0022-538X/97/$04.0010
Copyrightq1997, American Society for Microbiology
An Amino-Terminal Domain of the Sendai Virus Nucleocapsid
Protein Is Required for Template Function
in Viral RNA Synthesis
TINA M. MYERS†ANDSUE A. MOYER*
Department of Molecular Genetics and Microbiology and Department of Pediatrics, University of Florida, College of Medicine, Gainesville, Florida 32610
Received 13 June 1996/Accepted 15 October 1996
The nucleocapsid protein (NP) of Sendai virus encapsidates the genome RNA, forming a helical nucleocap-sid which is the template for RNA synthesis by the viral RNA polymerase. The NP protein is thought to have both structural and functional roles, since it is an essential component of the NP0-P (P, phosphoprotein),
NP-NP, nucleocapsid-polymerase, and RNA-NP complexes required during viral RNA replication. To identify domains in the NP protein, mutants were constructed by using clustered charge-to-alanine mutagenesis in a highly charged region from amino acids 107 to 129. Each of the mutants supported RNA encapsidation in vitro. The product nucleocapsids formed with three mutants, NP114, NP121, and NP126, however, did not serve as templates for further amplification in vivo, while NP107, NP108, and NP111 were nearly like wild-type NP in vivo. This template defect in the NP mutants from amino acids 114 to 129 was not due to a lack of NP0-P,
NP-NP, or nucleocapsid-polymerase complex formation, since these interactions were normal in these mutants. We propose that amino acids 114 to 129 of the NP protein are required for the nucleocapsid to function as a template in viral genome replication.
The nucleocapsid proteins (NP) of paramyxoviruses play important and unique roles in viral genome RNA replication. The viruses contain nonsegmented, single-stranded negative-sense RNA genomes that are tightly associated with the nu-cleocapsid protein, forming an RNase-resistant helical nucleo-capsid (RNA-NP), which is the template for RNA synthesis (16, 18; for a review see reference 24). The Sendai virus nu-cleocapsid is approximately 15 nm by 1mm, with 2,564 mole-cules of the NP protein (524 amino acids [aa]) associated with the genomic RNA in a ratio of 1 polypeptide per 6 nucleotides (6). The NP protein must interact with itself, an NP-NP inter-action, and with the viral RNA (RNA-NP) within the nucleo-capsid. Biochemical evidence for NP-NP binding was shown by the self-assembly of the NP protein into nucleocapsidlike par-ticles which apparently contained nonspecific cellular RNA (4). Previously, a soluble NP0-P complex was identified as the
substrate for encapsidation of nascent genome RNA during replication (21). The polymerase complex, consisting of the phosphoprotein (P) and the large (L) proteins, is associated with the nucleocapsid template via the P moiety and catalyzes both transcription and replication (3, 22, 27, 30, 31).
Deletion analysis of the Sendai virus NP protein has shown that the domains required for the NP protein to function in RNA replication in vitro and to self-assemble into nucleocap-sidlike particles are within the N-terminal 400 aa (4, 12). Of 22 deletion mutants, all those with deletions from aa 1 to 400 were inactive, while those containing any deletions from aa 400 to 524 were active in both RNA replication and self-assembly. While the C-terminal 124 aa of the NP protein were thus not required for genome synthesis in vitro, which represents the encapsidation of nascent RNA synthesized from a virus tem-plate containing the wild-type (wt) NP protein, they were re-quired for a template function in replication in vivo. Template
function has been defined as the ability of the genome RNA encapsidated with a mutant NP protein to serve as a template for further rounds of genome replication (12). A P binding site on NP assembled into nucleocapsids has been mapped to the C-terminal region of NP. Antibodies recognizing aa 405 to 524 caused P protein to be released from nucleocapsids and pre-vented exogenous P from binding (32). Furthermore, nucleo-capsids containing deletions in NP of aa 426 to 496 or 456 to 524 lost the P interaction, whereas those with deletions of aa 400 to 415 or 414 to 439 still bound P protein (3). These data suggest that the residues between 400 and 439 are required for an as yet undefined template function which is separate from binding the P protein.
The deletion analysis did not identify independent domains for the NP-NP, RNA-NP, and soluble NP0-P interactions,
which must reside within the N-terminal 400 aa of NP. This region is largely hydrophobic, with a few clusters of charged residues (aa 60 to 71, 107 to 129, and 391 to 397) (4, 12, 28, 33). Two of the charged clusters have been postulated to be RNA-binding domains (aa 60 to 72 and 107 to 115), since the basic nature of the majority of these amino acids could lead to an interaction with the phosphate backbone of the RNA (4, 28). Although there are no direct data, some of these residues are conserved among the parainfluenza virus NP proteins but not generally among other members of the paramyxovirus family (26).
To identify residues of the NP protein required for these various interactions, we selected clustered charge-to-alanine mutagenesis because of the likelihood of targeting surface res-idues, which are potential sites for protein or protein-RNA interactions (1, 2, 10, 35). Previously charge-to-alanine mutagenesis has been used to identify residues on the surface of proteins important for protein-protein interactions (1, 2, 34), for catalytic activities (14), and to create mutants having temperature-sensitive (ts) phenotypes (14, 17, 35). Based on this approach, we present evidence that charged residues in the
* Corresponding author.
† Present address: Department of Molecular Microbiology, Wash-ington University School of Medicine, St. Louis, MO 63110-1093.
918
on November 9, 2019 by guest
http://jvi.asm.org/
N terminus of the Sendai virus NP protein are required for the nucleocapsid to function as a template in viral RNA synthesis.
MATERIALS AND METHODS
Cells and viruses.Wild type Sendai virus and the Sendai virus defective interfering (DI) particle, DI-H (Harris strain), were propagated in the allantoic fluid of 9-day-old embryonated chicken eggs as described previously (21). Re-combinant vaccinia virus containing the gene for phage T7 RNA polymerase (VVT7) (15) was grown in Vero cells. Protein and RNA synthesis were per-formed in human A549 cells (American Type Culture collection).
Plasmids and antibodies.Plasmids pGEM-NP, pGEM-P/C, and pGEM-L were described previously (11). Plasmids pBS-NP and pTM1-GST-P, containing the glutathione S-transferase (GST) gene fused in frame to the Sendai virus P gene (GST-P), were described before (references 9, and 8, respectively). All of the viral genes were cloned downstream of the phage T7 promoter. A DNA clone of DI-H was obtained by reverse transcription-PCR of nucleocapsid RNA iso-lated from A549 cells coinfected with Sendai virus and DI-H as described pre-viously (5). The hepatitis delta virus ribozyme and T7 terminator, generously provided by L. A. Ball (University of Alabama) (29), were subcloned 39to the DI-H sequence to create pSPDI-H. The sequence of the DI-H clone was con-firmed by restriction endonuclease mapping and sequencing. Transcription of the DI-H clone by T7 RNA polymerase generates a full-length plus-sense DI-H RNA with the correct termini. Immunoprecipitation and immunoblotting used the following antibodies: a rabbit anti-Sendai virus (a-SV) antibody (7) and a rabbit anti-L (a-L) antibody specific for the Sendai virus L protein (21).
Construction of NP mutants.Charge-to-alanine mutagenesis targeted aa 107 to 129 of NP, generating six mutants containing the alanine substitutions shown in Fig. 1 and 2. PCR was used to create the mutants NP108, NP111, NP114, NP121, and NP126, using two overlapping complementary mutagenic oligode-oxynucleotide primers and two standard outside oligodeoligode-oxynucleotide primers (Table 1) with pGEM-NP as the template by the method of Higuchi et al. (19). The PCR products were digested with EcoRI and AflII and subcloned into those sites in pGEM-NP. For screening of mutant clones, each mutagenic primer introduced a new silent restriction site as shown in Table 1. The mutant NP146-6, containing a single charge-to-alanine substitution at aa 107 of NP, was created by using pBS-NP and an Oligonucleotide-directed in vitro Mutagenesis System Version 2.1 kit (Amersham) according to the manufacturer’s protocol. A
KpnI-AflII DNA fragment from pNP146-6 was subsequently subcloned into those sites
in pGEM-NP, creating pNP107. The mutations in each clone were confirmed by double-stranded dideoxy sequencing of the entire subcloned fragment.
In vitro RNA synthesis and encapsidation.Subconfluent A549 cells in 60-mm-diameter dishes (approximately 4.83106cells) were infected with VVT7 at a
multiplicity of infection of 2.5 PFU/cell and transfected with one or more plas-mids at 378C. Unless indicated otherwise, the amounts of plasmid transfected per
dish were as follows: wt or mutant pGEM-NP, 2mg; pGEM-P/C, 5mg; and pGEM-L, 0.5mg. Cytoplasmic cell extracts were prepared at 18 h post transfec-tion (p.t.) by lysolecithin permeabilizatransfec-tion at 48C, and DI-H RNA synthesis and encapsidation was assayed in vitro with the addition of [a-32
P]CTP and deter-gent-disrupted (dd), purified DI-H (2.2mg) as described previously (21). The nucleocapsid products were nuclease treated and purified by banding on CsCl gradients, and the RNA was extracted and analyzed by electrophoresis on 1.5% agarose-acid-urea gels. The replication products were quantitated with a PhosphorImager (Molecular Dynamics).
In vivo replication was measured by cotransfecting pSPDI-H (2.5mg) with the plasmids containing genes for the P, L, and wt or mutant NP proteins into VVT7-infected cells. DI-H RNA replication was measured either by making cell extracts and assaying encapsidation in vitro as described above or by Northern analysis. For Northern analysis, infection and transfection were as described above except that 100-mm-diameter dishes (approximately 1.83107
cells) were used and the amounts of plasmids transfected were increased proportionately. The cells were then incubated at either 32 or 378C for 36 or 22 h, respectively. Cell extracts were prepared, and the nuclease-resistant replication products were purified as described above. The extracted RNA was identified by blot analysis using a denatured [a-32
P]dCTP-labeled, random-primed DI-H probe (Pharma-cia), and the product RNA was quantitated on the PhosphorImager.
Portions of the in vitro extracts (10%) were analyzed by immunoblotting using ana-SV primary antibody and an alkaline phosphatase-conjugated secondary antibody. Enhanced chemiluminescence (ECL; Amersham Life Science) was used to detect the proteins in the in vivo replication extracts according to the manufacturer’s protocol.
Protein analysis.VVT7-infected cells transfected with the wt or mutant NP plasmid were labeled with Tran35
S-label (66mCi/ml) at 5.5 h p.t. for 30 min in methionine- and cysteine-free medium, and cell extracts were prepared imme-diately (pulse) or following an overnight chase with medium containing 10-fold excess methionine and cysteine. For overnight labeling, the cells were labeled in medium containing 0.13unlabeled methionine and cysteine. Nonidet P-40 cell extracts were prepared, and samples were immunoprecipitated with the appro-priate antibody (1ml) as indicated in the figure legends and collected with inactivated Staphylococcus aureus Cowan strain as described previously (7). For analysis of NP0-P complex formation, the wt or mutant NP and pTM1-GST-P (1
[image:2.612.75.278.70.219.2]mg) plasmids were transfected as indicated in the figure legends, samples of extracts (75ml) were either immunoprecipitated or incubated with glutathione-Sepharose 4B beads (15ml per reaction; Pharmacia Biotech) for 15 min at 48C and washed according to the manufacturer’s protocol, and the immunoprecipi-tated and bound proteins were analyzed by sodium dodecyl sulfate-polyacrylam-ide gel electrophoresis (SDS-PAGE) (9% gel).
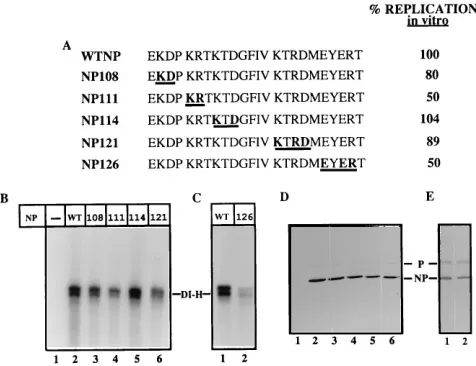
FIG. 1. Pulse-chase analysis and in vitro replication with the mutant NP107. (A) Amino acid sequences (aa 107 to 130) of wt NP and mutant NP107 proteins with the underlined letter changed to Ala. (B) VVT7-infected A549 cells were transfected in duplicate with no plasmids (2) or the wt (WT) or mutant NP (107) plasmid, as indicated at the top. The cells were pulse-labeled (P), and extracts were prepared immediately or following a chase (C) as described in Materials and Methods, immunoprecipitated witha-SV antibody, and analyzed by SDS-PAGE. The position of the NP protein is indicated. (C) VVT7-infected cells were transfected with no plasmids (2) or the P and L plasmids together with the indicated wt or mutant NP plasmid. Cytoplasmic cell extracts were prepared and incubated with dd DI-H in the presence of [a-32P]CTP. The nucleocapsid
prod-ucts were purified, and RNA was extracted and analyzed by gel electrophoresis as described in Materials and Methods. The position of the DI-H RNA is indicated, and the amount of product DI-H RNA was quantitated on a PhosphorImager and calculated relative to the value for wt NP as 100% (A).
FIG. 2. In vitro DI-H RNA synthesis with the charge-to-alanine mutant NP proteins. (A) Amino acid sequence (aa 107 to 130) of wt and mutant NP proteins with the boldface underlined letters changed to Ala and the percent replication relative to that of wt NP as 100%. (B and C) VVT7-infected cells were trans-fected with no plasmids (2) or the P and L plasmids together with the indicated wt or mutant NP plasmid. Cytoplasmic cell extracts were assayed for in vitro replication and quantitated as described for Fig. 1. The values in panel A represent this experiment, and similar results were obtained in multiple (two to four) experiments. NP121 was assayed relative to wt (not shown) in a separate experiment. (D and E) Immunoblot analysis of samples witha-SV antibody as described in Materials and Methods. The positions of the NP and P proteins are indicated. The sample numbers in panels B and C correspond to those in panels D and E, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.318.556.436.619.2]Self-assembly of NP proteins and polymerase binding.To measure self-as-sembly of the wt and mutant NP proteins, cells were infected and transfected as described above with the wt and mutant NP plasmids, and Nonidet P-40 cyto-plasmic cell extracts were prepared between 18 and 20 h p.t. The cell extracts were fractionated by sedimentation on separate CsCl step gradients as described previously (4) in an SW55 rotor at 36,000 rpm for 16 h at 48C. Fractions (0.714 ml) were collected from the top, and the pellet was resuspended in the last fraction. A portion of each fraction (25ml) was analyzed by immunoblotting with ana-SV primary antibody and an alkaline phosphatase-conjugated secondary antibody. The density of each fraction was determined with a refractometer.
To determine polymerase binding to nucleocapsids, VVT7-infected A549 cells were transfected with no plasmids (mock) or the wt or mutant NP plasmid (2mg) (self-assembled nucleocapsid), and multiple dishes were cotransfected with the P and L plasmids (5 mg of each) (polymerase). At 5.5 h p.t., the cells were incubated for 3 h with Tran35S-label (66mCi/ml) in methionine- and
cysteine-free medium, and cytoplasmic extracts were prepared (22). The mock and the self-assembled wt or mutant nucleocapsids were purified by pelleting through 30% (vol/vol) glycerol (5 ml) in an SW55 rotor at 50,000 rpm for 90 min at 48C, and the pellets were resuspended. Equal samples (100ml) of the polymerase extract were incubated for 1 h at 308C either with the purified mock or assembled nucleocapsid samples or with polymerase-free, purified DI-H RNA-NP. The samples were sedimented through glycerol (22), the pellets immunoprecipitated witha-SV anda-L antibodies, and the proteins were separated by SDS-PAGE (7.5% gel).
RESULTS
Effect of charge-to-alanine mutagenesis of the NP protein on Sendai virus RNA replication in vitro.We constructed charge-to-alanine mutations in the region from aa 107 to 129 (Fig. 1 and 2) as described in Materials and Methods to test for possible functions of this site in viral RNA replication. Initially, a single mutation (Glu to Ala, E107A) at aa 107 (NP107 [Fig. 1A]) was constructed. VVT7-infected cells were transfected in duplicate with the plasmids for wt NP or NP107, pulsed with Tran35S-label, and chased as described in Materials and
Meth-ods. Immunoprecipitation of the proteins showed that wt NP and NP107 were similarly expressed in the pulse (Fig. 1B, lanes 3 and 5) and both were relatively stable to an overnight chase (lanes 4 and 6). The other bands are vaccinia virus proteins that were nonspecifically immunoprecipitated with the anti-body as seen in the VVT7-infected but not transfected cell extracts (Fig. 1B, lanes 1 and 2).
To test the biological activity of the mutant, extracts of cells expressing wt NP or NP107 together with the P and L poly-merase proteins were incubated with dd DI-H and assayed for genome RNA synthesis and encapsidation in vitro as described in Materials and Methods. Analysis of the nucleocapsid RNA products showed that replication with the NP107 mutant was
actually somewhat better (141%) than that with wt NP (Fig. 1C, lanes 3 and 2, respectively). As expected, activity was dependent on the expression of the viral proteins, since no product was observed in an extract of VVT7-infected but not transfected cells (Fig. 1C, lane 1). Amino acid 107 is, therefore, not essential for the function of the protein in this assay.
Based on these results, we decided to change clustered groups of charged residues to Ala in the remaining five mu-tants (Fig. 2A) in an attempt to produce stable proteins with mutant phenotypes. The NP108, NP111, NP114, NP121, and NP126 proteins were all relatively stable, as shown by pulse-chase (data not shown) and immunoblot (Fig. 2D and E) analyses. These mutants were then tested for activity in DI-H RNA replication in vitro. Compared to replication of the DI-H template with wt NP protein (100% [Fig. 2B, lane 2]), mutants NP108, NP114, and NP121 gave nearly equal or equivalent product (Fig. 2B, lanes 3, 5, and 6). RNA synthesis and encap-sidation with NP111 and NP126 were somewhat more reduced but still significant, at 50% of levels for wt NP (Fig. 2B, lane 4, and Fig. 2C, lane 2, respectively). The proteins were all ex-pressed in these extracts, as shown by immunoblot analysis (Fig. 2D, lanes 2 to 6; Fig. 2E, lanes 1 and 2). These data show that the charged residues encompassing aa 107 to 129 in the NP protein can be changed in clusters to Ala without drasti-cally affecting the ability of the proteins to encapsidate nascent RNA in vitro.
NP0-P complex formation is conserved in the mutant NP
proteins.One of the requirements for Sendai virus RNA rep-lication is the formation of the encapsidation substrate, the NP0-P complex (13, 21). Since all of the mutants were active in
vitro to some degree, we expected that they were forming NP0-P complexes, but this needed to be confirmed based on
previous results with the measles virus nucleocapsid (MV N) protein (8). In this case, we showed that the MV N protein supported Sendai virus encapsidation in vitro, yet this activity did not depend on complex formation between the MV N and Sendai virus P proteins. To assay the NP0-P complex, we used
a fusion protein (GST-P) containing the GST protein linked to the N terminus of P protein and measured the cobinding of NP with GST-P to glutathione-Sepharose beads.
The wt and mutant NP plasmids were transfected individu-ally and together with the GST-P plasmid into VVT7-infected cells and incubated with Tran35S-label. Immunoprecipitation
[image:3.612.51.557.82.245.2]of cell extracts showed that significant levels of wt NP and
TABLE 1. Oligodeoxynucleotide primers
Mutant(s)
Primera
Mutagenic primer Standard outside Enzyme
NP108 SM212(1)-CATAGAGGCGGCGCCTAAGAGGACG NarI
SM213(2)-CCTCTTAGGCGCCGCCTCTATGTTG
NP111 SM214(1)-GACCCTGCGGCGACGAAGACAGAC BsoFI
SM215(2)-TGTCTTCGTCGCCGCAGGGTCTTTCTC
NP114 SM216(1)-AGGACGGCGACAGCCGGCTTCATTGTGAAGACG NaeI
SM217(2)-AATGAAGCCGGCTGTCGCCGTCCTCTTAGG
NP121 SM218(1)-GTGGCGACCGCGGCTATGGAATATGAGAGG SacII
SM219(2)-TTCCATAGCCGCGGTCGCCACAATGAATCCGTC
NP126 SM220(1)-GATATGGCATATGCGGCGACCACAGAATGG NdeI
SM221(2)-TGTGGTCGCCGCATATGCCATATCTCTGC
NP146-6 SM146(1)-CTACAACATAGCGAAAGACCC None
NP108, NP111, NP114, NP121, and NP126
SM030(1)-TAATACGACTCACTATAG SM123(2)-GGGAGCTCTGGGGCC
aThe plus or minus sign refers to the messenger sense or genomic sense, respectively, of the oligonucleotide, and the sequences are written 59339. The restriction
sites used for cloning and/or screening are underlined.
on November 9, 2019 by guest
http://jvi.asm.org/
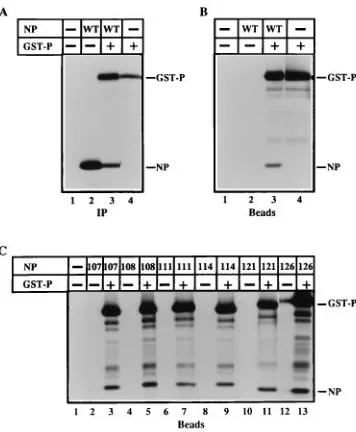
GST-P were expressed (Fig. 3A, lanes 2 to 4). As a positive control, GST-P expressed alone bound to the beads (Fig 3B, lane 4), while proteins from VVT7-infected but not transfected cells did not (Fig. 3B, lane 1). The faster-migrating bands (Fig. 3B, lane 4) are apparently proteolysis products containing GST. Wild-type NP protein formed a complex with GST-P, since NP cobound to the beads when coexpressed with GST-P (Fig. 3B, lane 3) but not when NP was synthesized alone (lane 2). The NP protein does not form a complex with GST alone (data not shown); therefore, the cobinding of NP with GST-P is specific for the P moiety of the fusion protein. Thus, this assay also measures NP0-P complex formation which had
pre-viously been demonstrated by blotting (20), coimmunoprecipi-tation (21), and cosedimencoimmunoprecipi-tation (13, 21). Similarly, each of the mutant proteins was synthesized (data not shown) and formed complexes with the P protein to the same extent as wt NP, as evidenced by their cobinding to beads in the presence, but not the absence, of GST-P (Fig. 3C).
The mutant NP proteins can self-assemble into nucleo-capsidlike particles. In addition to the NP0-P complex, NP
interacts with itself and self-assembles into nucleocapsidlike particles (4). We wanted to test if the differences in the encap-sidation ability of the mutants correlate with their degree of self-assembly. The individual wt or mutant NP proteins were expressed in cells, and extracts were banded on CsCl step gradients. Fractions were collected, and samples were analyzed by immunoblotting as described in Materials and Methods. Consistent with previous data (4), the majority of wt NP self-assembled into nucleocapsidlike particles that banded in frac-tion 5 at the 30 to 40% interface (Fig. 4B) like that of authentic nucleocapsids (4, 25). Unassembled NP protein was previously shown to sediment in fractions 1 to 4 under these conditions (4). VVT7-infected, mock-transfected cells showed no viral
protein (Fig. 4A), as expected. Each of the mutants banded similarly to wt NP protein (Fig. 4C to G), suggesting that each self-assembled into nucleocapsidlike particles. For NP126 (Fig. 4G), the densities of fractions 4 and 5 in this particular gradi-ent were close, and so some protein was found in fraction 4 as well. These data show that the substitution of Ala for the charged residues between aa 107 and 129 did not seem to disrupt the NP-NP interaction, and so the 50% decrease seen in replication in vitro for two of the mutants is apparently not due to this function of the protein.
Charge-to-alanine mutagenesis of the NP protein disrupts replication in vivo.One limitation of the in vitro replication-encapsidation assay is that it does not measure if the genome RNA encapsidated by the mutant protein can subsequently be used as a template for further rounds of replication. To mea-sure this template function, we used a DI-H clone (pSPDI-H) (see Materials and Methods) as described previously (12). Fol-lowing cotransfection of the DI-H plasmid with the NP, P, and L plasmids into VVT7-infected cells, plus-strand DI-H RNA is transcribed by T7 RNA polymerase and encapsidated by the expressed NP protein. The plus-strand DI-H RNA-NP is then replicated by the viral proteins to amplify the template. Rep-lication of the endogenous template with the wt proteins was detected following preparation of cell extracts by labeling in vitro with [a-32P]CTP (Fig. 5B, lane 2). The DI-H clone is not
replicated in the absence of the Sendai virus proteins (Fig. 5B, lane 1), as expected. Compared to replication with wt NP as 100%, the RNAs encapsidated by the mutants NP107, NP108, and NP111 showed somewhat reduced (71, 68, and 69%, re-spectively [Fig. 5B, lanes 3 to 5]), but still significant levels of
FIG. 3. Complex formation between GST-P and the wt and mutant NP proteins. VVT7-infected cells were transfected with no plasmids (2) or combi-nations of the wt or mutant NP plasmids and GST-P plasmid as indicated. The cells were incubated overnight with Tran35S-label, cytoplasmic cell extracts were
[image:4.612.89.267.68.284.2]prepared, and samples were either immunoprecipitated (IP) witha-SV antibody (A) or incubated with glutathione-Sepharose beads (B and C), and the bound proteins were analyzed by SDS-PAGE as described in Materials and Methods. The positions of the NP and GST-P proteins are indicated. The differences in the apparent migration of the NP121 and NP126 proteins in panel C are due to the use of data from separate gels.
FIG. 4. Wild-type and mutant NP proteins self-assemble into nucleocapsid-like particles. VVT7-infected cells were transfected with no plasmids (mock) or the wt or mutant NP plasmid. At 20 h p.t., unlabeled cell extracts were prepared and banded on separate CsCl step gradients, and fractions were collected from the top of the gradient as described in Materials and Methods. Sedimentation is from left to right. Samples were separated by SDS-PAGE and analyzed by immunoblotting witha-SV antibody. The positions of the wt and mutant NP proteins are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
replication of the amplified endogenous templates. DI RNAs encapsidated by mutants NP114 and NP126, however, were significantly impaired in template function (28 and 18%, re-spectively [Fig. 5B, lanes 6 and 8]), while RNA encapsidated by NP121 was completely inactive (Fig. 5B, lane 7). Immunoblot analysis on samples of the cell extracts demonstrated that the wt and mutant NP proteins were similarly expressed (Fig. 5C, lanes 2 to 8). Although the NP114, NP121, and NP126 proteins were capable of encapsidating the nascent RNA during the first round of replication using DI-H encapsidated with wt NP as a template (Fig. 2), the product RNAs encapsidated by these mutants were defective to various degrees as templates for further rounds of replication.
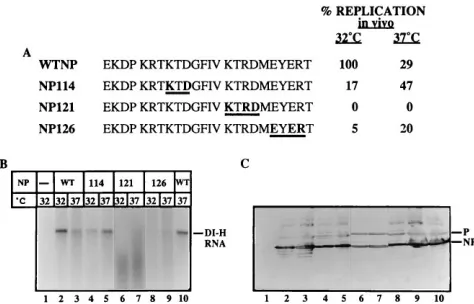
Clustered charge-to-alanine mutations in a variety of pro-teins have been shown to yield a proportion of ts mutants (14, 17, 35). We tested if the NP mutants defective in vivo, NP114, NP121, and NP126, have a temperature conditional phenotype by incubating the infected and transfected cells at 37 or 328C. Nucleocapsid product RNAs were detected by Northern anal-ysis with a DI probe as described in Materials and Methods. This method of analysis does not require the temperature shift between expression in cells (378C) and the assay (308C) that was necessitated by the in vitro experiments (Fig. 5). An un-labeled in vitro replication product from dd DI-H provides a marker (Fig. 6B, lane 10). There was no DI RNA in cells transfected only with the DI-H plasmid, showing that genome amplification was dependent on expression of the viral proteins (Fig. 6B, lane 1). The nucleocapsid RNA product produced with wt NP was reproducibly about threefold greater at 328C than at 378C (Fig. 6B, lanes 2 and 3). Replication with NP114, however, was about threefold greater at 378C than at 328C (Fig. 6B, lanes 5 and 4, respectively), suggesting that this mu-tant has a cold sensitive (cs) phenotype. In vivo replication with NP126 was significantly inhibited at both temperatures (5 and
20% [Fig. 6B, lanes 8 and 9]). Although these data are also suggestive of a cs phenotype for NP126, at these low levels of replication, this remains speculative. The mutant NP121 was completely inactive in the replication of genomic-length RNA at both temperatures (Fig. 6B, lanes 6 and 7). There were, however, some smaller RNA products produced with NP121 that may represent incomplete replication products which were not detected in the analysis in vitro. Immunoblot analysis showed that the wt and mutant NP proteins and P protein were all expressed (Fig. 6C). The amount of NP121 in this experi-ment was somewhat reduced, but this would not account for the lack of activity of this protein in vivo. The other bands are vaccinia virus proteins staining nonspecifically in the ECL as-say.
Binding of the RNA polymerase to the wt and mutant self-assembled nucleocapsidlike particles. One requirement for viral RNA replication is the recognition of and binding of the viral polymerase complex through the P moiety to the template (3, 22, 27, 30, 31). To determine if the template defects were due to the inability of the polymerase to bind to the mutant nucleocapsids, proteins representing each defective phenotype seen in replication in vivo, significant (NP111), reduced (NP126), and no (NP121) activity, were self-assembled by the expression in VVT7-infected and transfected cells in the pres-ence of Tran35S-label. The radiolabeled nucleocapsidlike
par-ticles were purified by sedimentation and incubated with ex-tracts expressing the radiolabeled P-L complex and, as a positive control, with purified polymerase-free DI-H (RNA-NP template). The samples were then sedimented through glycerol, and the pellets were collected and immunoprecipi-tated to monitor the proteins associated with the nucleocap-sids.
[image:5.612.62.298.69.243.2]The P and L proteins copelleted and, therefore, bound in the presence of both the viral RNA-NP template and the wt nucleocapsidlike particles (Fig. 7, lanes 6 and 1, respectively), with only a small amount of the polymerase proteins pelleting
FIG. 5. Template function of the DI-H RNA-NPs assembled in vivo with wt or mutant NP proteins. (A) Amino acid sequence (aa 107 to 130) of the wt and NP mutant proteins with the boldface underlined amino acids changed to Ala. The level of replication was assayed in vitro with each mutant template as described for Fig. 1. (B) VVT7-infected cells were transfected with the DI-H plasmid alone (2) or together with P and L plasmids and the indicated wt or the mutant NP plasmid. Cytoplasmic cell extracts were assayed for in vitro replica-tion of the endogenous template as described in Materials and Methods. The position of the DI-H RNA is indicated, and the amount of product DI-H RNA was quantitated on a PhosphorImager. Lane 3 is from a separate gel. The values in panel A represent this experiment, but similar results were obtained in mul-tiple experiments. (C) Immunoblot analysis of the extracts was performed as described for Fig. 2. A bubble in the blot (lane 4) blocked visualization of P protein.
FIG. 6. Northern analysis of DI-H RNA replication in vivo. (A) Amino acid sequence (aa 107 to 130) of the wt and the mutant NP proteins with the boldface underlined letters changed to Ala. In vivo replication with the wt and mutant NP proteins at 32 and 378C is shown relative to that of wt NP at 328C as 100%. (B) VVT7-infected cells were transfected in duplicate with the DI-H, L, and P plasmids together with the indicated wt or mutant NP plasmid and incubated at either 32 or 378C. Cytoplasmic cell extracts were prepared and analyzed by Northern blotting with a32P-DI-H probe as described in Materials and Methods.
An unlabeled in vitro reaction with dd DI-H and an extract from cells transfected with the NP, P, and L plasmids was included as a marker (lane 10). The position of the DI-H RNA is indicated. The values in panel A represent this experiment, but similar results were obtained in two to three experiments. (C) Immunoblot analysis witha-SV antibody on a portion of the samples, using ECL as described in Materials and Methods. The positions of the NP and P proteins are indicated. Lanes 6 and 7 are from a separate blot.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.317.554.452.604.2]in the absence of nucleocapsids (lane 5), in agreement with previous data (22). The polymerase complex also bound to each of the three mutant nucleocapsidlike particles (Fig. 7, lanes 2 to 4) at levels about equal to those for wt nucleocap-sidlike particles. The additional bands are vaccinia virus pro-teins that pelleted and were nonspecifically immunoprecipi-tated in this assay. Since all of these mutant particles still bind the viral polymerase, these data suggest that the template de-fect seen in NP121 and NP126 during replication in vivo is due to disruption of a separate, as yet unidentified function of NP.
DISCUSSION
Clustered charge-to-alanine mutagenesis of the Sendai virus NP protein has identified a region from aa 114 to 129 that was required for the nucleocapsid to function as a template in RNA replication (Fig. 5 and 6) but was not required for the initial round of RNA encapsidation (Fig. 2). It was somewhat surprising that changing a total of 13 charged aa to alanine in these mutant proteins had little effect on in vitro encapsidation (Fig. 1 and 2), since deletion analysis of the NP protein by Curran et al. (12) had shown that the entire region from aa 1 to 400 was required. In fact, a deletion of residues encompass-ing this region (aa 107 to 131) gave an unstable protein. These conflicting results are likely due to the more drastic effect that deletions could have on protein structure as opposed to the Ala substitutions. The mutant proteins encompassing aa 107 to 129 appear overall to be correctly folded, since each formed both NP0-P and NP-NP complexes and could support RNA
encapsidation in vitro (Fig. 2 to 4).
When RNA encapsidation and amplification were assayed entirely at 378C (Northern assay) as compared to encapsida-tion and amplificaencapsida-tion at 378C followed by an in vitro assay at 308C (in vitro labeling), some template function was restored to the mutant nucleocapsid containing NP114 (Fig. 5 and 6). In addition, NP114 replication at 378C was about threefold better than at 328C, suggesting that this mutant has a cs phenotype. In
fact at 378C, NP114 is twofold more active than wt NP. It appears that the conformation of NP114 at 308C inhibits its ability to function in the template, but an active structure is maintained at 378C. Both ts and cs mutants have been de-scribed previously in charge-to-alanine-scanning mutants (14, 17, 35).
One possibility for the observation that the nucleocapsids formed with the mutants NP114, NP121, and NP126 were defective as templates for replication (Fig. 5 and 6) could be that the RNA polymerase could not bind. Binding of the P-L complex to the template is mediated through the P protein (22, 27, 30, 31). We showed that the viral polymerase (P-L com-plex) did bind to NP121 and NP126 self-assembled nucleocap-sids at wt levels (Fig. 7), suggesting that this is not the template defect. Interestingly, it was shown previously that four dele-tions within the C-terminal 124 aa of NP had defective tem-plate functions similar to those exhibited by NP114, NP121, and NP126, and they also formed NP-NP complexes and en-capsidated RNA in vitro (12). A P binding site has been mapped within the C terminus of the self-assembled NP pro-tein between aa 440 and 524 (3, 32), and so the mutants containing deletions of aa 426 to 497 or 456 to 524 could not replicate in vivo because the polymerase could not bind. How-ever, the mutants with deletions of aa 400 to 415 or 414 to 439, like NP114, NP121, and NP126, could not replicate in vivo either yet bound the P protein.
In combination, these data suggest that at least two sites are required for template function not related to polymerase bind-ing. One is located in the C-terminal 400 to 439 aa, and the other is from aa 114 to 129. The similar phenotypes of mutants in both regions suggest that they may form a single domain in the mature protein. While aa 114 to 129 are quite conserved in the parainfluenza viruses, this is not the case for the other paramyxoviruses. Only aa 121 has a conserved basic charge in 13 different NP proteins (26). These data suggest that a basic amino acid is the crucial one in mutant NP121, which had altered two additional charged but nonconserved residues. In other NP proteins, different regions may be involved in the template function.
Several models can be envisioned for the role of these res-idues in template function, depending on whether these sites form one domain or two and whether the bases are exposed or protected in the nucleocapsid. If the bases are covered by the NP protein and aa 114 to 129 and 400 to 439 form a single domain in the NP protein, perhaps this domain is required for the transient displacement of NP to allow the viral polymerase access to the template during RNA synthesis (12). RNA syn-thesis does not appear to change the nuclease-resistant char-acter of the template; therefore, the displacement of NP must be minimal and/or something else may take its place. Perhaps the NP protein is transiently removed by its binding to the P subunit of the RNA polymerase, via aa 114 to 129 and 400 to 439, with the L subunit filling the gap. Following synthesis, the displaced NP protein would be returned to the template. It had indeed also been proposed that in vesicular stomatitis virus, the NS (P protein) protein may mimic the template RNA and act as a transient binding site for the N protein (23).
Alternatively, the mutations in the template function do-main may have resulted in an increased affinity of NP for either the RNA (NP-RNA interaction) or the adjacent NP proteins (NP-NP interaction) which might prevent the temporary dis-placement of the mutant NP protein from the RNA or from adjacent NP proteins. This increased affinity would not affect in vitro replication utilizing a viral RNA template that is encap-sidated by wt NP protein. If the bases are accessible on the surface of the nucleocapsid, where the NP protein does not
FIG. 7. The viral polymerase binds to wt and mutant self-assembled nucleo-capsidlike particles. VVT7-infected cells were transfected with no plasmids (2) or the wt or the mutant NP plasmid (nucleocapsidlike particles), or separately transfected with the P and L plasmids (polymerase), and incubated with Tran35
S-label, and cell extracts were prepared. The mock extract and the nucleocapsidlike particles were purified by pelleting through glycerol. Equal aliquots of the poly-merase extract were incubated with mock (lane 5), each of the purified nucleo-capsidlike particles (NUC; lanes 1 to 4), and purified polymerase-free DI-H (lane 6). The samples were pelleted through glycerol, and the bound proteins were analyzed by SDS-PAGE. The positions of the NP, P, and L proteins are indi-cated.
on November 9, 2019 by guest
http://jvi.asm.org/
need to be displaced, alternative, as yet undefined interactions which these NP mutations have disrupted must be occurring. One possibility is that the mutated residues are involved in the intra- rather than intermolecular interactions of NP and are required for NP assembled in nucleocapsids to undergo con-formational changes during viral RNA synthesis.
In summary, by charge-to-alanine mutagenesis, we have con-structed six stable mutant NP proteins which have identified a second region (aa 114 to 129) of the NP protein, in addition to the previously identified C-terminal 400 to 439 aa, which is required for template function of the nucleocapsid. Further studies will be necessary to determine the nature of the defect and if these two sites form one domain or fulfill separate functions.
ACKNOWLEDGMENTS
We thank Sherin Smallwood and Cheryl Zack for technical assis-tance and Sandra Horikami for helpful discussions and advice.
This work was supported by grant AI 14594 from the National Institutes of Health.
REFERENCES
1. Bass, S. H., M. G. Mulkerrin, and J. A. Wells. 1991. A systematic mutational analysis of hormone-binding determinants in the human growth hormone receptor. Proc. Natl. Acad. Sci. USA 88:4498–4502.
2. Bennett, W. F., N. F. Paoni, B. A. Keyt, D. Botstein, A. J. S. Jones, L. Presta,
F. M. Wurm, and M. J. Zoller.1991. High resolution analysis of functional determinants on human tissue-type plasminogen activator. J. Biol. Chem.
266:5191–5201.
3. Buchholz, C. J., C. Retzler, H. E. Homann, and W. J. Neubert. 1994. The carboxyl-terminal domain of Sendai virus nucleocapsid protein is involved in complex formation between phosphoprotein and nucleocapsid-like particles. Virology 204:770–776.
4. Buchholz, C. J., D. Spehner, R. Drillien, W. J. Neubert, and H. E. Homann. 1993. The conserved N-terminal region of Sendai virus nucleocapsid protein NP is required for nucleocapsid assembly. J. Virol. 67:5803–5812. 5. Calain, P., J. Curran, D. Kolakofsky, and L. Roux. 1992. Molecular cloning
of natural paramyxovirus copy-back defective interfering RNAs and their expression from DNA. Virology 191:62–71.
6. Calain, P., and L. Roux. 1993. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J. Virol. 67:4822–4830. 7. Carlsen, S. R., R. W. Peluso, and S. A. Moyer. 1985. In vitro replication of Sendai virus wild-type and defective interfering particle genome RNAs. J. Virol. 54:493–500.
8. Chandrika, R., S. M. Horikami, S. Smallwood, and S. A. Moyer. 1995. Mutations in conserved domain I of the Sendai virus L polymerase protein uncouple transcription and replication. Virology 213:352–363.
9. Chandrika, R., T. Myers, and S. A. Moyer. 1995. Measles virus nucleocapsid protein can function in Sendai virus defective interfering particle genome synthesis in vitro. Virology 206:777–782.
10. Cunningham, B. C., and J. A. Wells. 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244: 1081–1085.
11. Curran, J., R. Boeck, and D. Kolakofsky. 1991. The Sendai virus P gene expresses both an essential protein and an inhibitor of RNA synthesis by shuffling modules via mRNA editing. EMBO J. 10:3079–3085.
12. Curran, J., H. Homann, C. Buchholz, S. Rochat, W. J. Neubert, and D.
Kolakofsky.1993. The hypervariable C-terminal tail of the Sendai paramyxo-virus nucleocapsid protein is required for template function but not for RNA encapsidation. J. Virol. 67:4358–4364.
13. Curran, J., J.-B. Marq, and D. Kolakofsky. 1995. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol.
69:849–855.
14. Diamond, S. E., and K. Kirkegaard. 1994. Clustered charged-to-alanine
mutagenesis of poliovirus RNA-dependent RNA polymerase yields multiple temperature-sensitive mutants defective in RNA synthesis. J. Virol. 68:863– 876.
15. Fuerst, T. R., E. G. Niles, F. W. Studier, and B. Moss. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that syn-thesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA
83:8122–8126.
16. Galinski, M. S., and S. L. Wechsler. 1991. The molecular biology of the
Paramyxovirus genus, p. 41–82. In D. Kingsbury (ed.), The paramyxoviruses.
Plenum, New York, N.Y.
17. Hassett, D. E., and R. C. Condit. 1994. Targeted construction of tempera-ture-sensitive mutations in vaccinia virus by replacing clustered charged residues with alanine. Proc. Natl. Acad. Sci. USA 91:4554–4558. 18. Heggeness, M. H., A. Scheid, and P. W. Choppin. 1980. Conformation of the
helical nucleocapsids of paramyxoviruses and vesicular stomatitis virus: re-versible coiling and uncoiling induced by changes in salt concentration. Proc. Natl. Acad. Sci. USA 77:2631–2635.
19. Higuchi, R., B. Krummel, and R. K. Saiki. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367.
20. Homann, H. E., W. Willenbrink, C. J. Buchholz, and W. J. Neubert. 1991. Sendai virus protein-protein interactions studied by a protein-blotting pro-tein-overlay technique: mapping of domains on NP protein required for binding to P protein. J. Virol. 65:1304–1309.
21. Horikami, S. M., J. Curran, D. Kolakofsky, and S. A. Moyer. 1992. Com-plexes of Sendai virus NP-P and P-L proteins are required for defective interfering particle genome replication in vitro. J. Virol. 66:4901–4908. 22. Horikami, S. M., and S. A. Moyer. 1995. Alternative amino acids at a single
site in the Sendai virus L protein produce multiple defects in RNA synthesis in vitro. Virology 211:577–582.
23. Hudson, L. D., C. Condra, and R. A. Lazzarini. 1986. Cloning and expression of a viral phosphoprotein: structure suggests vesicular stomatitis virus NS may function by mimicking an RNA template. J. Gen. Virol. 67:1571–1579. 24. Kingsbury, D. W. (ed.). 1991. The paramyxoviruses. Plenum Press, New
York, N.Y.
25. Kolakofsky, D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555.
26. Miyahara, K., S. Kitada, M. Yoshimoto, H. Matumura, M. Kawano, H.
Komada, M. Tsurudome, S. Kusugawa, M. Nishio, and Y. Ito.1992. Molec-ular evolution of paramyxoviruses: nucleotide sequence analyses of the hu-man parainfluenza type I virus NP and M protein genes and construction of phylogenetic trees for all the human paramyxoviruses. Arch. Virol. 124:255– 268.
27. Morgan, E. M. 1991. Evolutionary relationships of paramyxovirus nucleo-capsid-associated proteins, p. 163–179. In D. Kingsbury (ed), The paramyxo-viruses. Plenum Press, New York, N.Y.
28. Morgan, E. M., G. G. Re, and D. Kingsbury. 1984. Complete sequence of the Sendai virus NP gene from a cloned insert. Virology 135:279–287. 29. Pattnaik, A. K., L. A. Ball, A. W. LeGrone, and G. W. Wertz. 1992. Infectious
defective interfering particles of VSV from transcripts of a cDNA clone. Cell
69:1011–1020.
30. Portner, A., and K. G. Murti. 1986. Localization of P, NP and M proteins on Sendai virus nucleocapsids using immunogold labeling. Virology 150:469– 478.
31. Portner, A., K. G. Murti, E. M. Morgan, and D. W. Kingsbury. 1988. Anti-bodies against Sendai virus L protein: distribution of the protein in nucleo-capsids revealed by immunoelectron microscopy. Virology 163:236–239. 32. Ryan, K. W., A. Portner, and K. G. Murti. 1993. Antibodies to paramyxovirus
nucleoproteins define regions important for immunogenicity and nucleocap-sid assembly. Virology 193:376–384.
33. Shioda, T., Y. Hidaka, T. Kanda, H. Shibuta, A. Nomoto, and K. Iwasaki. 1983. Sequence of 3,687 nucleotides from the 39end of Sendai virus genome RNA and the predicted amino acid sequences of viral NP, P and C proteins. Nucleic Acids Res. 11:7317–7330.
34. Wells, J. A., B. C. Cunningham, G. Fuh, H. B. Lowman, S. H. Bass, M. G.
Mulkerrin, M. Ultsch, and A. M. de Vos.1993. The molecular basis for growth hormone-receptor interactions. Recent Prog. Horm. Res. 48:253– 275.
35. Wertman, K. F., D. G. Drubin, and D. Botstein. 1992. Systematic mutational analysis of the yeast ACT1 gene. Genetics 132:337–350.