FOR COLON-SPECIFIC DRUG DELIVERY
Thesis submitted to
The Tamilnadu Dr.M.G.R.Medical University, Chennai, for the award of the Degree of
DOCTOR OF PHILOSOPHY IN
PHARTMACY
Submitted by L.Raju, M.Pharm.,
Under the guidance of Dr.P.Vijayan
May-2010
J S S.College of Pharmacy, Rockland’s, Ootacamund,
Certificate
This is to certify that the thesis entitled “Development of multiple-unit oral formulations for colon-specific drug delivery”, submitted by Mr.L.Raju, to The
Tamilnadu Dr.M.G.R.Medical University, Chennai, for the award of the Degree of Doctor of Philosophy in Pharmacy is the record of the independent research work carried out by him at J.S.S.College of Pharmacy, Ootacamund, under my supervision, during the year 2007-2010. I also certify that the thesis or any part thereof has not formed the basis for award of any other research degree, of this or any other University, previously.
(Dr.P.Vijayan)
Certificate
This is to certify that the thesis entitled “DEVELOPMENT OF MULTIPLE-UNIT ORAL FORMULATIONS FOR COLON-SPECIFIC DRUG DELIVERY”submitted
by Mr.L.Raju, to The Tamilnadu Dr.M.G.R.Medical University, Chennai, for the award of the Degree of Doctor of Philosophy in Pharmacy is based on the research work carried out by him under the supervision of Dr.P.Vijayan, Professor, J.S.S.College of Pharmacy, Ootacamund. The thesis or any part thereof has not been previously submitted for the award of any other research degree of this or any other University. .
Certificate
This is to certify that the thesis entitled “DEVELOPMENT OF MULTIPLE-UNIT ORAL FORMULATIONS FOR COLON-SPECIFIC DRUG DELIVERY”submitted
by Mr.L.Raju, to The Tamilnadu Dr.M.G.R.Medical University, Chennai, for the award of the Degree of Doctor of Philosophy in Pharmacy is based on the research work carried out by him under the supervision of Dr.P.Vijayan, Professor, J.S.S.College of Pharmacy, Ootacamund. The thesis or any part thereof has not been previously submitted for the award of any other research degree of this or any other University.
(Dr.M.J.Nanjan) Director
Declaration
I hereby declare that the thesis entitled “DEVELOPMENT OF MULTIPLE-UNIT ORAL FORMULATIONS FOR COLON-SPECIFIC DRUG DELIVERY”, submitted by
me to The Tamilnadu Dr.M.G.R. Medical University, Chennai, for the award of Degree of Doctor of Philosophy in Pharmacy, is the result of my original and independent research work carried out at J.S.S.College of Pharmacy, Ootacamund, under the supervision of Dr.P.Vijayan, Professor, J.S.S.College of Pharmacy, Ootacamund, The Nilgiris. The thesis or any part thereof has not formed the basis for award of any degree, diploma, associateship, fellowship, or any other similar title, of this or any other university, previously.
Ootacamund
The Nilgiris L.Raju
My most sincere pranams to lotus feet of ‘‘Living God’’ Sri Shivakumara Swamiji, Sidaganga Mutt,Tumkur and His Holiness Jagadguru Sri Shivarathri Desikendra Mahaswamigalavaru, Suttur Mutt, Mysore for their divine blessings to my endeavour. With immense pleasure, I express my sincere gratitude and respect to my guide Dr.P.Vijayan, Professor and Head, Department of Pharmaceutical Biotechnology, for his encouragement, valuable guidance and generous support during the course of this study.
It is my privilege to thank Dr.B.Suresh, Vice Chancellor, J.S.S.University, Mysore, Dr.K.Elango, Principal, and Mr.S.Puttarajappa, Superintendent, J.S.S.College of Pharmacy, Ootacamund for accommodating me as a research scholar and for the facilities extended to carry out the research work.
I am very grateful to Dr.M.J.N.Chandrasekar and Dr.B.Duraiswamy for their help, enormous encouragement and suggestions to carry out this project.
I owe my sincere thanks to Prof.Dr.M.J.Nanjan, Director, Research and PG.Studies, J.S.S.College of Pharmacy, for his encouragement during the study.
I am very much grateful to Quality Improvement Programme, All India Council for Technical Education for the award of fellowship to me and special thanks to Prof.Chinnasamy, Professor (emeritus) J.S.S.College of Pharmacy Ootacamund for his Moral support.
My sincere thanks to Dr.Gowthamarajan and Mr.R.Sureshkumar, Prof.Damodharan, Dr.M.N.Sathish Kumar, Dr.Meyyanathan, Dr.Dhanapal, and Dr.Anand Vijayakumar for their help and advice.
My thanks to Mr.G.N.K.Ganesh, Mr. V.Senthil, Mrs.Sreevidhya, Mr.Vadivelan, Mr.Jawahar, Ms.Suja, Mr.Vekatesh, for their cooperation.
encouragement.
Finally I am indebted to my family-my bellowed parents, my uncle Mr.Ramalingam,Mrs Saroja Ramallingam,Mathesh,Vino and my wife Mrs.Chitra for their constant Love, support and encouragement.
1. INTRODUCTION
Pharmaceutical research is increasingly focusing on drug delivery systems which enhance desirable therapeutic objectives while minimizing side effects, quantity of drug, formulation cost, storage and transportation. Oral delivery is the most preferred route of drug administration, especially for chronic therapies where repeated administration of the drug is required. Medication has been given by oral route from many thousands of years. Oral administration offers greater convenience, higher likelihood of compliance and reduced risk of cross-infection and needle stick injuries (1). Hence formulations of oral drug delivery continue to dominate more than half of the drug delivery market share. Despite these advantages, the oral route is not suitable to administer certain active pharmaceutical ingredients (API), due to their high susceptibility to digestive enzymes in the gastrointestinal tract, poor absorption, and their limited ability to transport across the intestinal epithelial barrier.
Ongoing research in the area of oral drug delivery has led to improved and profound insights into the physiology, biology and physical chemistry (pharmacokinetics, partitioning phenomenon) of organs, cells, membranes, cellular organelles and functional proteins (e.g., transporters) associated with absorption processes of xenobiotics in the alimentary canal. This comprehensive research has led to improvements in the design of newer formulation targeted for site specific activity (2).
form, taken orally, will be able to make the long path to the large bowel and allow precised regional treatment or cellular targeting within the colon. As a result, new strategies of drug delivery are being developed to overcome the concomitant obstacles encountered by oral delivery systems.
Colon, an area where drugs are free from the attack of numerous proteases, is thought to be an ideal location to direct drugs into the bloodstream and the immune system. The challenge in the design of oral drug delivery vehicles, targeting colon is that the formulation, should remain intact while passing through the upper gastrointestinal tract. The API should be protected from physical, chemical and enzymatic degradation and be able to release upon reaching the colonic segment. A number of serious diseases of the colon, like colorectal cancer, Crohn’s disease can be treated more effectively if the drugs were targeted to the colon.
Colon-specific drug delivery system could also be used in treating conditions in which a diurnal rhythm as in the case like asthma, rheumatic disease, ulcer disease, cancer and ischemic heart disease (3).The incidence of asthmatic attacks is greatest during the early hours of the morning. Hence a dosage form which remains in the large intestine longer than in the small intestine as in the case with colon-specific formulations could be used to prolong the drug activity.
colon as a site for drug delivery include the presence of feces which might entrap the drug, presence of bacterial enzymes and low absorptive capacity. However, these factors have been reported to be easier to deal.
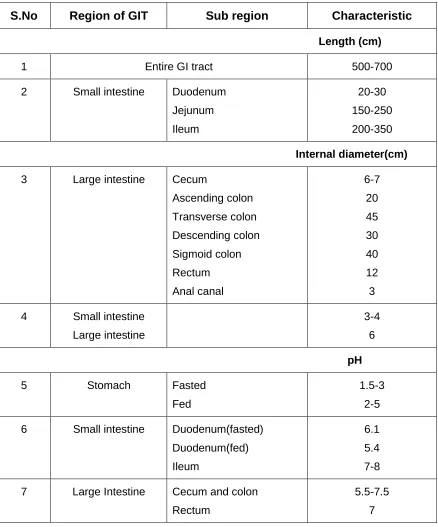
1.1. Anatomy, physiology and environment of GI tract
The large intestine extends from the distal end of the ileum to the anus. The human large intestine alone is about 1.5 m long shown in Table 1. Its caliber is higher near the cecum and gradually diminishes to the rectum, where it enlarges just above the anal canal (Figure 1). The colon is the upper 5 foot of the large intestine and mainly situated in the abdomen. The colon is a cylindrical tube lined by a moist, soft pink lining called mucosa. The pathway is called the lumen and is approximately 2 to 3 inches in diameter (6).The caecum forms the first part of the colon and leads to the right colon or the ascending colon (just under the liver) followed by the transverse colon, the descending colon, sigmoidal colon, rectum, and the anal canal(7).Unlike the small intestine, the colon does not have any villi. However, because of the presence of plicae semilunares, which are crescentic folds, the intestinal surface of the colon is increased to approximately 1300 cm and the physiology of the proximal and distal colon differs in several respects that can have an effect on drug absorption at the site. The physical properties of the luminal content of the colon also change, from liquid in the cecum to semisolid in the distal colon. The major functions of the colon are
Consolidation of the intestinal contents into feces by the absorption of the water and electrolytes and to store the feces until excretion.
Creation of suitable environment for the growth of colonic microorganisms such as Bacteroides, Eubacterium, and Entero bacteriaceae and
Absorption of water and Na+ from the lumen, concentrating the fecal content, and expulsion of content at a suitable time
1.2. pH of the colon
proximal small intestine and to a peak of pH 7.5 in the distal small intestine. The right, mid and left colon have pH values of approximately pH 6.4, pH 6.6, and pH 7.0 respectively.
S.No Region of GIT Sub region Characteristic
Length (cm)
1 Entire GI tract 500-700
2 Small intestine Duodenum
Jejunum Ileum 20-30 150-250 200-350 Internal diameter(cm)
3 Large intestine Cecum
Ascending colon Transverse colon Descending colon Sigmoid colon Rectum Anal canal 6-7 20 45 30 40 12 3 4 Small intestine
Large intestine
3-4 6
pH
5 Stomach Fasted
Fed
1.5-3 2-5 6 Small intestine Duodenum(fasted)
Duodenum(fed) Ileum
6.1 5.4 7-8 7 Large Intestine Cecum and colon
Rectum
[image:11.612.89.527.138.665.2]5.5-7.5 7
Figure 1. Anatomy and cross sectional view of the human colon
The pH of the colon is often lower than the small intestine, between pH 8 and pH 9.The change in pH along the GI tract has been used as a means for targeted colon drug delivery. Enteric coatings are employed to prevent the drug release in the stomach (8).These coatings are generally made from pH responsive polymers which remain unionized and intact at the low pH of the stomach, but dissolve at the higher pH of the small intestine. The principle has been extended to colonic delivery.
The first colon targeted pH responsive delivery system was developed, comprising a capsule coated with polymethacrylic acid methylmethacrylate ester co-polymer. The pH difference between the stomach and the small intestine has historically been exploited to deliver the drug to the small intestine by way of pH-sensitive enteric coatings .There is a fall in pH on the entry into the colon due to the presence of short chain fatty acids that arise from bacterial fermentation of polysaccharides (9). For example, lactose is fermented by colonic bacteria to produce large amounts of lactic acid, resulting in a drop in the pH.
1.3. Colonic microflora and their enzymes
as well as to break bonds between an inert carrier and an active agent (ie.prodrug gets metabolized to drug molecule). Over 400 distinct bacterial species have been found, largely from the genus Bacteroides.The upper regions of the GI tract have a very small number of bacteria and predominantly consist of gram-positive facultative bacteria. The concentration of bacteria in the human colon is 1011 to 1012 CFU/ml. The most important anaerobic bacteria are Bacteroides, Bifido-bacterium, Eubacterium peptococcus, Ruminococcus, Propioni-bacterium, and Clostridium (11).
1.4. GI and colonic transit
The transit of the drug through the GI tract depends on many factors like the fasted or fed state of the subject, quality and quantity of food, size and density of dosage form, concomitant administration of other drugs, and physical exercise.Thetransit time of different dosage forms in the GI tract vary depending on the size of the formulation and shown in Table 2. The effect of transit abnormalities has been examined using gamma scintigraphy study. Whole gut transit times are relatively long, ranging from 56 to 78 hour (h). Colonic transit time (estimated from the mouth to cecum) ranges from 50 to 70 h. The average mouth to anus transit time in humans is 24-72 h (12).
Table 2.Average transit time (h) of different formulations in human Gastric transit of single unit non-disintegrating dosage forms has been reported to vary from 15 minutes (min) to more than 3 h. The presence of food generally increases gastric residence. It is widely agreed that small intestinal residence time is fairly constant at 3-4 h (13). It also appears that the type of
Transit time (h)
Dosage form Stomach Small intestine Total
Tablets 2.7 ± 1.5 3.1 ± 0.4 5.8
Pellets 1.2 ± 1.3 3.4 ± 1.0 4.6
Capsules 0.8 ± 1.2 3.2 ± 0.8 4.0
dosage form and the presence of food do not affect the small intestinal transit. Therefore, considering the highly variable gastric transit and relatively constant intestinal transit times, it might take 4-12 h for the dosage form to reach colon following oral administration.
The mean colonic transit time in humans measured using radio-opaque markers and gamma-scintigraphy is reported to be 33 h in men and 47 h in women (14). Assimilation to the other regions of GI tract, colonic transit is more variable. Larger tablets of 9-12mm in size tend to move faster than smaller tablets of 3-6m in size (15).In comparison, tablets moved significantly faster than 0.5-1.8 mm pellets through the ascending colon. Pellets have advantage over other dosage forms on colonic delivery as their transit time in the large intestine is the longest and because they spread over a large area in the intestine
1.5. Drug absorption in the colon
Absorption of Drugs from the colon is by four main ways namely, Transcellular absorption,
Paracellular absorption,
Transcellular absorption followed by incorporation into chylomicron and transport ion into lymphatic system and
Active transport
because the colon having much lower surface area compared to the small intestine. This is partly compensated by the long residence time and slow movement of drugs in the colon. Most of the drugs that are poorly absorbed in colon are those which are primarily absorbed by paracellular route.
Since it is reported that many sustained-release dosage forms rely in part on colonic absorption to remain therapeutically effective for the desired duration of action, it is imperative that the extent of colonic absorption is established for 12 h and 24 h release formulations.
1.6. General Considerations for Design of oral Colonic Formulations
Formulations for colonic delivery in general are delayed release dosage forms which may be designed either to provide immediate or sustained / prolonged release on reaching the colon. The proper selection of a formulation approach depends on several important factors (19), which include,
Pathology and pattern of the disease,
Physicochemical and biopharmaceutical properties of the drug and Desired release profile of the active pharmaceutical ingredient (API). The most common physiological factor considered in the design of delayed release colonic formulations is pH gradient of the GI tract. In normal healthy subjects, there is a progressive increase in luminal pH from the duodenum pH 6.6±0.5 to the terminal ileum pH7.5± 0.4, a decrease in the cecum pH 6.4± 0.4, and then a slow rise from the right to the left colon with pH 7.0± 0.7(20).
presolubilized form or formulation should be targeted for proximal colon which has more fluid than in the distal colon.
1.7.Strategies for colon specific drug delivery a. Prodrugs
A prodrug is pharmacologically inactive derivative of a parent drug molecule that requires spontaneous or enzymatic transformation in vivo to release the active drug. For colonic delivery of drugs, prodrugs are designed to undergo minimal absorption and hydrolysis in upper GI tract. The drugs undergo enzymatic hydrolysis in the colon, releasing the active drug (21).A number of other linkages susceptible to bacterial hydrolysis specifically in the colon have been prepared where the drug is attached to hydrophilic moieties like amino acid, glucuronic acid, glucose, galactose, cellulose, coating materials over drug cores, etc. Polysaccharides are used as glucuronic prodrugs, which are specifically degraded by colonic-glucuronidases, and glycosidic prodrugs, which are specifically degraded by colonic glycosidases. The most widely used polysaccharide for this application is dextran.The action of bacterial glycosidase enzyme on the glycosidic bond permits the release of the attached drug. This triggers its pharmacological activity. The metabolism of azo compounds by the intestinal bacteria is one of the most extensively studied bacterial metabolic processes.
b. pH sensitive drug delivery
pH-sensitive enteric coatings have been used routinely to deliver drugs to the small intestine. These polymer coatings are insensitive to the acidic conditions of stomach, yet dissolve at the higher pH environment of the small intestine. This pH differential principle has also been attempted for colonic delivery purposes.
The pH of the human GI tract increases progressively from the stomach (pH 1 to pH 2) which increases to pH 4 during digestion, small intestine (pH 6 to pH 7) at the site of digestion, and increases to pH 7 to pH 8 in the distal ileum (22). The coating of pH-sensitive polymers to the tablets, capsules or pellets provides delayed release and protects the active pharmaceutical ingredient (API) from gastric fluid. The polymers used for colon targeting, should be able to withstand the lower pH values of the stomach and of the proximal part of the small intestine and also be able to disintegrate at the neutral of slightly alkaline pH of the terminal ileum and preferably at the ileo-cecal junction.
Polymers such as methacrylic resins (Eudragits), Eudragit L and Eudragit S are copolymers of methacrylic acid and methyl methacrylate are widely used (23). Colon targeted drug delivery systems based on methacrylic resins have been also used for delivery of insulin, prednisolone, quinolones, salsalazine, cyclosporine, beclomethasone dipropionate, and naproxen.
It has been shown that polymers with non esterified phthalic acid groups dissolve much faster and at a lower pH than those with acrylic or methacrylic groups. The presence of plasticizer and the nature of the salts in the dissolution medium influence the dissolution rate. The free carboxylic groups of Eudragit-S are partially methylated. The effectiveness of this product as a colon specific coating material has been established on human volunteers using in vivo
the inner layer determine the lag time. This system is found to release drug in the colon of the rat between the fifth and tenth hour. A multi-unit dosage form containing 5-Amino salicylic acid is used for treating ulcerative colitis (25).
c.Time-dependent drug delivery
Time-dependent dosage forms are formulated to release their API load after a pre determined lag time. Although it is not a site specific drug delivery system, it has been suggested that colonic targeting can be achieved by increasing a lag time equivalent to the mouth-to-colon transit time in the formulation. A nominal lag time of 5 h is usually considered sufficient, since small intestinal transit has been considered relatively constant at 3 to 4 h (26).
A number of systems have been developed based on this principle, with one of the earliest being the Pulsincap device (27).This device consists of a non disintegrating half capsule shell sealed at the open end with a hydrogel plug. The plug hydrates on contact with GI fluid, and swells to an extent that it is expelled from the capsule body, thus releasing the drug. By altering the composition and size of the hydrogel plug, it is possible to achieve drug release after varying lag times.
A pulsed system, called the Time-Clock System, has been developed (28). This system comprises of a solid dosage form coated with a hydrophobic surfactant layer to which a water-soluble polymer is attached. The surfactant improves adhesion to the core. The thickness of the outer layer determines the time required to disperse the drug in an aqueous environment. After the dispersion of the outer layer, the core becomes available for dispersion. Studies performed on human volunteers showed that the lag time is not affected by gastric residence time.
d.Pressure controlled drug delivery
temperature. In the upper GI tract, PCDCs are not directly subjected to the luminal pressures since sufficient fluid is present in the stomach and small intestine. Due to the reabsorption of water in the colon the viscosity of luminal content increases. As a result, increased intestinal pressure directly affects the system via colonic peristalsis. In response to the raised pressure, PCDCs rupture and release the drug in the colon.
e. Bacterially triggered drug delivery
The resident gastrointestinal bacteria provide a further means of effecting drug release in the colon. These bacteria predominantly colonize the distal regions of the gastrointestinal tract where the bacterial count in the colon is 1011 per gram, as compared with 104 per gram in the upper small intestine. Moreover, 400 different species are present. Colonic bacteria are predominantly anaerobic in nature and produce enzymes that are capable of metabolizing endogenous and exogenous substrates, such as carbohydrates and proteins that escape digestion in the upper GI tract. Therefore, materials that are recalcitrant to the conditions of the stomach and small intestine, yet susceptible to degradation by bacterial enzymes within the colon, are utilized as carriers for colonic drug delivery. Enzymatically degradable polymers have an interesting application providing colon- specific drug delivery and rely mainly on the principle
the use of prodrugs breakdown by bacterial enzymes within the colon and
the use of tools (coatings/ matrices) susceptible to colonic bacteria.
f. Osmotically controlled drug delivery
Osmotically controlled drug delivery is one of the approaches to target the drugs to the colonic region. The OROS-CT (Alza Corporation) can be used for the treatment of disease or to achieve systemic absorption that is otherwise unattainable (31). The OROS-CT system can be single osmotic unit or may incorporate as many as 5-6 push-pull units, each 4mm in diameter, encapsulated within a hard gelatin capsule. Each push-pull unit contains osmotic push-pull units. Because of its drug-permeable enteric coating, each push pull unit is preventing from absorbing water in the acidic aqueous environment of the stomach and hence no drug is delivered (32).As the unit enters the small intestine, the coating dissolves in the higher pH environment, water enters the unit causing the osmotic push compartment to swell and concomitantly create a flowable gel within the compartment. Swelling of the osmotic push compartment force drug gel out of the orifice at a rate precisely controlled by the rate of water transport through the semipermeable membrane. OROS-CT units can maintain a constant release rate for up to 24h in the colon.
g. CODES technology
coating will dissolve in the small intestine. Upon entry into the colon, the polysaccharide inside the core tablet will dissolve and diffuse through the coating. The bacteria will enzymatically degrade the polysaccharide into organic acid. This lowers the pH surrounding the system sufficient to affect the dissolution of the acid-soluble coating and subsequent drug release.
1.8. Dosage forms
a. Single unit dosage forms
Single unit dosage forms are defined as oral dosage forms that consist of single units, with each unit containing one dose of the API and intended to be administered singularly. Numerous such dosage forms were been developed and available in the market. These monolithic matrix-based tablets are the most common single unit dosage form used for controlled drug delivery.
b.Pills
The pill was one of the most common dosage forms until the middle of the 20th century. Pills were compounded in pharmacies by thoroughly mixing the powders in a mortar, adding necessary quantity of excipients, and triturating the mixture in a pestle until a plastic mass resulted. The mass was then rolled into cylinders and divided either on the pill tile or by means of the pill cutter. Finally, the cut segments were rolled until required spherical form. The combined technique of extrusion and spheronization is the advancement to the traditional pill-making process (34).
c.Tablets
One of the most commonly used methods for developing controlled release tablets for therapeutic agents is by formulating the tablets in a matrix system. A matrix tablet is a compressed dosage form containing an active ingredient, matrix agent, fillers, lubricants and other excipients. Matrix systems are highly resistant to release inconsistencies and dose dumping since they are relatively simple systems and are robust to process and ingredient variations, production methods and end use conditions (35).
d. Multiple unit dosage form
The various approaches in formulation of multiple unit dosage forms are
The placement of a drug in an insoluble matrix where the eluting medium penetrates the matrix and the drug diffuses out of the matrix and into the surrounding pool for absorption,
Enclosing the drug particles with a polymer coat. In this case, the portion of the drug that has been dissolved in the polymer coat diffuses through an unstirred film of liquid into the surrounding fluid and.
Eroding pellets from which the drug is released as the pellet matrix erodes or dissolves.
Multiple unit dosage forms for oral delivery of API offer the following advantages over the other oral dosage forms.
The gastric emptying time is less variable since the pylorus can be passed even in the fed state.
The dosage forms are more homogeneously distributed within GI Tract. The stagnation at the ileo-caecal junction is less likely to occur than with
larger single units.
Slower transit of small particles through the colon which prolongs the contact between the formulation and the absorptive surface.
Coated dosage forms can generally contain higher dosage of the drug than conventional systems.
Pellets with different release rates of the same API can be formulated in a single dosage form, which can freely disperse in the gastrointestinal tract as subunits. This maximizes the drug absorption and reduces peak plasma fluctuation.
gelatin capsules or compressed into tablets, which rapidly disintegrate into multiple units after administration.
Owing to their smooth surface morphology, narrow size distribution, spherical shape, low friability and improved hardness pellets can be easily coated; they have good flow properties which ensure reproducible die or capsule filling and consequently good content uniformity. Dust formation is minimised.
Compaction and drug layering are the most widely used pelletization techniques in pharmaceutical industry (38). Of the compaction techniques, extrusion and spheronization is a predominant method. Other pelletization methods, such as globulation, balling and compression are also used in the development of pharmaceutical pellets although in a limited scale (39). Regardless of which manufacturing process is used, pellets have to meet the following requirements
They should be near spherical and have a smooth surface. The particle size range should be as narrow as possible.
The pellets should contain as much active ingredients as possible to keep the size of the final dosage form within reasonable limits.
Advantages of pellets
Pellets prove more advantageous than other pharmaceutical formulations technically, physiologically and in biopharmaceutical aspects.
Pellets offer great flexibility in pharmaceutical solid dosage form design and development.
They flow freely and pack easily without significant difficulties, resulting in uniform and reproducible fill weight of capsules and tablets.
Pellets composed of different drugs can be blended and formulated into a single dosage form. This approach facilitates the delivery of two or more drugs, chemically compatible or incompatible, at the same sites or different sites in the GI tract.
Even pellets with different release rates of the same drug can be formulated into a single dosage form.
The pelletized product can freely disperse in the GI tract as a subunit, thus maximizing drug absorption and reducing peak plasma fluctuation.
Local irritation derived from higher local concentrations of a drug from a single unit dose, can be avoided.
Controlled release oral solid dosage forms are usually intended either for delivery of the drug at a specific site within the GI tract or to sustain the action of drugs over an extended period of time through the application of coating materials (mainly different polymers), providing the desired function or through the formulation of matrix pellets to provide the desired effect.
Targeting of oral drug delivery into any region of GI tract is feasible. Disadvantages of pellets
Proportionally higher need for excipients Large number of process variables Multiple formulation steps
Higher cost of production Need of advanced technology
1.9. Extrusion and spheronization
Wet-mass extrusion and spheronization
Extrusion-spheronization process involves a multiple-step compaction process comprising
Dry blending of API and excipients Wet granulation
Extrusion
Charging the extrudates into the spheronizer Drying
Screening (for achieving the desired size)
Figure 2. Extrusion and spheronization process mechanism
1.10. Coating processes
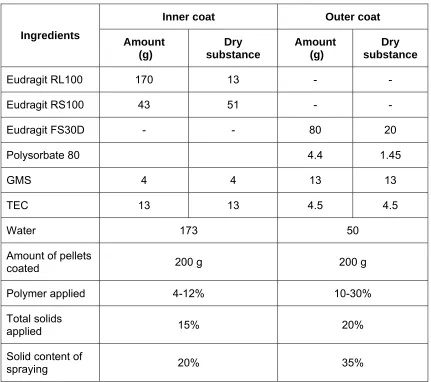
Coating of dosage forms are performed with the intention of improving the technical or biopharmaceutical properties. Various polymers are utilized for coating in accordance with their purpose. Solid formulations of the desired API are coated with pH resistant polymers, so as to carry the formulation until colon without any loss while in transit through GI tract. Such formulations may be used to deliver drugs to the distal ileum.
The formulations for colonic delivery are designed to withstand both low and slightly basic pH values for several hours(41).During this time, they pass through the stomach and the small intestine and reach the large intestine, where the coat disintegrates and the drug release process is initiated. The polymers used for this purpose are commonly acrylic acid derivatives or cellulose derivatives such as cellulose acetate phthalate or ethyl cellulose.
Formulations based on pH responsive polymers (Eudragit S,Eudragit L, Eudragit FS 30D and Eudragit P4135) have been investigated in order to target the Ileum colon (42).Before applying the polymer, it is either dissolved in an organic solvent or dispersed in water as pseudo latex. The formation of coatings from solutions involves conversion of a viscous liquid into a viscoelastic solid, which is accomplished in three steps of solvent removal (43).
1) Evaporation of solvent at the surface of the coating,
2) Diffusion of solvent from the wet coating on the surface, and 3) Slow diffusion of residual solvent in the dried coating.
polymer particles. Moreover, it is generally accepted that there are two stages in the process of coalescence of aqueous dispersions .The first one involves the formation of a dry, transparent, apparently continuous film, which occurs concurrently with the evaporation of water. The second stage, although not a totally separate phenomenon, may be considered to be the gradual completion of coalescence. This occurs on ageing of the film. During the ageing process, the individual polymer particles completely fuse into a homogeneous, continuous film as the water finally evaporates from the interstitial spaces. This process, known as curing, is dependent on the time and temperature of the coating and post-coating processes.
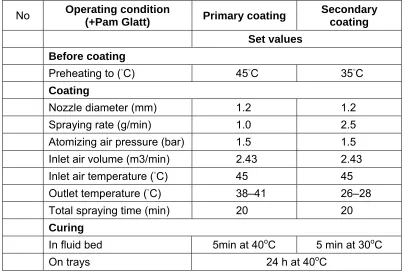
Fluidized bed coating is one of the particle coating techniques that has become increasingly important for industrial applications (44). In a fluidized bed, particles are suspended and circulated by an air flow. Depending on the size, shape, and density of particles, the air flow rate needs to be set in the range between the minimal fluidization velocity and the pneumatic transportation. The coating solution is delivered into the fluidized bed chamber through an atomizing nozzle. The solution is atomized into droplets and then spread onto the surfaces of the fluidized particles. Evaporation of the coating solvent leaves a solid layer of solute on the surface. The evaporation rates are usually slow and depend on numerous parameters, such as the temperature, relative humility of air, air flow rate, coating solvent and particle properties (size, size distribution, surface roughness).
1.11. Coating of pellets
Pellets of varying diameter can be manufactured by several methods. Pellets usually range in size from 0.5-2 mm depending on the application. Pellets are agglomerates of fine powders or granules of bulk drugs and excipients. They consist of small, free-flowing, spherical or semi-spherical solid units, typically from about 0.5 mm to 1.5 mm and are intended usually for oral administration. Pellets spread out over a large area of intestine and make more effective for the treatment of local diseases of colon and may improve absorption possibilities for large molecules. Multiple unit delivery systems are also statistically more reliable than single-unit delivery systems. In addition, the pellets are advantageous solid dosage form for colonic delivery as their transit time in the large intestine is the longest. The use of pellets as a drug vehicle for reservoir-type controlled-release dosage forms for oral administrations has gained importance. They owe their popularity to the reduced risk of systemic toxicity due to dose dumping, minimal potential side effects by reducing peak drug fluctuation, improved flow properties and the flexibility in formulation development (45).
The two methods that are widely reported in the literature for the preparation and coating of pellets are fluidized-bed processing and pan-coating.
Stomach Jejunum Ileum
Colon
1.12. Multiparticulate tablets
Multiparticulate tablets are composed of coated subunits which are embedded in excipients mixture. Once the coated subunits are developed, different dose strengths can be prepared just by varying the tablet size, keeping the composition same, with no additional developmental efforts (46).
The main challenges in multiparticulate tablet manufacture are To ensure uniformity in content and weight.
To ensure sufficient hardness and low friability without damaging the coatings.
Uniformity of the API content and tablet weight is mainly based on well flowable tabletting mixtures with narrow particle size distribution. Highly flexible coatings provide elastic properties which prevent cracking when compression forces are applied. Film elasticity can be increased by adding a plasticizer or by combining brittle polymer with the very soft grade polymer. Film flexibility as a factor for mechanical stability is also related to the coating thickness. Coatings with high thickness support elastic properties. They provide protective effects to the film coating whereas sharp-edged and abrasive crystalline materials damage the coating with increase in compression force. During development of multiparticulate tablets comparative dissolution tests should be conducted to check for possible differences between the release rates of the uncompressed tabletting mixture versus the tablets.
1.13. Experimental design
a. Statistical design of experiment (DOE)
Experimental design is defined as the statistical strategy practiced to carry projects efficiently and systematically to give reliable and coherent information.
product, or both).They are powerful tool for improving the efficiency of experimentation. Through an iterative process, they allow us to gain knowledge about the system being studied with a minimum number of experiments. Inclusion of replicate test conditions allows the estimation of random, experimental variation. Statistical analysis of data generated from the experiment clearly establishes the relationship between the measured parameter of interest (response) and the process parameters (input factors) being studied (47). The factors may have individual, simple effects on the response (referred to as main effects) or may have effects that are interdependent (referred to as interaction effects). Since the designed experiments are generated on the basis of statistical theory, confidence in the results obtained and conclusions drawn are clearly defined.
The information is then used to achieve the goal of the project with maximum efficiency. Thus use of budget (i.e., the resources of money, equipment, raw material, manpower, time, repeatability, etc..) optimized to reach the objectives quickly with best precision.
Experimental work of the project was carried out in the following order Screening
Quantitative factor study Response surface modeling Optimization
Validation
Different types of screening designs, such as fractional factorial and Plackett-Burman screening designs, have been used for preformulation evaluations.
1. performing the statistically designed experiments, 2. estimating the coefficients in a mathematical model,
3. predicting the response and checking the adequacy of the model (48). This design is suitable for exploration of a quadratic response surface and constructs a second order polynomial model, thus helping in optimizing a process using a small number of experimental runs. Other RSM designs include 3-level factorial design, central composite design, Box-Behnken design (BBD) (49) and D-optimal design.
b.Box-Behnken Design
A modified central composite experimental design which is an independent, rotatable or nearly rotatable quadratic design (contains no embedded factorial or fractional factorial design), in which the treatment combinations are at the midpoints of the edges of the process space and at the center.
LITERATURE REVIEW
A review of the literature was carried on oral modified release formulation,
formulation and coating techniques, colonic drug delivery system, statistical
designing of experiments and the polymers used for the development of these
systems.
Marvola et al., 1999, (52) described the use of a multiple-unit system for drug release in the colon using enteric polymers. Film coated matrix pellets were
prepared with enteric polymers as both binders and coated material. It was found
that drug release from the formulation occurs in the distal part of the small
intestine and the colon with enteric polymers dissolving at pH 7.
B.Song et al., 2007, (53) studied the effect of freeze dried and oven dried pellets obtained from extrusion-spheronization technique. On compaction change
in voidage and crushing strength of tablets were occurred and affected the
dissolution rate.
Khan et al.,1999,(54) studied aqueous coating of various combinations of two pH-dependent polymers Eudragit L100-55 and Eudragit S100 on
lactose-based placebo tablets for colonic delivery. The authors demonstrated that the
release profiles of the drug could be manipulated by changing the ratio of
Eudragit L100-55 and Eudragit S100 within the pH range of 5.5 to 7.0.
Libo Yang et al., 2001, (55) investigated the feasibility of the manufacturing process with achieving in vivo site specificity, design rationale, and pressure-controlled colon delivery capsules.
Helton Santos et al.,2005,(56) studied the compaction and compression of xanthan gum pellets and evaluated the drug release from tablets made from
pellets prepared by extrusion-spheronization method. The formulations included
xanthan gum, at 16% (w/w), diclofenac sodium or ibuprofen, at 10% (w/w),
among other excipients An amount of 500 mg of pellets fraction 1-1.4 mm were
compacted in a single punch press. Physical properties of pellets and tablets
2. SCOPE AND OBJECTIVES
Multiple unit matrix pellets is of great practical importance in releasing the
drug in the colonic region. In order to develop a reliable colonic drug delivery
system, the transit time of dosage forms through the GI tract in different pH
conditions need to be well understood. The transit through the GI tract is highly
variable and depends on many factors. Gastric transit of single-unit
non-disintegrating dosage forms have been reported to vary from 15 minutes to more
than 3 hours. However, it is widely agreed that the small intestinal residence time
is fairly constant at 3–4 h (94). The mean colonic transit time in humans is
reported to be varying from 33 h in men and 47 h in women. Scientists have
developed pH-dependent systems, time-dependent systems and bacterially
degradable systems to deliver drugs to the colon.
A colonic delivery system which is based either on pH dependence or
transit time would not be reliable because of the inherent variability of pH and
emptying times from the GI tract. Advantage can be taken on relatively constant
transit time of the small intestine (3–4 h) and high pH of the distal small intestine
(pH 5–pH 7.5) by combining pH characteristics of different polymers and their
transit time in the small intestine to develop a reliable colonic delivery system.
The multiple unit matrix pellets should be an ideal drug delivery system, since
they reach, scatter over a larger area and release the drug following oral
administration. This will make the pellets more effective for the treatment of local
diseases of colon and may improve site-specific of drug delivery in colon.
It was proposed to develop multiple unit matrix pellets for oral use in
• Capsule and
• Multiparticulate tablet dosage form.
An extrusion and spheronization technique was proposed to be adopted
in formulating pellets. The pellets containing selected drug candidate was coated
with a polymer mixture of Eudragit RL100 and Eudragit RS100 for sustained
pellets have to be protected in upper GI tract, an enteric protection would be
given by coating them with Eudragit FS30D.This polymer dissolves only above
pH 6.8 and expected to dissolve in the ileum region. This makes the pellets to
expose the inner coat for diffusion of the API from the core matrix in a sustained
manner. Ibuprofen (95), furosimide (96) and budesonide (97) were selected as
model drugs in this study on their basis of absorption pattern in GIT.
Ibuprofen is well absorbed throughout the GI tract and is therefore
selected as a model drug in relation to study of colon-specific formulations.
Furosemide is well absorbed in the stomach. Small amounts may be
absorbed in the small intestine. However absorption in the large intestine is
negligible. It is proposed to use this drug as an unabsorbed model drug in
studying colon-specific formulations.
Budesonide, a second generation glucocorticoid and potent drug
candidate for treating IBD has first pass metabolism. It has the highest affinity for
glucocorticoids receptor as compared to other steroids (hydrocortisone,
prednisolone, and dexamethasone) and it does not reduce the cortisol levels.
The pharmacokinetic profile of budesonide favors a high topical efficacy because
of rapid uptake by mucosal tissue and enhanced receptor binding properties. In
light of these facts, a dosage form capable of delivering budesonide in a
sustained release manner during its transit through the lower part of GIT can be
envisaged to provide a new paradigm in treating IBD more effectively.
It was proposed firstly to optimize the formulation parameters by using
ibuprofen as model API.
The study was proposed to be carried in the following phases.
Phase-I
• Preformulation studies
Phase-II
Development of multiple unit matrix pellets and tablets Stage-I. A
• Formulation of core pellets by extrusion spheronization technique
• Coating of pellets
Stage-I. B
• Formulation of multiparticulate tablets
Stage-II. Characterization of the pellets and tablets a. Characterisation of uncoated core pellets
• Percentage yield
• Physical evaluation of pellets
• Pellet size, shape and surface characteristics
• Particle size distribution
• Uniformity of drug content
• Friability
• Morphological study by scanning electron microscopy
b. Characterisation of coated core pellets
• Physical evaluation of pellets
• Pellets shape and surface characteristics
• Uniformity of drug content
• Friability
• Flow rate and angle of repose
• Bulk density and compressibility
• Fluid uptake study
• Fill volume of pellets
c.Characterization of the multiparticulate tablets
• Average weight and weight variation
• Thickness and diameter
• Shape and surface characteristics
• Friability
• Disintegration
• Morphological study by scanning electron microscopy
Phase III
In vitro dissolution study of the pellets
• In vitro drug dissolution release profile of the selected multiple unit matrix pellets.
In vitro dissolution study of the multiparticulate tablets
• In vitro drug dissolution release profile of the selected multiparticulate tablets.
Stability study as per the ICH guidelines
Selected pellet formulation and multiparticulate tablet formulation will be
subjected to stability studies at different temperature and humidity conditions as
prescribed by the international conference on harmonization (ICH).
• 250C with 60% relative humidity
• 450C with 75% relative humidity
Phase IV
Pharmacokinetic studies
To carry out a comparative bioavailability studies in a cross over design in
wister strain albino rabbits and to evaluate the following pharmacokinetic parameters
• Determination of Tlag • Cmax
• Tmax
• Area under the curve (AUC)
the upper surface and the surface of fractured tablets revealed that particles
remained as coherent individual units after compression process.
Viness Pillay et al., 1999, (57) investigated the cross linking of sodium alginate, low ethoxylated pectin and their novel binary mixture with calcium ions
through ionotropic gelation method to pelletize diclofenac sodium, using
‘‘environmentally benign’’ solvents. Crosslinked pellets of the above polymers in
2% (w/v) aqueous calcium chloride solution were prepared and evaluated.
The average size of the different pellets was 1.3 mm and drug entrapment
capacity was optimized by reducing the calcium chloride solution. In vitro drug release profile was estimated in simulated human GI tract condition. Negligible
drug release occurred in pH range of 1–4, and the drug was released as a
controlled manner in pH 6.6.
Chunsheng Gao et al., 2006, (58) developed meloxicam-loaded colon-specific pellets coated with Eudragit FS 30 D by drug layering on nonpareil
seeds. The pellets were coated with a copolymer of methyl acrylate, methyl
methacrylate and methacrylic acid. The obtained pellets with 15% (w/w) coating
level had a spherical form and a smooth surface. The in vitro drug release from the pellets was pH-dependent with sufficient gastric resistance. In vivo study on beagle dogs showed the onset of meloxicam absorption from the coated pellets
with 15% (w/w) Eudragit FS 30D was significantly delayed.
Amol Paharia et al., 2007, (59) prepared and evaluated Eudragit coated pectin microspheres for colon targeting of 5-fluorouracil (FU). Prepared by
emulsion dehydration method using different ratios of FU, pectin and emulsifier
concentrations. The yield of the preparation and the encapsulation efficiencies
were high for all pectin microspheres. Evaluated for surface morphology, particle
size and size distribution, swellability, percentage drug entrapment, and in vitro
drug release in simulated GI fluids. The release profile of FU from
Eudragit-coated pectin microspheres was found to be pH dependent.
vinyl alcohol and poly ethylene glycol. They adopted a central composite
statistical design for investigating the effect of the polymer blend ratio, the film
coat thickness and the plasticizer concentration on the drug release. The
blending ratio of the polymers and the film thickness were found to have a major
influence on the drug release.
Karin Krogars et al., 2000, (61) investigated the effects of three independent variables (amounts of Eudragit S, citric acid and spheronising time)
on pellets size, shape and drug release. They studied with statistical central
composite design and .utilized extrusion–spheronization pelletization for
preparing pH-sensitive matrix pellets for colon-specific drug delivery.
He Wei et al., 2008, (62) studied the in vitro and in vivo studies of pectin ethyl cellulose coated pellets containing 5-fluoro uracil for colonic targeting.
Found that 1:2 ratio of pectin ethyl cellulose coated pellets with 30% total weight
gain produced more satisfactory drug release profiles in the simulated GI and
colonic fluids. Most of the coated pellets were eliminated from the stomach in 2 h,
moved into the small intestine after 2–4 h, and reached the large intestine after
4 h. After oral administration 5-FU started appearing in the plasma and reached
peak plasma concentration at 7 h.
Brunner et al., 2005, (63) evaluated the GI transit, release and absorption of budesonide from a newly designed multimatrix formulation tablets for the
release of drug throughout the colon, assessed the formulation of multimatrix
budesonide tablets appear suitable for colonic drug delivery. Transit parameters
and low systemic bioavailability warranted further studies with this newer
formulation.
Vivek Ranjan Sinha and Rachana Kumria., 2003, (64) studied a comparison of the enteric coating polymers. Eudragit, cellulose acetate phthalate
with shellac and ethyl cellulose were used as carriers for colon specific drug
delivery. Lactose based indomethacin tablets were prepared and coated with one
of the coating polymers to varying coat thickness. The coated formulations were
conditions. From the dissolution data obtained, it was found that the dissolution
rate varied with the type and concentration of the polymer applied. Comparative
dissolution data revealed that, of all the various polymers and coat thicknesses
used, a 3% (m/m) coat of shellac was most suitable for colonic drug delivery. It retarded drug release by 3–4h (the usual small intestinal transit time) in
simulated small intestinal fluid, where after a rapid drug release was noticed.
Ingunn Tho et al., 2001, (65) investigated the possibility of producing pectin-based pellets by extrusion-spheronization method and identified the
factors influencing the process and the characteristics of the resulting product.
Three types of pectin with different degrees of amide and methoxyl substitution
were studied in combination with different granulation liquids like water, calcium
chloride solution, citric acid solution, microcrystalline cellulose and ethanol.
Pellets were prepared using controlled twin screw extruder, spheronized and
dried. The products were subjected to image analysis, sieve analysis,
disintegration and dissolution tests. The results were evaluated by multivariate
analysis. Different additives, either in the granulation liquid or in the powder
mixture, influenced the ability of the extruded mass to form pellets. However, the
different pectin types responded to modifications of pellets, nearly spherical
pellets are obtained by using ethanol as granulating agent. Pellets produced with
ethanol were mechanically weak and tend to disintegrate. Addition of
microcrystalline cellulose as an extrusion aid generally resulted in improvements
in shape and size.
Siew Lee F et al., 2000, (66) studied the potential of organic-based amylose-ethyl cellulose film coatings for oral colon-specific drug delivery.
Amylose, in the form of an amylose-butan-1-ol dispersion and ethyl cellulose,
dissolved in ethyl lactate, ethanol, or propanol and plasticized with dibutyl
sebacate, were mixed in various proportions and applied using a fluidized bed
coater to achieve a range of film thicknesses on 5-aminosalicylic acid pellets.
Drug release from the coated pellets was assessed under gastric and small
intestinal conditions in the presence and absence of pepsin and pancreatin.
testing with human feces in the form of slurry was used. Under upper GI tract
conditions, the rate and extent of drug release were found to be related to the
thickness of the coating and the ratio of amylose to ethyl cellulose within the film.
Drug release data revealed that the ratio was more important than coat thickness
in controlling drug release, irrespective of the solvent used for coating. Coatings
with a thick film and or low amylose content were relatively impermeable and able to
delay drug release under conditions simulating the upper gastrointestinal tract.
Purushotham Rao et al.,2003,(67)investigated the potential use of pectin in combination with two added hydrocolloids, hydroxyl-propyl-methyl cellulose
and hydroxyethyl cellulose in varied concentrations and coated with ethyl
cellulose and cellulose acetate phthalate. The results of in vitro drug release and roentgen graphic studies on healthy human volunteers revealed that pectin,
hydroxyethyl cellulose base coated with ethyl cellulose and cellulose acetate
phthalate was found to be a promising carrier for to colon. Naproxen was used as
a model drug in their study.
Mohini Chourasia et al., 2006, (68) investigated, guar gum microspheres containing methotrexate for local release in the colonic region. Guar gum
microspheres were prepared by the emulsification method using glutaraldehyde
as a cross-linking agent. Surface morphological characteristics were investigated
using scanning electron microscopy. Particle size, shape, and surface morphology
were significantly affected by guar gum concentration, glutaraldehyde concentration,
emulsifier concentration (Span 80), stirring rate, stirring time, and operating
temperature. Guar gum microspheres showed adequate potential in achieving local
release of drug in the colonic region.
Steckel et al., 2004, (69) produced pellets comprising equal amounts of MCC and chitosan without additional binder. Furthermore, they succeeded to
produce pellets with acceptable quality with increased chitosan concentration
using a 0.1N acetic acid solution as granulation liquid. The authors postulated
that partial dissolution of chitosan at the particle surface increased the wet mass
the mixture, a higher amount of granulation liquid was needed for successful
extrusion / spheronization.
Kopecek et al., 1992, (70)examined bioadhesive water-soluble copolymers for colon specific delivery of mesalamine and hydrogels degradable by microbial
enzyme in the colon for delivery of peptides. They found that both systems have
potential for colon specific drug delivery.
Morshita et al., 1993, (71) compared the insulin delivery of two formulations containing Eudragit L100 and Eudragit S100 respectively. Formulation containing
Eudragit S100 showed optimum delivery of insulin in the ileum at pH 7.
Yuen et al., 1993, (72) investigated the GI transit and absorption of theophylline from a novel multiparticulate controlled release formulation under
fed and fasted conditions. The drug pellets were prepared using extrusion and
spheronization technique. They were coated with methylcellulose-ethyl cellulose
mixture to control the drug release. Presence of food delayed the gastric
emptying but there was no influence on the intestinal transit time. The delay in
gastric emptying was associated with a delay in drug absorption. The overall rate
and extent of drug absorption, however, was essentially unaffected by the
presence of food. The pellets were less dispersed in the stomach than in the
small intestine or colon. In the small intestine 47% of the drug was released,
whereas only 14% of the drug was released in the stomach. The remaining 39%
of the drug was taken up from the colon, which acts as a significant site of
absorption.
Sekigawa et al., 1995, (73) described a coated dosage form suitable for oral administration of drugs in the large intestine. It was prepared by coating a
core containing the active ingredient. First, with chitosan having specified degree
of deacetylation and a defined degree of polymerization and then top coating with
an enteric-soluble polymer such as hydroxypropylmethyl cellulose acetate succinate.
Ciftci et al., 1996, (74) studied in vitro and in vivo evaluation of PLGA microspheres containing 5-FU prepared by a solvent evaporation method. They
more than 70% w/w of 5-FU. The in vitro study revealed that the drug release depends upon the amount of entrapped drug, molecular weight of polymer and
pH of dissolution medium. The release mechanism was diffusion controlled and
followed a square root of time relationship. The in vivo distribution of the microspheres was characterized initially with uptake by different organs of the
mononuclear phagocyte system, mainly the Kupfer cells cytoplasm and near the
liver sinusoids.
Milojevic et al.,1996, (75)studied the preparation and in vitro evaluation of amylase film coated 5- amino salicylic acid pellets for colon-specific drug
delivery. However, under simulated GI conditions, the coating of amylase had a
swelling effect and allows the drug release. The different range of cellulose and
acrylate based copolymers were assessed, of which commercial ethyl cellulose
was found to have better control for effective swelling.
Krishnaiah et al., 2003, (76) studied the colon-specific drug delivery systems based on a guar gum using in vitro and in vivo methods. In vitro drug release studies have shown that guar gum in the form of compression coat
applied over indomethacin core tablets protects the drug from being released
under conditions simulating mount to colon transit. Studies using pH 6.8
phosphate buffered saline containing 4% w/v rat caecal content have
demonstrated the susceptibility of guar gum to the colonic bacterial enzyme
action with consequent drug release. Gamma-scintigraphic studies in human
volunteers showed that entering the ascending colon, the tablets started to
release the tracer indicating the breakdown of the gum coat by the enzymatic
action of colonic bacteria.
Nykanen et al., 1999, (77) investigated the influence of organic acids as excipients on the drug release from methacrylate enteric matrix granules. Ibuprofen
was used as a model drug. They studied the bioavailability in healthy volunteers.
Khan et al., 1999, (78) studied the release profiles of a theophylline immobilized calcium pectinated beads carried by Eudragit S 100 coated capsules
in different dissolution medium. It was found that the drug release rate in the
phosphate buffer was significantly faster than that in Hank’s solution.
Bhalla and Shripad., 1994, (79) prepared spheroids of diclofenac sodium by spheronization technique and these pellets were coated with ethyl cellulose.
The pellets were evaluated for size distribution, drug content and in vitro release profile. The formulation showed good stability in terms of drug content, content
uniformity and in vitro release. When exposed to accelerated conditions of storage the formulation showed a sustained in vivo blood level pattern as compared to the conventional formulations.
Vanarase and Nagarsenkar., 1995, (80) prepared prochiorperazine pellets coated with ethyl cellulose using extrusion and spheronization technique.
The pellets were evaluated for drug content, surface area, specific surface,
sphericity and in vitro release. It was concluded that the release of
prochiorperazine could be retarded with increasing amounts of ethyl cellulose
application. The amount of drug released from these pellets of different size,
coated to the same extends, however, depended upon the specific surface
provided by these pellets.
Pramila and Shrivastava.,1996,(81) formulated senidazole pellets for improved therapy in intestinal amoebiasis using the polymer, Eudragit S 100
.They determined the angle of repose for the coated pellets using the fixed funnel
method. Size distribution was determined by sieve analysis. The pellets were
also evaluated for friability, sphericity and drug content.
Brabander et al., 2002, (82)prepared matrix pellets combining microcrystalline waxes pregelatinised starches and hydrolyzed starches. Ibuprofen, sodium
salicylate, benzoic acid, sodium benozoate and chloroquine phosphate were
used as model drugs. It was reported that increasing the wax concentration the
rate of drug release decreased and assumed that the drug release rate was
Lindner et al., 1994, (83) investigated the use of powdered cellulose instead of microcrystalline cellulose in the extrusion spheronization process. The
aim of the study was to asses differences between two types of powdered
cellulose using two fractional factorial designs. Water content and amount of
binder were found to be most important while types of cellulose and twin screw
speed had only negligible influence on the extrusion process and the resulting
pellets. Pellets obtained with powdered cellulose showed higher porosities and
faster dissolution rates compared to those made with microcrystalline cellulose.
Lucy and Lai., 1991, (84) studied the effects of spheronization speed and residence time on the size and sphericity of microcrystalline cellulose lactose
spheroids. It was found that the combination of speeds ranging from 1000-2000
rpm and residence time between 5-15 min might be used to produce spheroids
with a model fraction in a range of 0.7-1.0 mm. The effects of varying MCC
content and the amount of water required for spheronization was also studied.
Sousa et al., 2002, (85) studied the physical characteristics of pellets prepared by extrusion and spheronization method by using water as mass
forming agent. The result showed that the amount of water, the extrusion,
residence time in spheronizer and drying process influenced the physical
characteristics of the pellets.
Vonk et al., 1997, (86) prepared pellets of MCC and lactose using waster as binder liquid in two high-shear mixers Gral 10 and 25. Growth of pellets was
investigated by measuring the pellet size distribution drug the process. On the
basic of the experiment the destructive nucleation growth mechanism was
defined. Pelletization was started with the formation of large primary nuclei.
Small secondary nuclei were formed due to break up of the primary nuclei. Due
to densification, the secondary nuclei became stronger and growth proceeded
exponentially by coalescence.
Fukui et al., 2000, (87) developed enteric coated timed-release press-coated oral tablets for colonic release of diltiazem hydrochloride. They press-coated
diltiazem hydrochloride. The in vitro results showed acid resistance and timed release of diltiazem hydrochloride at both pH 1.2 and pH 6.8.
Semde et al., 2000, (88) studied the influence of cellulosic and acrylic polymeric coatings on theophylline pellets containing 10% calcium pectinate. The
coated pellets were found to decreases the release in the media without
enzymes than in the media containing enzymes. The results showed that the
combination of pectin and the insoluble Eudragit polymers in aqueous dispersion
intended for specific-delivery of drug to the colon.
Gazziranga et al., 1995, (89) designed and evaluated oral tablets (Chronotropic) to achieve time and / or site release of antipyrine. The prepared
enteric-coated tablets were found to retard the release of drug with site
specificity. Results indicated a delayed drug release for a specified time.
Turkoglu and Ugurlu., 2002, (90) reported the tablets containing 5-amino- salicylic acid for colonic delivery. Tablets were compression coated with
different ratios of pectin and HPMC. Drug dissolution studies were carried out in
pH 1.2 and 6.8 buffers using a pectinolytic enzyme. They concluded that pectin
alone was not sufficient to protect the core tablets and HPMC addition was
required to control the release of drug.
Lamprecht et al., 2003, (91) designed pH sensitive microspheres for the colonic delivery of the immunosuppressive drug tacrolimus. Eudragit P-4135F,
a pH-sensitive polymer for colonic delivery was used to prepare tacrolimus
microparticles using an oil/oil emulsification or an oil/water emulsification technique
combined with a solvent extraction or evaporation. Although the pH-dependent drug
release was similar for all types of microspheres, it was found that an encapsulation
rate of oil/water systems was superior to the oil/oil emulsification.
Akhgari et al., 2005, (92) statistically optimized the pH dependent methacrylic acid polymer coated in different ratios on indomethacin pellets for
colonic drug delivery. They found that factorial design is a suitable tool for