JOURNALOFVIROLOGY, July1993, p.4358-4364 0022-538X/93/074358-07$02.00/0
Copyright C)1993,AmericanSocietyforMicrobiology
The Hypervariable C-Terminal
Tail of the Sendai
Paramyxovirus
Nucleocapsid
Protein Is
Required
for
Template
Function
but Not for
RNA
Encapsidation
JOSEPHCURRAN,' HORSTHOMANN,2CHRISTIANBUCHHOLZ,2SYLVIE
ROCHAT,1
WOLFGANG NEUBERT,2 ANDDANIELKOLAKOFSKY1*
Department of Genetics andMicrobiology, University of Geneva SchoolofMedicine, CMU, 9 Ave deChampel, CH1211Geneva, Switzerland,1and Max Planck InstituteofBiochemistry, Department
of Virology, Am Klopferspitz 18a, D-8033 Martinsried, Gennany2 Received 18February 1993/Accepted 20 March 1993
Theparamyxovirusnucleocapsid proteins(NPs)arerelatively well conserved,exceptfor theC-terminal20%o (orca.100aminoacids), referredtoasthetail. We haveexamined whether thishypervariabletail isrequired
forgenome synthesis, bothinvitro,where synthesisispredominantlyfrom theinput templates, and invivo,
wheremultiplerounds ofamplification occur.Intheseviruses,genomesynthesis andassemblyof thenascent chainarecoupled.We find thatthe tail isrequiredinvivo butnotinvitro. Closer examination of theinvivo systemshowedthat the tailless NP couldencapsidatethegenomechainbut thatamplificationdid notoccur.We
interpret these results as indicating that the tail isnot required for RNAassembly but is required for the
templatetofunction in RNAsynthesis.Relativelysmall deletions within the conserved N-terminal 80%oofthe
protein,onthe other hand, rendered the proteinnonfunctional ineithersystem.Thepossible functionsof the tail in RNAsynthesisarediscussed.
The nonsegmented genomes of paramyxoviruses (ca. 15
kb)arefoundpredominantlyashelicalnucleocapsids (NCs), inwhichthegenomeRNAistightlyassociatedwithca.2,600
copies of the viral nucleocapsid protein (NP [8, 18]). Two
other viralproteins, L (large) and P (phosphoprotein), are more loosely associated with NCs, and togetherthis
com-plexcansynthesize mRNAs from theminus-strandgenome. P and Ltogether are thought to constitute the viral poly-merase,whereas NPis consideredtobepartof thetemplate,
because thepolymerase willnot copynaked genome RNA (11, 12). Duringgenome replication,NP also assembles the nascent antigenome chain, and thisconcurrent assemblyis
presumably required to maintain the processivity of the
polymerasein traversingthetemplate (28). TheNP
respon-sible for this assemblyappears to actas anNP-P complex, possiblyin association with the P-Lpolymerase (15).
ThesequencesofnumerousparamyxovirusNPshavenow been predicted (reference 29 and references therein), and comparisonsof themcoupled with proteasestudiessuggest that NP can be divided into two domains. The N-terminal
80%of theprotein is relativelywellconservedamongrelated
viruses, whereas the C-terminal 20% is poorly conserved. This hypervariable C terminus appears to be a tail at the
surface of aglobular N-terminal body, because the Sendai virus (SEN)NP(62 kDa), forexample, ishypersensitive to trypsindigestion,leavinga48-kDaN-terminalcore(13, 24). The tailalsocontainsmostof theprotein'sphosphorylation
sites(16)andantigenic sites (2, 10). The tail's hypervariabil-ity suggests that it may notbe functionally important, and largepartsof thisregionwererecently foundtobe
dispens-ableforbindingto Pwithaninvitro blottingassay(14). Systems to study SEN genome replication, both in vivo and in vitro, have recently been described (3, 15). The in vivo system uses copy-back defective interfering (DI)
viri-*Corresponding author.
ons (freed of their helper virus by UV inactivation) to naturallyinfectcells,andthehelperfunctionsforreplication
areprovided bytransfection ofplasmids expressing NP, P, and L. The in vitro system uses core DI genome NCs (washed free of their P and L) as templates and crude extractsof transfected cells as asource of the viral helper proteins. We have studied whether the hypervariable C terminus of NP is required for its function in genome replication byusing both of thesesystems.
MATERIALS AND METHODS
Construction of DNA subclones. The construction of
pGEM-NPwth, pGEM-P/C, pGEM-L, and some of the NP deletion mutants has been previously described (3-5, 14).
Most of the NP deletion mutants detailed in Fig. 5 were cloned by fusion polymerase chain reaction. The PIV1 pGEM-NP clone (clone 4-31) was a kind gift from Yumi Matsuoka(21). The mutantpGEM-NPBalwasgeneratedby
fusing the uniqueBalIrestriction site (Fig. 1) to ablunted BamHI site in the downstreampolylinker.TheC-terminally
truncated protein expressed from this clone would contain two additional (non-NP) codons followed by an amber codon. pGEM-NPNco was constructed by insertion of a
three-way-stop oligomer, 5'CTAACTAGTI1G, into the blunted NcoI site. Thisduodecamer isperfectly palindromic
andcan be self-annealed to providedouble-stranded DNA for insertion intoanyblunted site. It contains stop codons in allthreereading frames,inboth orientations (oneis
under-lined), and itsSpeI restriction site (in boldface) allowseasy
screeningofclones.
Invitro and invivo RNAsynthesis.DIH4Uv amplification
in vivo was performed as described previously (3). RNA
synthesisinvitrowasperformed essentiallyasdescribedby
Curranetal. (6),with thefollowingmodifications. NP-RNA
(RNP) templates were prepared from egg-grown defective virus by first pelleting the virus through a cushion of25%
4358
Vol.67, No.7
on November 9, 2019 by guest
http://jvi.asm.org/
co IT
92% (CR1) 47%
--- mL
mO z<0
co z
'9 L
C-0
11-28% 77% 16% 65%
CR2AMmCR3
II
%activity
98
±10
29 ± 4
<2
+ - + +
P1l Eq1 EEE t nDE DitDi1 nK iaR R1aDx R 491
SEN 1 R 1 EB E tn D EDv sDsi R Ri a mR1aE R R 491
bPI3 D tqt1 svt1 iBsiKtEqRn i R D R 1 nR R 481
hPI3 DD R t q at soDniKt Eq q n i R D R1nxR 481
_- -_+
FIG. 1. Sequence comparison of the SEN proteins and PIV1
NPs.Thetop boxshowsthehomologiesoverthe entire length of the
proteins.Thenumbersabove refertoresidueposition; the percent-agesbelowrefertoamino acid identities. TheN-terminal80% ofthe
protein ishighly conserved (CR1), whereas the C-terminal20% is
more divergent. Below, an enlargement of this divergent domain
reveals two blocks of higher homology, CR2 and CR3. Three restrictionsitesusedinthesestudiesto generateC-terminal
trunca-tions are also shown above. These mutants are diagrammed just
below, and their percent activity relative to NP' in genome
amplificationinvivo(Fig. 2) isindicated. Atthe bottom is shown a
four-wayalignmentoftheprimarysequencesof the CR2 domainsof SEN, PIV1 (PI1), hPIV3 (hPI3), and bPIV3 (bPI3). Positions in
which a positive or negative residue is conserved in all four sequencesareindicatedinboldface andwithanasteriskbetweenthe
two pairs. The plus and minus signs refer to conserved charged residueswithineachpair.
(vol/vol) glycerol in TNE (10 mM Tris [pH 7.4], 50 mM NaCl,1mMEDTA),resuspendingthe viralpellet in 150 mM NaCl-50 mM Tris (pH 7.4)-10 mM EDTA-0.6% (vol/vol)
NonidetP-40(lysis buffer), and thenbanding theNCs twice on 20 to40% (wt/wt) CsCl gradients (38,000 rpm for 2 h at 12°C in an SW41 rotor) to remove the endogenous
poly-merase. This material was then diluted threefold in water,
andtheNCswerepelleted by spinningat50,000rpmfor 90
minat12°CinanSW60rotor.Templateswereresuspended in20%glycerol-water (50 ,u/10ml of startingallantoicfluid) andwerestoredas10-,ul aliquotsat-70°C.Normally,2,ulof templatewas usedperreaction.
Cytoplasmic extracts were prepared essentially as
de-scribed previously (6), exceptthat CV1 cellswerereplaced
with A549 cells (a human epithelial linewhich gavehigher
levelsofgenomereplication invitro) and theactinomycinD
levelswere increased to afinal concentration of 20 ,ug/ml. Thiselevated concentrationwas requiredto inhibit all
T7-derived DNA-dependent RNA synthesis in the extracts (24a). Cells were infected with the vaccinia recombinant vTF7-3(9)atamultiplicity of infectionof 2 PFUpercell 30
min
before transfection and were harvested at 24 h postin-fection by solubilizing the monolayer in lysis buffer. NC and RNA products were separated on a 20 to 40% CsCl gradient. The NCs were phenol extracted and analyzed directly on a 1.5% agarose-formaldehyde gel.Antibodies and probes. The monoclonal antibodies NP877 and P 1.180 were a kind giftfrom Claes Oervell, Stockholm, Sweden. The monoclonal antibody NP W16 (which
recog-nizes both Sendai
NP'W
andNPBal
[10])
was akind giftfrom Alan Portner.The L monoclonal antibody was raised against aC-terminal peptide and has been reported previously (7).RESULTS
The precise sequence of the C-terminus of the SEN NP has been the subject of some confusion. The sequences of the Z (27) and Enders (23) strains were reported first, and these werebasically identical except for asingle-base inser-tion or deleinser-tion within codon 493. The C termini were consequentlydifferentin sequence from this point, as well as different in length (strain Z was 524 amino acids [aaJ and Enders was 517 aa). Shioda et al. (27
[corrigendum])
cor-rected their sequence atthis site to agree with that of Morgan etal. (23), yielding one sequence of NP, which we refer to as PQQ-517 (PQQ are the last 3 aa of this sequence of 517 residues). In 1990, however, Middleton et al. (22) reported that the NP gene of their Z straincorresponded
to that initially reported by Shioda et al., and essentially the same protein sequence was then reported by Neubert et al. (25)forthree separate variants of the Fushimi strain. In this last
case, moreover, the predicted C termini (all GGI-524) were
confirmed by the sequencing of tryptic peptides. It then seems likely that GGI-524 represents the
correct
C terminus of SEN NP, and we provide some evidence below thatthis is so. In retrospect, the original 1983 sequence contained the GGI-524 C terminus because it also contained a nearby compensatory insertion or deletion (26a). Perusal of other paramyxovirus NPs in the data base indicates that this sort of problem is not unique.The SEN andPIV1 NPs. To gain insight about the C-ter-minus of NP, we compared these regions in different pairs of paramyxoviruses. Comparison of very closely related pairs, such as parainfluenza virus 4 (PIV4) A and B or human and bovine PIV3 (hPIV3 and bPIV3, respectively) was uninfor-mative; there were too few differences here. Comparison of measles virus and canine distemper virus was also uninfor-mative, because there was
almost
no homology in this region, presumably because they were not sufficiently re-lated. However, comparison ofPIV1
and SEN, which are only slightly less related than the bovine and human strains of PIV3, was informative.PIV1 NP is also 524 aa long and ends with the sequence GGI (19, 21). When aligned with SEN NP, the N-terminal 426 residues are 92% identical (conserved region 1 [CR1]), and the remaining C termini are relatively poorly conserved (47% identical). However, there are two blocks with stron-ger conservation within this tail (Fig. 1): a highly charged region in the middle (CR2, residues 463 to 488, 77% identi-cal) and a negatively charged region followed by a hydro-phobic one, representing the tip of the tail (CR3, residues 508 to 524, 65% identical). These two regions stand out because they are flanked by nonconserved regions.
We first examined the NP requirements for genome syn-thesis in vivo, where different NP plasmids were
cotrans-fected
(along,with
pGEM-P and pGEM-L) into cellsinfected with DIH4 . Genome replication here is dependent on NP,-7,r-IV .Wl% II
on November 9, 2019 by guest
http://jvi.asm.org/
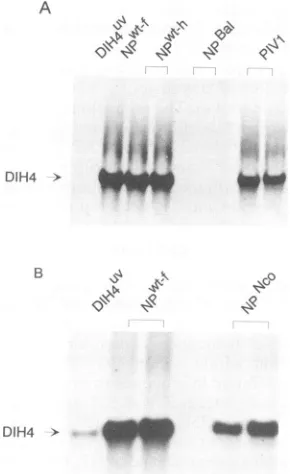
[image:2.612.61.291.73.336.2]4360 CURRAN ET AL. A
DIH4 >
B I'll v00
DIH4 > __
FIG. 2. DIH4 amplification in vivo with various NP proteins.
CV1 cell monolayersinfected with vTF7-3 were transfected with pGEM-P/C and pGEM-L plus different forms of NP as indicated above eachpanel. PIV1 referstoNP from hPIV1 andNPl-h and NPwt-freferto NPfrom the Harris and Fushimi strains of Sendai
virus, respectively. After 48 h of incubation, NC bands were isolated fromaCsCl gradient and phenol extracted,and the RNA
wasresolvedon a1.5% agarose-formaldehyde gel.The RNAwas
then transferredtonitrocellulosemembranes andprobedwitha(+) riboprobe transcribed from theplasmid pEX5'. The lanes marked
DIH4Uvarecontrols in which thehelper plasmidswereomitted and
indicate the level oftheinputDI in eachexperiment. Duplicatelanes represent separatebutparalleltransfections.
P, and L (3). After 48 h of incubation, NCswere isolated from these cells and their DI RNA levelswereestimatedby
Northern(RNA) blotting. Thebackground level dueto the
infectingor input genomeswas determined in each
experi-ment by omitting the helper plasmids (lanes DIH4Uv, Fig. 2). Each experiment also contained, as a reference, the
wild-typeNPgenefrom either the Harris(h)orthe Fushimi
(f) strain,which supportedgenomereplicationtovery sim-ilar levels(cf. singlelane NPwt-f withduplicatelanesNPwt-h, Fig. 2A).
Wefound that thePIV1NPgene(lanesPIV1)could in fact
replacethat of SEN and stillyield goodlevels ofreplication, i.e., 53% + 7% of NPWt (as determined in a
Phosphor-Imager). Thus, either the tail is unimportant here or only the conserved regions are important. Several C-terminal
truncations, NPPSt(or
NP1-518),
NpNco(NP1-498),
andNpBal (NP'-456),werethen tested. Theseresultsareshown inFig.2 andare summarized inFig. 1. Removing onlythe
hydro-phobic tipof the tail(residues519to524; NPPSt) produceda protein which was basically wild type in activity (Fig. 1).
NPNCo, which is missing the entire CR3, continued to
supportgenomereplication,butatathreefold reduced level. Residues 499 to 518 are therefore not essential for NP function inthis systembut contributetoactivity. NPBal,on the other hand, appearedtobetotally inactive,eventhough it was expressed at levels similar to NPWt as judged by
Western blotting (immunoblotting) (data not shown). Resi-dues 456 to 498, which include highly charged CR2, then
appeartobe essential forgenome replication.
We havecontrolled NP levels in all experiments, andwe report results
only
for those mutantproteins
which were expressed to nearwild-type levels. Two constructs, eventhough
they
weremade morethan once,missing
theC-ter-minal 98aa andone inparticular containing aframeshift in
codon 493 to emulate
PQQ-517 expressed proteins
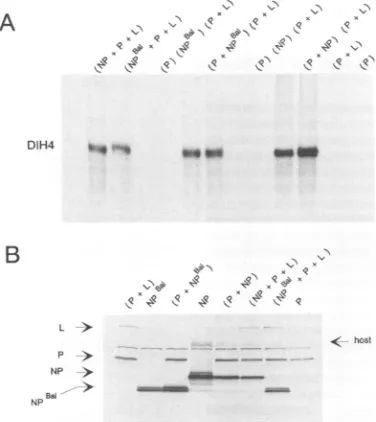
which were toounstable for conclusionstobe drawn.Differences between the in vivo and in vitro systems. In starkcontrasttoitsinabilitytosupportDIH4 RNAsynthesis
in vivo, NPBal
appeared
to be almostas active as NPWt in vitro (Fig. 3A). NPBaI, moreover, shared another property withNPWt,whichsuggestedthat itsactivityin vitrowasreal.Intransfected cell extracts, P is foundas a
complex
notonlywith Lbut also with unassembledNP(NP°), because
antibodies to one
protein
will cross-select the other fromcoexpressed extracts (15). P-L is competent by itself for
transcription (6)
butrequires
P-NP°aswell forreplication,
the latter
presumably being
involved innascentchainassem-bly. An
important
property of the in vitro system is thattransfected cellextracts areactiveonlywhen either all three
proteins
arecoexpressed [single
lanes(NP
or NPBal+ P + L), Fig. 3A; the parentheses indicate whichproteins
werecoexpressed]
orPand Larecoexpressed
inoneculture and P and NP are expressed in another and then combined[duplicate
lanes(P
+ NP orNPBal)
and(P
+L)] (15).
Inparticular,
NP is inactive unlesscoexpressed
with P[lanes
(P), (NP),
and(P
+L)],
eventhough
its level in theextract(like
those of P andL)
is verysimilar,
whether or not coexpression occurred(Fig. 3B).
As shown inFig. 3A,
NPBal
wasjust
asdependent
oncoexpression
withPfor itsactivityin vitroasNPWt.The
experiment
shown inFig.
3 has beencarriedoutthree times withbasically
the sameresult;
NPBal appearstobe 40to 100% asactiveasNPWt in vitro.The remarkable difference in
activity
ofNPBalin vivo and invitro mayreflect differences in what each systemactually
carriesout.
Multiple
rounds ofreplication
occurinvivo;
weoften find a 50-fold increase in DI genome levelsover the
input template by
Northernblotting (e.g., Fig.
2A).
Because it is essential here tocompletely
UV inactivate thehelper
genomes in the DI
stock,
relatively long
illuminations are carriedout to ensurethis,
with the consequence that(i)
most of theDIgenomes(probably
>90%)
arealso UV inactivated and cannot function astemplates,
but(ii)
all will still be competenttohybridize
the Northern blotprobe.
The50-fold increasewe see then morelikely
represents a muchlarger
amplification
of the non-UV-inactivatedinput.
Invitro,
onthe other
hand, only
asmall amountofhighly
radiolabelled DI RNAis made fromarelatively large
amountoftemplate
(and
in a more limitedperiod
oftime),
andsoit ispossible
that only
synthesis
from theinput templates
isbeing
mea-sured. If so, all thatis
being
measured in vitro may be theability
of NPto assemble thenascent chain(and
drive thepolymerase
acrossthetemplate).
Inthein vitro test, this NP neednotact, onceassembled,
as anessential componentof thedenovo-madetemplate.
If
NPBal
can supportreplication only
from theinput
templates
carrying
NPWt,
genomeamplification
in vivo would be linear rather thanexponential,
and this very limitedsynthesis might
notbevisibleby
blotting against
theback-ground
of the totalinput
DIs. It is therefore important tospecifically
measure the genomes made de novo in vivo.Directly
radiolabelling
the transfected cultures is the bestapproach,
but it isimpractical
here because of the small J. VIROL.:4
l3p. 14p>,I\ 1\
,\R.,
0 Ng 0 9
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.108.253.76.313.2]\.'71~
K1L":
\K
e'1~q(h%"
x. x
qx qx 4Rx
\ -\. .,\
9 9 9
A: (-)probe
DIH4 b4 " we_
(+) DIH4 >
B
0 100 1.18+8 +.04
, 'l s9 I 4
zoi @ @ M£@ x
L
p NP NP
[image:4.612.366.512.79.373.2]< host
FIG. 3. NPBa' supports DIH4 amplification in vitro, but only when coexpressed with P. RNA synthesis was carried outwith [32P]CTP in cytoplasmic extracts prepared from vTF7-3-infected A549 cells transfected with various combinations of pGEM plas-mids, asindicated above each panel. Parentheses indicateextracts
from a 5-cm petri dish in which the various constructs were
expressedorcoexpressed. Each petri dishwasusedtoprepare100 .1l ofextract,35 pl of whichwasaddedtoeach reaction. The final reaction volumewasadjustedto115 ,ul withanextractfrom cells infected only with vTF7-3, so that all reactions contained equal
amounts of cell extract. As negative controls, reactions were
performed in the absence of NP [lane (P+ L)]orin thepresenceof onlyP [lane (P)]. (A) Autoradiograph of the NC RNA from each
reaction thatwas isolatedfromaCsCl gradient and analyzedon a
1.5%agarose-formaldehyde gel. Duplicate lanes in the middle of the
figure represent separate but parallel transfections. (B) Equal
amountsof the various cellextracts(10 ,ul)weremonitored for NP, P,and L levelsby Western blotting withacombination of NP (W16),
P(1.180),and L monoclonalantibodies. NotethatNPBa'appears as adoublet band.
amount of synthesis expected with NPBal. We therefore
madeuse of another efficient system forgenome amplifica-tion recently developed, similar to that used above but in whichreplication isinitiated fromplasmid DNA rather than natural virus (DIH4Uv)infection (2a, 2b); therenowshould be no backgroundof input NCs. A(+)-DIH4 RNA is first transcribedbythe T7polymerase,andthis is thenassembled
andreplicated bytheNP,P,and Lhelperfunctions(lanes 3
and 4, Fig. 4A; the duplicate lanes represent separate transfections). When NP'W is replaced with NPBal, a very much smallerbut reproducible amountof (+)-DIH4 RNA is
found in the NC band regionof the CsCl gradient (lanes 5
and6, Fig. 4A). Autoradiographyis unsuited forquantitating signalsof suchunequal intensity, so aPhosphorlmagerwas also used, and these numbers are shown below the lanes. Note that there isnodetectablebackgroundin thisregion of the gradient in the control transfections (minus any NP plasmid; lanes 1 and 2, Fig. 4A), presumably because the
unencapsidatedT7 transcripts pellethere. Figure4Ashows
that NPBaI canin factencapsidate (+)-DIH4RNA in vivoto
a very small level, and this level could represent linear
amplification. When the same samples are examined for
(-)-DIH4 RNA,which should arise onlyif viralreplication
B: (+) probe
(-)DIH4 >
0 100 0.11
+11 + .11
FIG. 4. Genomereplication initiated from plasmid DNA. DIH4 genome replication was carried out as before, except that the
DIH4' infectionwassubstitutedby the addition of 5 p.gofplasmid
DNA which expresses (+)-DIH4 RNA via T7 polymerase (2b).
RNAs were recovered from the 1.31-g/ml region of the CsCl gradients and were probed on Northern blots for both (+)- and (-)-DIH4 RNAs (with riboprobes to the central region of these
RNAs).Thefigure showsan8-hexposureofthegels, whichwere
alsoquantitatedinaPhosphorImager,and thesenumbersareshown below the lanes. In eachcase,theaveragevalue of theNP' sample was set to 100. This value was three times as strong for the (+)-DIH4 RNA as that for the (-)-DIH4 RNA; however, this difference is dueatleast inpart tothe differentriboprobesused.
can occur, there isnowvirtuallyno PhosphorImager signal when NPWt isreplaced with NPBaJ (Fig. 4B), noris there a
PhosphorImager signalafterprolongedexposureof thegelto film (not shown). This experiment is consistent with the notion that NPBal iscapableofencapsidatingDIH4 RNA in
vivo, but the NPBal-RNA complex thus formed cannot be
used, or is used much less efficiently, for (-)-DIH4 RNA
synthesis.
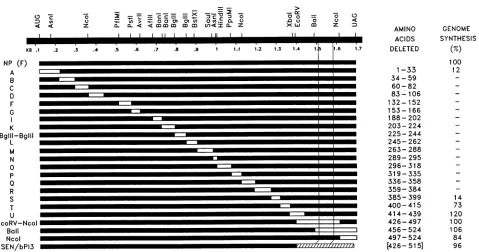
Mapping boundaries ofregions requiredfor assemblyand
amplification. Because NPBal is still active forgenome
syn-thesis invitro,wetestedaseries of deletionmutants tomore closely define the regions required for this activity; the results are presented in Fig. 5. Deletion of residues 400 to 415,414to439,and 426to497, like NPBal(missingresidues 456to524),had littleor noeffectonactivityrelativetoNPWt. None of the C-terminal 125 residues of NP thenappeartobe essential foractivityinvitro,which ispresumablyrestricted
to nascent chainassembly. On the other hand, deletion of residues385to399 reducedactivity sevenfold,and deletions of short regionsbetween residues 34 and 384(mutantsB to R, Fig. 5)-including mutant N, which is missing only 7
A
>
> jmmwm&....
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.89.277.84.295.2]4362 CURRAN ET AL.
D v
-e - - - _ _ Y -_ _, >2
O6 = =L- = CECE =Ex= 7 aC V 3 0
01(/
0 = ID > :- a a CJ ) (1) aO 0.C t;
z X X < < mmm m m tn.<Z L Z [it
KB .1 .2
NP (F)
A B C D F G
K
Bglll-BgllI
L M
N
0
p
Q
R S T
0o
.0 u
II
a
m
. . . .3
.5 .6 .7 .8 .9 1 1.1 1.2 1.3 1.4 1.
.I
5
o~ 0
0 <
z
l1.6 1.7
- -
~~~~-
m--!
-~~~~
-I-Eli
m~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~I
-- I - Eli mi
--I
li!m
--~l
- - Eli mi
-- I - Eli mi
--I - Eli m
I
-I=I
u
-EcoRV-Ncol Ball Ncol
SEN/bPI3 a2,,,z7777zz77/7777
AMINO ACIDS DELETED
1-33
34- 59 60-82 83-106 132- 152 153- 166 188-202
203-224 225-244
245-262
263-288 289- 295 296-318
319- 335
336-358 359-384
385- 399 400-415
414- 439 426-497
456-524 497- 524
[426-515]
GENOME SYNTHESIS
(%) 100
12
14
73
120 100 106
84
96
FIG. 5. The activity of various deleted NPs in vitro. A series of constructs containing deleted NP genes (reference 14 and Materials and Methods)were cotransfected into vTF7-3-infected A549 cells, and their cytoplasmic extracts were tested for theirability to support the synthesisofDIH4genome RNA in vitro (Materials and Methods). The regions deleted are indicated as openboxeswithin thegene, andtheir precisepositions are shown on the right. The hatched box of constructSEN/bPI3represents residues 426 to 515 of bPIV3. Thethin vertical lines on therightindicate the position of CR2. For genome synthesis, the numbers represent percent activity relativetoNPwt; a minussign indicatesnodetectable activity (<5%). NP levels in each extract were monitored by Western blotting andwere foundto besimilar tothose ofNP'W (not shown).
aa-led to NPswhich showed no activity above background
(i.e., <5%). Only mutant A, missing residues 1 to 33, showedsomeactivity above background. This suggests that CR1 is not only highly structured but that this structure depends on interactions which cover the entire region. Furthermore, changing the Leu-Val pair at either positions 229and 230 or 237 and 238 to Arg-Glu totally inactivated the protein in vivo, whereas changing Glu-489 to Gly or Pro reduced activity only two- and fourfold, respectively (not
shown).NPthen appearstobe composed ofaglobular head whichcantolerate almost no changes, with a C-terminal tail which forms a separate structure and which can accommo-date sequencechanges more easily. Only the globular head appears to be required for the RNA encapsidation, which naturally occurs concomitantly with genome synthesis.
Last, wenote thatthe phenotype ofNPBai isnot unique. Deleting residues 400 to 415 and 414 to 439, in particular, also eliminates activity in vivo but not in vitro. However, unlike
NPBal,
both ofthese mutants still contain the highly charged CR2 (residues 465 to 491). The residues betweenposition400 and CR2 are thus also important for activity in vivo. However, because residues 427 to 463 are very poorly conserved (Fig. 1), these residues may be important mostly
toposition the highly charged domain relative to the globular head,asopposed to containingsequences which themselves
are important for replication. Computer predictions of the
protein's secondary structure support this notion; residues 400 to 463 are predicted to contain several beta turns,
whereasCR2 itself is alpha helical. Interestingly, a chimeric
NP containing the entire CR1 of SEN (which is very well conserved with that of bPIV3) and the C-terminal tail of bPIV3 is also inactive in vivo (not shown) but is active in
vitro(Fig.5). However, its CR2 would be 10 residues closer
toCR1 than theNPWt CR2.
DISCUSSION
The central finding of this work is that thehypervariable C-terminal tail of NP is required for genome synthesis in vivo butnotin vitro. We have argued that in vitro, genome
synthesisis restrictedto thevastexcessoftemplates added
to the reaction, because diminishing their level 10-fold has
noeffect on netsynthesis(not shown). NP here then needs
onlytoassemble the nascent RNA during genomesynthesis.
Invivo,however,amuch smalleramountof DItemplates is usedtostarttheinfection, and genomesynthesis is detected
onlyafterallowing for multiple rounds of amplification. The
plasmid-expressed NPin the invivo system mustthen also beabletofunction aspartof thedenovo-made template. If so, theC-terminal tail ofNPisnotrequired fornascentchain assembly but presumably is required for the template to be functional. This is consistent with the finding that trypsin-treatedNCs, containingNPwhich ismissing the C-terminal 12 kDaof sequence(or roughly 100aa), aremorphologically
verysimilartountreated NCs(13, 24).Thebasicproperties
ofNPresponsible for the helical structure of the NC,orits formation, therefore donotdependontheC-terminal tail. In
addition, Buchholz et al. (la) have found an excellent correlation between the ability of these NP mutants to
assemble into NC-like structures(when expressed
indepen-dently)and theirabilitytosupport genomesynthesis in vitro. The tail accumulates changes more quickly during evolu-tion than the trypsin-resistant core. Yet two regions, one
highly charged in the middle of the tail and the other J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.67.546.75.327.2]negatively charged and then hydrophobic at the tip, are more conserved. Because deletion of this latter region (CR3) reduces genome amplification by only threefold, it presum-ably is conserved (in part) for functions other than RNA synthesis, e.g., for virion budding, in which it might interact with theamphipathic matrix protein (which has a net posi-tivecharge) (1). The 27-aa region, which is highlighted by sequence comparisons and appears essential for multiple roi nds of RNAsynthesis (CR2), contains 15 of 16 charged residues which aremostly conserved between SEN, PIV1, hPIV3, and bPIV3, with the negative charges distributed mostly at the N-terminal end (Fig. 1). This negatively charged end of CR2 has already been noted by Parks et al. (25a)as a commonregion of all paramyxovirus NPs. In the aboveviruses, this end also contains conserved Ser and Thr residues which could bephosphorylation sites, and so CR2 could beeven more negatively charged. However, a more detailed mutational analysis of CR2 will be required to detern ine whether these sequences aredirectly involved in multiple round genome synthesis.
What function th tailservesis unclear fromthiswork, but thereareseveral p ;sibilities of interest.
(i) The RNA within NCs is resistant toRNaseatanysalt concentration(13, 20), presumablybecause it isprotected by thesurrounding NP. During RNA synthesis, NPNCmaybe locally displaced from the template RNA so that the bases mayberead, and theC-terminaltail may berequiredfor this. Forexample, the tailmightsubstitute for thetemplate RNA boundby the N-terminal core,asuggestion previouslymade for the poorly conserved N-terminal half of the vesicular stomatitis virusphosphoprotein (17).
(ii)NPclearly playsa moreactive role in RNAsynthesis,
besides that ofsimply beingastructuralprotein.In the PolR
mutantsof vesicular stomatitis virus(26) and the Z strain of SEN (29), the polymerase reads through the leader-NP
junction athigh frequency, even in the absence of concur-rent assembly. In both viruses, this phenotype maps to
NPNC and not to the P-L polymerase in reconstitution
experiments (2c, 26). NPNC can then condition how the
polymeraserespondstojunctions,and the tailmightevenbe involved in this. The Z strain NP is also unique in that it
migrates anomalously fast during sodium dodecyl
sulfate-polyacrylamide gelelectrophoresis, andit maynotbe coin-cidental that this mobility determinant also maps to the
C-terminaltail
(unpublished observations).
(iii)Invivo,NP al (coexpressedwith P andL)appearsto
assemble RNAnonspecifically, because very abundant and
sharpNC bands formon CsClgradient centrifugation
inde-pendently of genome replication, and this does not occur
withNPWt.IntheCsClgradientsof theexperimentshown in
Fig. 4, for example, the visible
1.31-g/ml
bands from thesamples containing NPBal were severalfold stronger than those that contained
NPWt,
even though they containedrelativelyminuscule amountsof DIH4 RNA. In a separate
study,Buchholzet al. (la)have examined similarstructures with the electron microscope. Theywerefound to be very similarto naturally formed NCs and to contain RNA. The
C-terminal tail of NP may then also
play
a role in thespecificityof RNAassembly.
(iv)It isalsopossiblethat the tailsimplyregresentsasite
or sites atwhich Por Linteractswith NPN duringRNA
synthesis. We have begun to
investigate
functional NP-Pinteractions by trying to rescue inactive chimeras between SEN and bPIV3 P by
complementation
with NPchime-ras-sofar,without success.
ACKNOWLEDGMENTS
WethankJean-Baptiste MarqandCatherine Studer for excellent technicalassistance. We areparticularly gratefultoPhilippeCalain and Laurent Roux forprovidingtheimprovedDIH4vectorandfor
discussions.
This workwas supported by the Swiss National Science Fund. EMBOprovidedashort-termfellowshiptoH.H.
REFERENCES
1. Blumberg,B.M.,K.Rose,M.G.Simona,L.Roux,C.Giorgi, and D.Kolakofsky. 1984.Analysis of the Sendaivirus M gene andprotein.J. Virol. 52:656-663.
la.Buchholz,C.J.,etal.Submitted forpublication.
2. Buckland, R., P.Giraudon, and F. Wild. 1989. Expressionof measles virusnucleoproteininEscherichia coli:useof deletion
mutants tolocate theantigenicsites.J. Gen.Virol. 70:435-441. 2a.Calain, P., J. Curran, D. Kolakofsky, and L. Roux. 1992. Molecularcloning of natural paramyxoviruscopy-back defec-tiveinterferingRNAsand theirexpressionfromDNA.Virology
191:62-71.
2b.Calain,P.,and L. Roux.Submitted forpublication.
2c.Curran, J.Unpublishedobservations.
3. Curran, J.,R.Boeck,and D.Kolakofsky.1991.The Sendaivirus Pgene expressesbothanessentialprotein andan inhibitorof RNAsynthesis byshufflingmodulesviamRNAediting.EMBO J. 10:3079-3085.
4. Curran, J.,and D.Kolakofsky.Scanning independentribosomal initiation of the Sendaivirus Xprotein.1988.EMBOJ. 7:2869-2874.
5. Curran, J., and D. Kolakofsky. 1989. Scanning independent ribosomalinitiation oftheSendai virusYproteinsinvitro andin vivo. EMBO J. 8:521-526.
6. Curran, J., J.-B. Marq,and D. Kolakofsky. 1992. The Sendai virus nonstructural C proteinsspecificallyinhibit viral mRNA synthesis. Virology189:647-656.
7. Einberger, H.,R.Mertz,P. H.Hofschneider,and W.J.Neubert. 1990. Purification, renaturation, and reconstituted protein
ki-nase activity of the Sendai virus large (L) protein: Lprotein phosphorylatestheNPandPproteinsinvitro. J.Virol. 64:4274-4280.
8. Fitch, J. T., and A. J. Gibbs. 1970. Observations on the
structure of the nucleocapsids of some paramyxoviruses. J. Gen.Virol. 6:141-150.
9. Fuerst, T. R.,E. G.Niles,F. W. Studier,and B. Moss. 1986. Eukaryotic transient-expression systembasedonrecombinant vaccinia virus that synthesizes bacteriophage T7 RNA
poly-merase.Proc.Natl.Acad. Sci. USA 83:8122-8126.
10. Gill,D.S., S.Takai,A. Portner, and D. W.Kingsbury. 1988. Mapping of antigenic domains of Sendai virus nucleocapsid protein expressedinEscherichia coli. J. Virol. 62:4805-4808. 11. Gotoh,H.,T.Schioda,Y.Sakai,K.Mizumoto,and H.Shibuta.
1989. Rescue of Sendai virus from viral
ribonucleoprotein-transfected cellsbyinfection with recombinantvaccinia viruses carryingSendaivirus LandP/Cgenes.Virology 171:434 443. 12. Hamaguchi, M.,T.Yoshida, K.Nishikawa, H.Naruse,and Y.
Nagai.1983.Transcriptive complexof Newcastle diseasevirus. I. Both Land P proteins arerequired to constitute anactive complex. Virology128:105-117.
13. Heggeness, M. H.,A. Scheid, and P. W.Choppin. 1981. The relationship of conformational changes in the Sendai virus nucleocapsidtoproteolytic cleavageof the NPprotein. Virol-ogy114:555-562.
14. Homann, H. E., W.Willenbrink, C. J. Buchholz, and W. J. Neubert.1991. Sendaivirusprotein-proteininteractions studied by a protein-blotting protein-overlay technique: mapping of domains on NPprotein required for binding to P protein. J. Virol. 65:1304-1309.
15. Horikami, S.M., J. Curran,D.Kolakofsky,and S. A.Moyer. 1992. Complexesof Sendai virus NP-P and P-Lproteins are requiredfordefectiveinterferingparticlegenomereplicationin vitro. J. Virol. 66:4901-4908.
16. Hsu,C.-H.,and D. W. Kingsbury. 1982.Topography of
phos-phateresidues inSendaivirusproteins. Virology120:225-234.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.181.414.71.228.2]4364 CURRAN ET AL.
17. Hudson, L. D., C. Condra, and R. A. Lazzarini. 1986.Cloning and expression of a viral phosphoprotein: structure suggests vesicular stomatitisvirus NS may function by mimicking an RNAtemplate. J. Gen. Virol. 67:1571-1579.
18. Lamb, R. A., B. W. J. Mahy, and P. W. Choppin. 1976. The synthesis ofSendaivirus polypeptides in infected cells. Virol-ogy69:116-131.
19. Lyn, D., D. S.Gill, R. A. Scroggs, and A. Portner. 1991. The nucleoproteinsofhumanparainfluenzavirus type 1 and Sendai virus share amino acid sequences and antigenic and structural determinants. J.Gen. Virol. 72:983-987.
20. Lynch, S., and D. Kolakofsky. 1978. Ends oftheRNAwithin Sendaivirusdefectiveinterferingnucleocapsidsare notfree. J. Virol. 28:584-589.
21. Matsuoka, Y., and R. Ray.1991. Sequenceanalysis and expres-sion of the human parainfluenza type 1 virus nucleoprotein gene. Virology 181:403-407.
22. Middleton, Y., M. Tashiro, T. Thai, J. Oh, J. Seymour, E. Pritzer, H.-D.Kienk,R.Rott,andJ. T.Seto. 1990. Nucleotide sequenceanalysesof the genesencoding the HN,M, NP,P, and Lproteins oftwohost range mutants of Sendaivirus.Virology 176:656-657.
23. Morgan, E.M., G. G. Re, and D. W. Kingsbury. 1984.Complete sequence ofthe Sendaivirus NP gene from a cloned insert. Virology135:279-287.
24. Mountcastle,W.E., R. W. Compans,H.Lackland, and P. W. Choppin.1974.Proteolytic cleavageofsubunits of the
nucleo-capsidoftheparamyxovirussimian virus 5. J. Virol. 14:1253-1261.
24a.Moyer,S. Personal communication.
25. Neubert, W. J., C. Eckerskorn, and H. E. Homann. 1991. Sendai virus NP genecodes for a 524 amino acid NPprotein. Virus Genes5:25-32.
25a.Parks,G. D.,C. D.Ward, and R. A. Lamb. 1992. Molecular cloningof the NP and L genes ofsimian virus 5: identificationof highlyconserved domains inparamyxovirusNP and Lproteins. Virus Res. 22:259-279.
26. Perrault, J., G. M. Clinton, and M.A. McClure. 1983. RNP templateof vesicularstomatitis virus regulates transcriptionand replicationfunctions. Cell35:175-185.
26a.Shioda,T.Personalcommunication.
27. Shioda, T., K. Hidaka, H.Shibuta, A. Nomoto, and K. Iwasaki. 1983.Sequenceof3,687 nucleotides from the 3' endof Sendai virusgenomeRNA and thepredicted aminoacid sequences of viralNP,P and Cproteins. Nucleic Acids Res. 11:7317-7330. (Corrigendum, 12:3726, 1984.)
28. Vidal, S., and D. Kolakofsky. 1989. Modified model for the switch from Sendai virustranscriptiontoreplication. J. Virol. 63:1951-1958.
29. Yuasa, T., H. Bando, M.Kawano, M. Tsurudome, M. Nishio, K. Kondo, H. Komada, and Y. Ito.1990.Sequenceanalyses ofthe 3' genome end and NP gene of human parainfluenza type 2 virus: sequence variation ofthe gene-starting signal and the conserved 3'end.Virology179:777-784.
J. VIROL.