 have
already been described (28). In June 1989, HIV-1 was
isolated from a sixth sample (L6) by cocultivation with
HIV-1 seronegative donor peripheral blood mononuclear
cells. Unlike all other isolates from this patient, the virus
derivedfrom L6, i.e., V6, wastypically arapid/high isolate (3) and induced large syncytia. It alsogrew well on estab-lishedcell lines.The DNA usedtocharacterize V6wasfrom first-passage virus.
PCR. The nef-U3/R amplification primers used for poly-merase chain reaction (PCR) were located in highly
con-served regions flanking these sequences. The DNA plus
strand primer NL1 (5'-CCAGCATGCAGTAGCTGAGGG
GACAGATAG) and DNA minus strand primer NL2
(5'-CCAGTCGACCAGAGTCACACAACAGACGGG) mapped
topositions 8278to8300 and 131to109,respectively,onthe
HIV-1Brusequence (37)(Fig. 1). The primers carried SphI
and SalI restriction sites (underlined), respectively. DNA
amplification conditions have been previously described
(28). Approximately 1 ,ug of total DNAwas used.
Denatur-ation, annealing, and elongation temperatures and times
were 94°C and 30 s, 55°C and 30 s, and 72°C and 1 min,
respectively.
Cloning and sequencing. PCR material was purified on a
low-melting-point agarose gel, phosphorylated, and
blunt-end ligated into dephosphorylated, SmaI-cleaved, M13mp8
replicative-form DNA (Amersham). Aftertransformation of
Escherichia coli TG1, plaqueswerescreenedin situ by using
amixture of three oligonucleotides, Si, S2, and S3, all of which were located in highly conserved regions within the
nef-U31R sequence. The sequences were Si, 5'-AGTC
CCCAGCGGAAAGT; S2, 5'-GGAAGTAGCCTTGCGCG;
andS3,5'-GCTGCTGTATTGCTACT, which mappedtothe
minusstrandat9047to9031,8753to8737,and8538to8522,
respectively (Fig. 1). Twenty M13 recombinants fromeach
sample were sequenced by the dideoxy chain termination
method (33) using M13 universal primer and the three
oligonucleotides, Si, S2, and S3.
Polyacrylamide gel blotting. After separation on a 3.5%
acrylamide-TBE (89mMTris-borate, 2 mM EDTA) gel,the
PCR-amplified products weredenaturedinsituby using0.2 MNaOH-0.6 MNaCl for30 minfollowedby30 min in 7%
formaldehyde. The DNAwasthen transferredtoa
nitrocel-lulose filterbyastandard method. The filterwas hybridized
withanequimolar proportion of5' 32P-labeled oligonucleo-tides Si, S2, and S3.
Construction ofexpressionvectorandsubcloning. Thelarge
HindIII-SspI fragment of the pBC12/PL/SEAP vector (5),
which encodes the entire human secreted alkaline
phos-phatase (SEAP) cDNA, was cloned into the HindIII and
SspI sites ofpUC18. This vector,
pUC/SEAP,
contains allthe pUC18 polylinker sites 5' tothe SEAP gene, and was
constructed so as to delete the simian virus 40 early
en-hancerfromthe original plasmid. The enhancer, promoter,
and TAR sequencesofHIV-1were amplifiedfrom 10to50
ngofrecombinantM13 DNAunderstandardconditions.The
amplificationprimerswere NL2andNL3
(5'-GAGAGGTC
GACCGGAGTATTACAAAGACTGCTGA, positions 8987 to9010)(Fig. 1). TheSall restrictionsite used insubcloningofthe HIV-1
promoter/TAR
fragments is underlined. After10 cycles, amplified DNA from 22 M13 recombinants and
from pBRU-2(kindlyprovided by Keith Peden)wascleaved
by HindIIl and Sall and ligated into pUC/SEAP
through
the same sites (HindIll cleaved within the Rregion, 3' to
the TAR region). The sequences ofthe resulting series of
23 constructions, named pTAR/SEAPO to pTAR/SEAP22,
were all checked by double-stranded plasmid sequencing
(20) using the M13 reverse sequencing primer. No
differ-ences between the subclone and the original recombinant
M13 sequenceswere identified.
Transfection and SEAP assays. SW480 cells(human colon
carcinoma cells) were cotransfected by
pTAR/SEAP
andpSV2tat Bru (28) by the calcium phosphate method (8),
whereas Jurkat-tat cells (31) were transfected by pTAR/
SEAPplasmids byusingthe DEAE-dextranprocedure(15). Sixty-hour posttransfection culture supernatants were
clearedbycentrifugation (15,000 x g)andheated for10min
at65°Cto inactivate any endogenous alkalinephosphatase.
SEAPactivities weredeterminedon50 ,ulof supernatantas
previously described (5). The valuesgivenarethemeansof
at least two independent transfections ofSW480 cells and
wereconfirmedby transfection oftheJurkat-tat cell line(see
Fig. 3).
Nucleotide sequence accessionnumbers. TheGenBank
ac-cession numbers for the HIV-1 nef and LTR sequences
presented hereare M58193 to M58283.
RESULTS
What is a HIV-1 sequence? Twenty recombinant M13
clones were sequenced for each sample (i.e.,
Li,
L3, L6,V2, V3, andV6). None of the 60 sequences from
Li,
L3, andL6 were identical over the 851-bp locus analyzed. The
maximum nucleic acid sequencedivergence noted between
anypairwas3.1%.Of the three in vitroquasispecies, V3was
the most homogeneous and V6 was the least. A major
species representing 35%of all sequences could beidentified
within V3. Nonetheless,evenwithin V3 upto 2.4% nucleic
acid sequence divergence was noted between any pair. As
was observed previously (28) the invivo quasispecies
(Li,
L3, andL6)were morecomplex than the in vitro
quasispe-cies (V2, V3, andV6).
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.66.296.70.220.2]EVOLUTION OF HIV-1 nefAND LTR SEQUENCES 227
PCR analysis of the nef-U3IR region from a molecular clone (pNL4.3; see reference 30 for sequence) indicated that there were no hot spots for the enzyme within this region. A study of 20 clones indicated that 1 clone in 4 carried a single base substitution per 851-bp sequence due to Taq polymer-ase errors (data not shown). This value was concordant with those derived from analyses of 300-bp segments of the HIV-1
tat and env genes (17, 28). Since the minimum number of substitutions within any quasispecies, using any sequence as a reference, was always greater than 35, the ratio of natural
to artifactual substitutions would be 7:1 [i.e., 35:(20/4)] or
more.
A comparison of thenef-U31R quasispecies in vivo and in
vitro (i.e., comparisons of L3 with V3 and L6 with V6)
showed that there were no common nucleotide sequences, indicating once again that the in vitro culture of HIV-1, whether from early (L3 and V3) or late (L6 and V6) stagesin
disease, leads to the selection of low-abundance forms. These in vitro forms are presumably in the original periph-eral blood mononuclear cell quasispecies. However, the resolution of the analysis (1 clone of 20) must have prohib-ited their identification.
What is a nef sequence? The nef protein sequences are shown in Fig. 2 for each of the three lymphocyte samples, Li, L3, and L6. Only two sequences, L1.14 and L1.16, were present at 10% of their original quasispecies. All other sequences were present at 5%, the resolution of the study.
Of the 60 sequences only 4 were common tothe Li and L3
quasispecies. All the L6 sequences were unique. None of the
V3 or V6 nef protein sequences could be found in the
parental L3 or L6 samples, respectively. Thus,nosingle nef
protein sequence could possiblyhavereflected the
complex-ity of nef sequences present in any sample.
The mutations were not randomly distributed but were
clustered in the amino and carboxy termini. Apart from a
few substitutions, the regionbetweenaminoacids 40and 130
was highly conserved. Overall the internal sequence
varia-tion among nef proteins was between 1 and 4.5%. While
there does not appear to be any obvious selection for a
particular nef sequence, some progression may be noted at
specific positions. Thus, threonine15 ispresentin 30% of all
Li sequences, in 45% of L3 sequences, and in 95% of L6
sequences.
Sequences L3.14 and L6.01 appeared particularly
diver-gent at their amino acid termini. These substitutions maybe
explained by G--A hypermutation of RNA genome during
DNA synthesis by the HIV-1 reverse transcriptase (36a).
There were fewcases ofobviously defectivenef sequences.
L6.01 encoded a mutated initiator methionine codon
(ATG-*ATA) aswell as an in-phase stop codon atposition
57. Sequences L1.13 and L1.21 also encoded in-phase stop
codons. Finally, clones L1.09 and L3.21 carrieddeletionsof
154 and 68 residues within their nef sequences. These
deletions, 462 and 204 nucleotides, respectively, were
mul-tiples of 3 andwere considered authentic for the following
reasons. (i) Polyacrylamide gel blot analysis of the original
PCR-amplified materialbyusing oligonucleotide probes
cor-responding to sequences within the deletions showed that
there were unclonedPCR products lackingthese sequences
(data not shown). (ii) No sequence homology around the
deletions that argued against a PCRartifactcould be found.
(iii) In the positivecontrol (amplificationof the same region
from a molecular clone) no similarly deleted products could
be identified. In all the loci that have been studied in the
laboratory by PCR no deletions have ever been found.
Occasionally a deletion was found at the 5' or 3' end of a
sequence. Invariably the neighboring amplification
primer
was alsodeleted, whichargues morefora
cloning
artifact.Functionalanalysis of the HIV promoterregion.The
essen-tialtranscriptional control sequences of the HIV-1 LTR map
to the noncoding region between bases 636 and 801 in the
locus amplified. If just this region was
analyzed
at thenucleicacid sequence level a verydifferent
picture
wouldbeobtained. Thus, the Li and L3 quasispecies were
complex
while that for L6 was
relatively
simple, amajor formbeing
present at 65%. Again there were substantial differences
among the Li, L3, and L6 subsets. While a number of
sequences were common to Li and L3, none of the L6
sequences could be found in either Li or L3
(data
notshown).
Whenalltheenhancer/promotersequences from the L and Vquasispecies weretaken together, mostofthe mutations
mappedtotheTARregion, notably in the baseof the stem.
All but twoofthemutationswerepoint mutations.As
usual,
transitionsgreatly outnumberedtransversions. The few
mu-tations in the upstreamregion
invariably
mapped
just
outsideofthe NFKB and Spl sites
(Fig.
3).Twenty-twoclones wereanalyzed atthe functional level
afterthe TARregion wasPCR
amplified
and subclonedinto the pUC/SEAP expression vector. The mutants and theirrelative trans-activation efficiencies with respect to HIV-1
Bru tat and LTR are givenin
Fig.
3.Therelativedegrees
oftransactivation variedfrom0.2to2 times thatof theHIV-1
Bru tat and LTR control.These differenceswere notdueto
experimental errors. Those
pTAR/SEAP
plasmids
whichyielded a reduced relative transactivation
efficiency
weretested at least three times
by
using
two differentplasmid
preparations. In these
experiments
thebasallevelof SEAP activity for eachplasmid
was the same. These levelswerecomparable with thatof theHIV-1 BruLTRreference. The
relative transactivation efficiencies varied
by
+0.05. All theclones
representing
morethan5% ofaquasispecies
showedrelative transactivation ratios
comparable
with that ofthereference (i.e., between 0.7 and
1.3).
Clones 7 and 12 encodeda G-*A substitution at
position
20 at the baseof the
bulge
which reducedthetransactivationefficiency byafactorof5.
However,
deletion ofasingle
basein the stem atposition 42
(clone
17)
hardly
affectedtransac-tivation. TwoC->T
substitutions,
oneinthebulge
andonein the loop, didnotinfluence transactivation. A smallerregion
at thebase ofthestemmay be
important,
for substitution atposition
7 or53 either decreasedor increased theefficiency
oftransactivation.
Interestingly,
clone13,
whichproved
tobe the most
efficiently
transactivated sequence and whichwas derived fromthe L3
quasispecies,
was notidentified inthe L6quasispecies whatsoever.
DISCUSSION
After
having
assimilated all the sequence data it is verydifficultto
speak
ofasingle,
distinct HIV-1 sequence eitheratthe nucleic oraminoacid level
(11, 12,
34).
The 851 basessequenced,
approximately
9% of the genome, were notamong the most variable
regions
of the HIV-1 genome.Extrapolating
from thisdata,
it ispossible
toestimate,
atleast in this individual case, that any two
complete
viralgenomes in vivomay differ
by
upwards
of 10to20 bases. Itmay not be
assumed, however,
that the HIV-1 reversetranscriptase
misincorporation
rate is of the sameorder,
because the
precise
relationship
between sequences and thenumberofcycles
separating
them is unknown.Given the number of
unique
sequences involved it is notVOL.65, 1991
on November 10, 2019 by guest
http://jvi.asm.org/
*.
. hihi
.~ ~~~ *i. hi hhhhhh
-2 CY Cy C 2 yCY2 a vC
]a . pp P PPP
fihil
01 j
I I 9H .
CyCYC yCpCYvor Icoay av o CY CY CY CyCyCYCya CY CYCy
2ko 4 A 2.-.1 A 2 222 22
I * a .
S
XvvvvvIvvvv.vsvvvvvvvvv* vv vvvv
o
CY
.,
OgXXp-XX J z .> - X J Jz JX X W Xx - . W - JKJ J X JJJ XJ W J
VAKK Ki FX KFX -X
S I . H~~~~~~~~~~~~~~~~b. H H H - w H
E~~~~~~~~~~~1
II
o;~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~t
I
1
RV . A ..x
:2 . . zz
3
.
"
.~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~C
^l * g .YS~~~~~~~~~~~~~~~~~~~~~~~~~u" "S
0~~~~~~~~~~~~~~~~~~~,DHD* - *Z-,D Z .0Z Dz ZoZZDZ
228
I
on November 10, 2019 by guest
http://jvi.asm.org/
EVOLUTION OF HIV-1 nef AND LTR SEQUENCES
A
CD CD E- C E U1 E40
LI (4CD 4 C,4Ut
14 )
I v
I V 04
V U)
o +w0r
4
A
I
4CD
41
LI
A A
E-4 E o o
4414
c
:
A
E4C.))14 U E4
* I
.
E4
E4 E-4
E-4
E-4
E-4
_: U UUc
E-E-4
n*1
E4
LI)
U
E4
in
U)
ol
P
In4
zw
w
1-4
1-4 1-4
8)
-p1 E
hO
V
1-4
C)
E-4
E
E14
E4
E4
C,
EqUq
4z VOL. 65, 1991
229
7
oc~ ~ ~ C)
(.
+ U
0
144
4.1 <
E- I I I
'C
U
Un< o~~~~~~
1~~~~~~~11
-C __
o
> o
C'C
u
:3 8 0 tr
441 1&
w -4*- ur-w ff n o o o o Ln un un
-4(n(
- - -4 u4 S7 MuM e(M0 n%Nn %a %
o,4 _>_
H~~~~~~~~~~~-4 -4 -4H -4 -4 -4 - N C1 Cl
I
(N -W r- 0%
lw lw lw lw
sr
1-+4 +
on November 10, 2019 by guest
http://jvi.asm.org/
230 DELASSUS ET AL.
surprisingthat any twoquasispecies aredistinct. Howeverit
is not possible to conclude that the differences are
signifi-cant. The sampling of 20 genomes, all present at low
frequencies,from a largepool wouldinvariably give different populations with perhaps different structures. These data
caution against using a single molecular clone to represent
HIV-1. Only a few sequential changes at particular sites,
such asThr-15--lAla-15, may possibly have any meaning. If
the four such sites are considered, then there was little
evolution of the nef protein sequence over the 4 years
coveredin this study.
Onceagain thein vitro data did not reflect the in vivo data.
Comparing either the nef or the LTR sequences, the
rela-tivelyabundantforms recovered in vitro represented minor
formsin vivo. In the case of the L3 and V3 and L6 and V6
pairs there were no common nucleic acid sequences (data notshown). The minimum number of differences was 1 to 2
nucleotides. This extends our previous study based on a
314-basesegment encoding the first exon of the tat gene (28).
Perhapsbecause the segment was smaller it was possible to
findtheminorform in the in vivo quasispecies. The new data
from thenef-U3/R region indicated that in vitro coculture of
HIV-1resultedin the isolation of very minor forms. V6 was
isolatedfrom the patient during late-stage disease
(CD4+
=10/mm3) and was typically a rapid/high syncytium-inducing
isolate. Despitethe fact that patients with AIDS have little or no cell-mediated immunity (21), the virus isolated still did notreflectthe major species in vivo. Once again these data
caution against the extrapolation of in vitro data to a
descriptionof HIV-1-associated pathogenesis.
Thedistortionof the population of genomes in culture may
be due to statistical factors, methods, or the nature of the
lymphocytes.
Recentlyit has been shown that, in the contextof viruscoculture, seronegative donor cells secrete a soluble
factor capable of inhibiting HIV-1 replication (7). Another
nonnegligible factor may be the presence of substantial
proportions of HNK-1 suppressor cells, particularly in
pa-tientswithAIDS(23). Clearly a detailed study of the factors
influencingtheisolationofHIV-1, perhaps by using a genetic
fingerprinting analysisas in this study, is urgently called for. Among all the 120 nef protein sequences within this study
only a few were obviously defective. Thus, sequences
L1.15, L1.21, and L6.01 encoded in-phase stop codons;
L6.01 encodeda mutatedstart codon; and L1.09 and L3.21
carried large deletions. The L1.09 and L3.21 proviruses
should be completely defective for replication, since the
polypurine tract and 5' inverted repeat were also deleted.
Though nef was, at one time, described as a negative
regulatory element, no obviously defective nef protein
se-quences
predominated.
These protein sequences in vitro orin vivo did not present any amino acid substitutions that
have not hitherto been observed (30). However, the
distri-bution ofamino acid substitutionsamong the Li,L3,and L6
(Fig. 2) or V2, V3, and V6 sets of sequences (data not
shown)appeared nonrandomand was essentially confined to
the amino- and carboxy-terminal regions. This would, if
anything, suggest thata
nef
gene product is selected both invitroand in vivo andthatthe central region between residues
40 and 130 encodes the most important functional domain.
These data are not irreconcilable with the observation that
nefmutant virusesareviable(14, 27, 36). The contribution of
neftothe HIV life cyclemay simply be subtle and
uname-nable to analysisinshort-term experiments.
That none of the LTR sequences were defective for
transcription is understandable. A functionally defective LTRwouldbe incapable of producing virus and cannot be
complemented. Itisinteresting to note that,while therewas
only a 10-fold difference in relative efficiencies of
transacti-vation, the correspondingtatgene products varied
by
morethan 100-fold in the same assay. This may simply reflect the
fact that adefectivetat maybecomplemented intranswhile
an LTR-defective genome may not. Clone
pTAR/SEAP13
(representing 5%ofL3 sequences) was twice as efficient as
any of its homologs, as well as the Bru
tat-U31R
pair, atdirecting transcription. It is intriguing that this genome did
not outgrow its siblings. A number of possibilities present
themselves. However, a coupleof trivial explanationscould
be (i) that the lymphocyte(s) harboring the sequence as a
latentprovirus was neveractivated by antigenor(ii)that the
genome harbored a defect in some other gene, thus
prohib-iting viral replication. In the other clones, themutations that
greatly reduced the transactivation efficiency were
concor-dant with the deletions and substitutions that have already
been described for the
Spl
sites (24) and the TAR region(29). In the initial study no selection for more-efficient tat
gene products wasobserved. The same conclusion can now
be drawn from the analyses presented hereof the
promoter/
TAR region. Taken together, these data indicate that the
apparent emergence of rapid/high syncytium-inducing
iso-lates (3, 9, 35) with declining CD4 cellnumbersis not due to
modulation of the transactivating system. Thus, differences
between theso-calledslow/lowandhigh/fast virusesmustbe encoded elsewhere within the genome.
Acomparison ofthe sequences fromthe V6 quasispecies showed that for both nef and U3/R there were two major
forms (36 and18% for nef and 45 and36% for U3/R). Despite
these comparable frequencies the sequences encoding the
major nef sequence did not at the same time encode the major U3/R sequence. Even when single point mutations were eliminated, thus simplifying the analysis, the same
conclusion held. This suggests that the nef andU3/R
quasis-pecies were evolving independently of each other. Since
they are located in cis, the only explanation for their being
uncoupled would be efficient recombination between ge-nomes.
The efficiency of recombination has been estimated to be of the order of 20 to 50%per cycle (4, 6, 22, 26). In view of this it is indeed likely that HIV gene sequences may be uncoupled throughout the genome. Consequently the HIV genome would resemble that of a segmented virus despite being continuous. This would once again emphasize the plasticity of the HIV and allow individual genes to evolve independently.
In conclusion these data greatly extend the notion, already described for a few molecular clones (13, 32), that the phenotype of any given clone may differ from that of another. As a consequence the biological properties of a given isolate will be made up of the sum of the properties of a myriad of subtly different genomes. In addition the ex-treme heterogeneity of the HIV-1 quasispecies either in vivo or in vitro cautions against extrapolating from sets of data that are toolimited.
ACKNOWLEDGMENTS
We thank Bryan Cullen for the gift of the plasmid
pBC12/PL/
SEAP, and Birgitta
Asjo,
John Sninsky, and Andreas Meyerhans for providing viral isolates, chromosomal DNA, and valuable help in PCR.Sylvie Delassus and
Remi
Cheynier were supported byl'tcole
Polytechnique and la Fondation pour la RechercheMedicale,
re-spectively. This work was supported by grants from Institut Pasteur andl'AgenceNationale de Recherches sur le SIDA.J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
EVOLUTION OF HIV-1 nef AND LTR SEQUENCES 231 REFERENCES
1. Ahmad, N., and S. Venkatesan. 1988. Nef protein of HIV-1 is a transcriptional repressor of HIV-1 LTR. Science 241:1481-1485.
2. Asjo,B., J. Albert, F. Chiodi, and E. M.Fenyo.1988. Improved
tissue culture technique for production of poorly replicating human immunodeficiency virus strains. J. Virol. Methods 19: 191-196.
3. Asjo, B., L. Morfeldt-Manson, J. Albert, G. Biberfeld, A.
Karlsson, K. Lidman, and E. M. Fenyo. 1986. Replicative
capacity of human immunodeficiency virus from patients with varying severity of HIV infection. Lancet ii:660-662.
4. Beemon, K., P. Duesberg, and P. Vogt. 1974. Evidence for crossing-over between avian tumor viruses based on analysis of viral RNAs. Proc. Natl. Acad. Sci. USA 71:4254-4258.
5. Berger, J., J. Hauber, R. Hauber, R. Geiger, and B. R.Cullen. 1988. Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 66:1-10.
6. Blair, D. G. 1977. Genetic recombination between avian leuko-sis and sarcoma viruses. Experimental variables and the fre-quencies of recombination. Virology 77:534-544.
7. Brinchmann, J., G. Gaudernak, and F. Vartdal. 1990. Cell-mediated suppression of HIV-specific cytotoxic T lymphocytes. J. Immunol. 144:2961-2966.
8. Chen, C., and H. Okayama. 1987. High-efficiency transforma-tion of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745-2752.
9. Cheng-Mayer, C., D. Seto, M. Tateno, and J. A. Levy. 1988. Biologic features of HIV-1 that correlate with virulence in the host. Science 240:80-83.
10. Dingwall, C.,I.Ernberg, M. J. Gait, S. M. Green, S. Heaphy, J. Karn, A. D. Lowe, M. Singh, M. A. Skinner, and R. Valerio. 1989. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc. Natl. Acad. Sci. USA 86:6925-6929.
11. Domingo, E., J. J. Holland, and P.Ahiquist. 1988. RNA genet-ics. CRC Press, Inc., Boca Raton, Fla.
12. Eigen, M. 1973. Self organization of matter and the evolution of biological macromolecules. Naturwissenschaften 58:65-523. 13. Fisher, A. G., B. Ensoli, D. Looney, A. Rose, R. C.Gallo, M. S.
Saag, G. M. Shaw, B. H. Hahn, and F. Wong-Staal. 1988. Biologically diverse molecular variants within a single HIV-1 isolate. Nature (London) 334:444 447.
14. Fisher, A. G., L. Ratner, H.Mitsuya, L. M. Marselle, M. E. Harper, S. Broder, R. C. Gallo, and F. Wong-Staal. 1986. Infectious mutants ofHTLV-III with changes in the 3' region and markedly reduced cytopathic effects. Science 233:655-658. 15. Fujita, T., H. Shibuya, T. Ohashi, K. Yamanishi, and T. Taniguchi. 1986. Regulation of human interleukin-2 gene: func-tional DNA sequences in the 5' flanking region for the gene expression in activated T lymphocytes. Cell 46:401-407. 16. Gallo, R., F. Wong-Staal, L. Montagnier, W. A. Haseltine, and
M. Yoshida. 1988. HIV/HTLV gene nomenclature. Nature (London) 333:504.
17. Goodenow, M., T. Huet, W. Saurin, S. Kwok, J. Sninsky, and S. Wain-Hobson. 1989. HIV-1 isolates are rapidly evolving quasi-species: evidence for viral mixtures and preferred nucleotide substitutions. J. Acquired Immune Defic. Syndr. 2:344-352. 18. Guy, B., M. P. Kieny, Y. Riviere, C. LePeuch, K. Dott, M.
Girard, L. Montagnier, and J. P. Lecoq. 1987. HIV Ff3' ORF encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature (London) 330:266-269.
19. Hammes, S. R., E. P. Dixon, M. H. Malim, B. R. Cullen, and W. C. Greene. 1989. Nef protein of human immunodeficiency virus type 1: evidence against its role as a transcriptional inhibitor. Proc. Natl. Acad. Sci. USA 86:9549-9553.
20. Hattori, M., and Y. Sakaki. 1986. Dideoxysequencing method using denatured plasmid template. Anal.Biochem. 152:232-238. 21. Hoffenbach, A., P. Langlade-Demoyen, G. Dadaglio, E. Vilmer,
F.Michel,C. Mayaud, B. Autran, and F. Plata. 1989.Unusually high frequencies of HIV-specific cytotoxic T lymphocytes in humans. J. Immunol. 142:452-462.
22. Hu, W. S., and H. M. Temin. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudo-diploidy and high rate ofgenetic recombination. Proc. Natl. Acad. Sci. USA87:1556-1560.
23. Joly, P., J. M.GuilHon,C. Mayaud, F. Plata,I.Theodorou, M. Denis, P. Debre, and B. Autran. 1989.CD8+ T cells inhibit HIV replication in naturally infected CD4+ T cells. J. Immunol. 143:2193-2201.
24. Jones, K. A., J. T. Kadonaga, P. A. Luciw, and R.Tjian. 1986. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Spl. Science 232:755-759.
25. Kim, S., K. Ikeuchi, R. Byrn, J. Groopman, and D. Baltimore. 1989. Lackofnegative influence on viral growth by thenef gene ofhumanimmunodeficiencyvirus type 1. Proc. Natl. Acad. Sci. USA86:9544-9548.
26. Linial, M., and D. Blair. 1982. Genetics of retroviruses, p. 649-783. In R.Weiss, N. Teich, H. Varmus, and J. Coffin (ed.), RNA tumor viruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
27. Luciw, P. A., C. Cheng-Mayer, and J. A. Levy. 1987. Mutational analysisof the humanimmunodeficiency virus. Theorf-B region down-regulates virus replication. Proc. Natl. Acad. Sci. USA 84:1434-1438.
28. Meyerhans, A., R. Cheynier, J. Albert, M. Seth, S. Kwok, J. Sninsky, L. Morfeldt-Manson, B. Asjo, and S. Wain-Hobson. 1989.Temporalfluctuations in HIV quasispecies in vivoarenot reflected by sequential HIVisolations. Cell 58:901-910. 29. Muesing, M. A., D. H. Smith, and D. J. Capon.1987. Regulation
of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell 48:691-701.
30. Myers, G., A. B. Rabson, J. A.Berzofsky, T. F. Smith, and F. Wong-Staal. 1990. Humanretroviruses and AIDS. Los Alamos National Laboratory, Los Alamos, N.M.
31. Rosen, C. A., J. G. Sodroski, and W. A. Haseltine. 1985. Location of cis-actingregulatory sequencesinthe human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 41:813-823.
32. Sakai, K., S. Dewhurst, X. Ma, and D. J. Volsky. 1988. Differ-encesin cytopathogenicity andhost cell range among infectious molecular clones in human immunodeficiency virus type 1 simultaneously isolated from an individual. J. Virol. 62:4078-4085.
33. Sanger, F., S. Nicklen, and A. R.Coulson. 1977. DNA sequenc-ing with chain-terminatsequenc-ing inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
34. Steinhauer, D. A., and J. J. Holland. 1986. Rapid evolution of RNA viruses. Annu. Rev. Microbiol. 41:409-433.
35. Tersmette, M., R. E.Y. de Goede, B. J. M. Al, I. N. Winkel, R. A. Gruters, H. T.Cuypers, H. G. Huisman, and F. Miedema. 1988. Differentialsyncytium-inducingcapacity ofhuman immu-nodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency
syndrome(AIDS) andAIDS-related complex.J.Virol. 62:2026-2032.
36. Terwilliger, E., J. G. Sodroski, C. A. Rosen, and W. A. Hasel-tine. 1986. Effects ofmutationswithin the 3' orfopenreading frame region of human T-cell lymphotropic virus type III (HTLV-III/LAV) onreplication andcytopathogenicity.J. Virol. 60:754-760.
36a.Vartanian,J.-P., A. Meyerhans, B. Asjo, and S. Wain-Hobson. Submitted for publication.
37. Wain-Hobson, S., P.Sonigo, 0. Danos, S.Cole, andM. Alizon. 1985. Nucleotide sequence of the AIDS virus, LAV. Cell 40:9-17.
38. Zapp, M. L., and M. R. Green. 1989. Sequence-specific RNA binding by the HIV-1 rev protein. Nature (London) 342:714-716.
VOL.65,1991