Received 12 June 1997/Accepted 18 November 1997
Differential viral gene expression during both productive and persistent infections of Hz-1 virus in insect
cells was elucidated. Despite more than 100 viral transcripts being expressed during productive viral infection,
massive viral gene shutoff was observed during viral persistency, leaving the 2.9-kb persistence-associated
transcript 1 (PAT1) as the only detectable viral RNA. Persistence-associated gene 1 (pag1), which encodes
PAT1, was cloned and found to contain no significant open reading frames. PAT1 is not associated with the
cellular translation machinery and is located exclusively in the nucleus. Further experiments showed that
PAT1 is functional in the establishment of persistent Hz-1 viral infection in the cells. All the evidence
collectively indicates that PAT1 is a novel nuclear transcript of viral origin. Our results showed that although
PAT1 and XIST RNA, a mammalian X-inactive specific transcript, are transcribed by different genes, they have
interesting similarities.
Persistent viral infection has been reported to occur
natu-rally in insects and cultured cells. Changes in rearing
temper-ature, the presence of high humidity, a decrease in food
qual-ity, superinfection with different viruses, and/or other stimuli
may activate persistent infections (6, 11, 21, 22, 34, 46).
How-ever, due to the difficulty in establishing persistent viral
infec-tions in laboratory stocks of insects, persistent viral infecinfec-tions
are usually studied only after unexpected viral activation from
previously healthy-looking insects or insect cells (21, 34, 46).
Hz-1 virus (also called Hz-1 baculovirus or Hz-1V) was
orig-inally identified in a persistently infected IMC-Hz-1 cell line
from the ovarian tissue of Heliothis zea (15). It is capable of
establishing persistent infection in various insect cells (6, 10,
36). Such persistent infection may be reactivated to resume
productive infection upon infection by heterologous viruses
(6). Persistently infected cell cultures are resistant to
superin-fection by homologous viruses due to the induction of
apopto-sis (26). Like baculoviruses (2, 44), Hz-1 virus is rod shaped
with a circular, double-stranded 228-kb DNA genome (9, 20).
It was previously referred to as the type species of the
subfam-ily Nudibaculovirinae in the famsubfam-ily of Baculoviridae (45).
Re-cently, Hz-1 virus and other nonoccluded baculoviruses were
removed from the baculovirus family, and they are temporarily
unclassified (44).
Hz-1 virus can establish both productive and persistent
in-fections in several lepidopteran cell lines (6). Upon productive
infection, more than 100 different viral transcripts can be
de-tected (10), and the infected cells eventually die by necrosis
(26). However, a very small proportion of the infected cells,
usually less than 0.2%, grow and become persistently infected
clones. In these cells, only one 2.9-kb viral transcript was
de-tectable in Sf9, Sf21, and TN368 cells that were persistently
infected with Hz-1 virus (reference 10 and data not shown),
and this RNA species was named persistence-associated
tran-script 1 (PAT1).
While little is known concerning persistent viral infections in
insects, there are several relatively well-studied examples in the
herpesvirus system. For instance, approximately 12 genes are
expressed during latent infection by Epstein-Barr virus, but
more than 50 viral genes are expressed in its lytic phase of viral
growth (24, 31). Herpes simplex virus probably expresses more
than 70 genes during productive viral infection, but only 3
related latency-associated transcripts (LATs) are detectable
during latent viral infection (39, 42). The study of similar
differential viral gene expression in viruses other than
mem-bers of the herpesvirus family would provide useful
informa-tion for understanding the molecular basis of persistent or
latent viral infections in eukaryotic cells.
Previously, we localized the region transcribing PAT1 within
the EcoRI-M fragment of the Hz-1 viral genome (10). In the
present study, persistence-associated gene 1 (pag1), which
en-codes PAT1, was cloned and characterized. We found that
PAT1 is not associated with the cellular translation machinery
and, interestingly, that it is located exclusively in the nucleus,
where PAT1 functions in the establishment of persistent Hz-1
viral infection.
MATERIALS AND METHODS
Cell lines.Cell line TN368 was derived from Trichoplusia ni (18), and the other two cell lines, Sf21 and Sf9, were from Spodoptera frugiperda. Persistently in-fected cells, TNP3 cells (10), were derived from TN368 cells; and SfP2 cells were derived from Sf21 cells. All cells were maintained at 26°C in TNM-FH medium supplemented with 8% fetal bovine serum (Gibco BRL Life Technologies).
DNA sequencing, primer extension, and RNase protection analysis. Progres-sive deletion clones were constructed in both directions from the viral genomic EcoRI-M fragment by using an exonuclease III-mung bean nuclease technique (17). The nucleotide sequence was determined directly from double-stranded plasmid DNA by the dideoxynucleotide chain termination method (12). Both strands were sequenced at least twice. Computer open reading frame (ORF) analysis of pag1 was done by using the Sequence Analysis Software Package of the Genetics Computer Group (GCG) (University of Wisconsin Biotechnology Center). Clusters of direct repeats were evaluated by self-comparison analysis of the pag1 sequence with a Dotplot program from the same software package.
The transcription start site was determined by primer extension. A 35-bp
* Corresponding author. Mailing address: Institute of Molecular
Biology, Academia Sinica, Nankang, Taipei, Taiwan, Republic of
China. Phone: 886-2-2788-2697. Fax: 886-2-2788-2697 or
886-2-2782-6085. E-mail: [email protected].
† Present address: Department of Biology, National Cheng Kung
University, Tainan, Taiwan, Republic of China.
2233
on November 9, 2019 by guest
http://jvi.asm.org/
primer from nucleotide 1109 (39) to 1143 (59), antisense to PAT1, was used. Total RNAs (25mg) extracted from both productively infected TN368 cells and persistently infected TNP3 cells were used as templates for the experi-ments.
The 39 end of PAT1 was mapped by RNase protection assays. Plasmid pHzEM-C, which contains only subfragment C of the EcoRI-M viral genome fragment, was used (Fig. 1A). Antisense probes were transcribed, using T3
polymerase, from pHzEM-C, which was restricted with HpaI and hybridized to the total RNAs (30mg) from persistently infected cells. After RNase A and T1digestion, the protected fragments were fractionated on 6% polyacrylamide–urea gels.
Cloning of overlapping cDNA fragments by RNA PCR (reverse transcriptase PCR).cDNA fragments were amplified by RNA PCR. By using paired primer sets, the H, M1, M2, M, and T fragments (see Fig. 3) were amplified, cloned, and sequenced. The region covered by the P fragment (see Fig. 3) is the promoter
on November 9, 2019 by guest
http://jvi.asm.org/
region, which cannot be generated by reverse transcriptase PCR and thus served as a negative control.
Analysis of the promoter region of pag1.Progressive deletions from both ends of the 59regulatory region of pag1 were generated by PCR and further verified by DNA sequencing. For DNA deletion analysis of the region specifying the 59-end sequence of PAT1, five progressive deletion fragments were synthesized and ligated separately upstream of an intact luciferase-coding region (see Fig. 4), and then 53105 Sf9 cells were cotransfected with 1mg of plasmid DNA containing each of the above-mentioned deletion constructs and a construct containing a chloramphenicol acetyltransferase (CAT)-coding sequence driven by the Drosophila actin promoter (0.25mg). The latter construct was used as an internal control to normalize the efficiency of transfection.
The 59regulatory region further upstream from the transcription start site, within positions2727 to129, was also analyzed by progressive deletion analyses and ligated separately upstream of a full-length lacZ coding sequence in the plasmid pTSV-2 (28). Sf9 cells (53105) were then cotransfected with two different plasmids: plasmid pTSV-2, containing progressive deletions of the pag1 promoter region driving an intact lacZ coding sequence (1mg), and a construct containing the CAT-coding sequence driven by the Drosophila actin promoter (0.25mg). The latter construct was again used as an internal control to normalize the efficiency of transfection. LacZ expression was determined by assayingb -ga-lactosidase activity as previously described (28). Briefly, 40ml of cell lysate was mixed with 160ml of reaction cocktail (containing 25 mM Tris-HCl [pH 7.5], 125 mM NaCl, 2 mM MgCl2, 12 mMb-mercaptoethanol, and 0.3 mM 4-methylum-belliferyl-b-D-galactoside) and incubated at 37°C for 30 min, and then 100ml of
the sample was aspirated into 2 ml of glycine-carbonate reagent and the
fluo-rescence was read with the TKO-100 Mini Fluorometer (Hoefer Scientific In-struments).
Polysome fractionation.The procedures for isolation and analysis of poly-somes described by Schmidt and Merrill (37) were used. TNP3 cells (107) were lysed and subjected to sucrose gradient centrifugation. One-tenth aliquots of the fractionated RNA were assayed by Northern blotting.
Nuclear localization.Cells were first fractionated into nuclear and cytoplasmic fractions by the procedure of Summers and Smith (43). Nuclear RNA was extracted from the nuclear pellets with guanidinium thiocyanate. Five micro-grams of each of the resultant RNAs was serially diluted and slot blotted. Two in vitro-transcribed probes containing either pag1 (Fig. 1, EcoRI-M fragment) or actin sequences from the genome of Drosophila (16) were used for hybridization by both Northern and slot blotting.
Preparation of RNA probes for fluorescence in situ hybridization.An RNA probe was made for fluorescence in situ hybridization of the persistently infected or stable transfected cell lines as follows: the 0.7-kb subfragment E (restricted by KpnI) of the EcoRI-M fragment (Fig. 1A) was cloned into plasmid pBluescript KSM1(Stratagene), and a 0.7-kb RNA probe labeled with digoxigenin-11-UTP was produced by in vitro transcription with T3 RNA polymerase (Boehringer Mannheim Biochemicals).
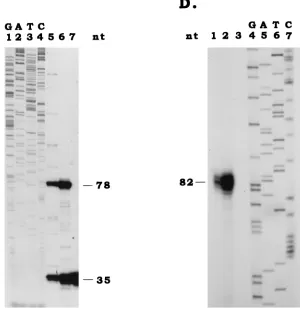
[image:3.612.159.459.73.384.2]PAT1 detection by fluorescent in situ hybridization.Sf9, SfP2, and SfPAG1-1 cells were resuspended in fresh medium at a concentration of 106cells/ml, and then 50ml of this suspension was pipetted onto the center of the premarked area on an in situ PCR glass slide (Perkin-Elmer Applied Biosystems). Slides were then incubated at 26°C for 1 h, followed by 4% paraformaldehyde fixation in phosphate-buffered saline (PBS) for 30 min. After fixation, paraformaldehyde FIG. 1. Location and sequence of pag1. (A) Map and location of PAT1 coding region. The first line represents percentages of the viral genome, the EcoRI map of the linearized 228-kb Hz-1 viral genome (9) is shown in the second line, the KpnI map of the EcoRI-M fragment is shown in the third line, the region sequenced is shown in the fourth line, and the orientation and transcriptional region of PAT1 are shown in the fifth line. The letters above the lines denote the restricted viral DNA fragments, and those below the lines denote restriction sites (Kpn, KpnI; Eco, EcoRI). (B) Nucleotide sequence of pag1. The putative AP1 consensus sequence and putative GATA, CAAT, and TATA motifs are underlined. CAGT, the conserved transcription start sequence for baculovirus early genes, is boxed. The transcription start site is indicated by an arrow and the transcription termination site is marked by an asterisk. (C) Primer extension was used to determine the transcription start site of PAT1. Lanes 1 to 4, sequence ladders. The extended (78-bp) and the primer (35-bp) bands derived from the total RNAs extracted from productively infected TN368 cells (lane 5), persistently infected TNP3 cells (lane 6), and healthy parental TN368 cells (lane 7) are shown. (D) RNase protection was used to map the 39end of PAT1. A32P-labeled RNA probe was transcribed by using T3 polymerase from subfragment C of viral EcoRI-M fragment. Before in vitro transcription, subfragment C was digested with restriction enzyme HpaI to generate an EcoRI-HpaI single-stranded antisense RNA probe (see panel A). Lanes 4 to 7, are sequence ladders. Three closely associated bands were protected for productively infected TN368 cells (lane 1) and persistently infected cells TNP3 (lane 2). The major protected 82-bp band is marked. No protection was observed for RNA extracted from uninfected TN368 cells (lane 3). nt, nucleotides.
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 2. DNA sequence analysis of pag1. (A) Computer ORF analysis of pag1. ORFs are indicated as open boxes. ATG initiation codons are indicated by a vertical line, and termination codons are indicated by a vertical line bisecting the horizontal. The orientations of the top three frames are the same as for PAT1. The orientations of the bottom three reading frames are opposite to those for PAT1. The transcriptional region of PAT1 is indicated by an arrow. (B) Relative locations of clustered repeats (a, b, and c) on PAT1. (C) Sequences and positions of three major clustered direct repeats (a, b, and c). Only those repeats larger than 10 bases are included.
on November 9, 2019 by guest
http://jvi.asm.org/
was inactivated by washing the slides twice (2 min each) in PBS. The labeled RNA probe in hybridization buffer (23SSC [13SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 5% dextran sulfate, 0.2% bovine serum albumin, and 50% formamide) was applied on each monolayer cell and assembled into AmpliCover discs and AmpliCover clips (Perkin-Elmer Applied Biosystems). Each assembled slide was transferred to a GeneAmp in situ PCR System 100 set for a 40°C soak program and incubated overnight. After hybridization, the slides were washed three times with 23SSC at 65°C. For the detection of the labeled probe, slides were incubated with 0.25% Triton X-100 in PBS for 5 min and blocked for 30 min with 10% fetal bovine serum (Gibco BRL Life Technologies) in PBS containing 0.25% Triton X-100. Subsequently, the slides were incubated with 0.4mg of antidigoxigenin fluorescein-conjugated antibodies per ml diluted in 0.25% Triton X-100 in PBS at room temperature for 1 h and then washed three times in PBS containing 0.25% Triton X-100. Finally, slides were incubated in PBS containing 1mg of DAPI (49,69diamidino-2-phenylindole) (Molecular Probes) per ml for 5 min and washed briefly with PBS.
Establishment of stably transfected cell lines SfPAG and SfPKN.Two plas-mids, pPAGN and pKN, were used to separately transfect Sf9 cells. Plasmid pPAGN contains two genes. The first is a full-length pag1 gene driven by its own promoter and is derived from the EcoRI-M fragment of Hz-1 virus (10) (Fig. 1). The second is a neomycin resistance gene driven by the heat shock promoter of Drosophila (41). Plasmid pKN contains only a neomycin resistance gene driven by the hsp70 promoter (41). Cellfectin (Gibco BRL Life Technologies) was used to transfect Sf9 cells (23105cells per well of a 24-well plate) with 1mg of either pPAGN or pKN. Twelve hours after transfection, the cells were replaced with fresh medium and incubated at 26°C for 24 h. Following this, the cells were maintained for 10 days in TNM-FH medium containing 2 mg of G418 per ml. The stably pag1-transfected clones SfPAG1-1, SfPAG1-2, SfPAG2-1, and SfPAG2-2 were established and isolated from different transfection experiments.
Challenge of stably or transiently pag1-transfected cells with Hz-1 virus.
Stably transfected pag1 cells were seeded at 43104cells per well onto 96-well plates. Cells were challenged with Hz-1 virus (multiplicity of infection [MOI]5
1). The number of persistently infected cell colonies was determined at 10 days after viral infection (dpi). In transient-transfection assays, Sf9 cells were trans-fected with pPAGN and pKN plasmids separately. Twelve hours after transfec-tion, the cells were infected with Hz-1 virus (MOI51), and the number of persistently infected colonies was again calculated at 10 dpi. The criteria we used to score persistently infected colonies were as follows: (i) the cells survived productive viral infection; (ii) the cells grew and formed a colony; (iii) the colony contained more than five cells at 10 dpi (colonies containing fewer than five cells/clone have little chance of survival and are thus difficult to propagate and assay regardless of whether they are persistently infected by Hz-1 virus); and (iv) the cells were resistant to superinfection with Hz-1 virus.
Simultaneous detection of viral DNA and antigen in cells infected by Hz-1 virus.Parental Sf9 cells, productively Hz-1 virus-infected Sf9 cells (MOI52; 18 h postinfection), persistently infected SfP2 cells, and newly established per-sistently infected SfPAG1-1P cells were used. These cells were derived from the infection of SfPAG1-1 cells with Hz-1 virus. After establishment, cells were mixed and passed four times before in situ analyses. They were first fixed in ethanol-acetic acid (3:1) at220°C for 30 min. One drop of cell suspension was added to 70% ethanol-cleaned dry slides. After spreading of the cells, the slides were dried at 60°C for 1 h followed by dehydration in ethanol at increasing concentrations (70, 80, 90, and 100%). DNA was denatured by incubation in 23
SSC–70% formamide (pH 7.0) for 2 min at 70°C. After being rinsed for 2 min in 23SSC, the slides were dehydrated again as described above.
A 0.3-kb HincII subfragment derived from HindIII-K of the viral genome (9) was cloned into plasmid pBluescript KSM1(Stratagene Cloning System), and a 0.3-kb RNA probe labeled with digoxigenin-11-UTP was produced from this plasmid by in vitro transcription with T3 RNA polymerase (Boehringer
Mann-heim Biochemicals). Probes diluted in hybridization buffer (23SSC, 5% dextran sulfate, 0.2% bovine serum albumin, and 50% formamide) were then denatured at 75°C for 5 min, chilled on ice, and placed on the slide, and then the Ampli-Cover discs and AmpliAmpli-Cover clips (Perkin-Elmer) were reassembled. For hy-bridization, slides were placed at 40°C overnight in the GeneAmp in situ PCR System 100.
After hybridization, slides were washed four times for 5 min each in 23
SSC–50% formamide at 60°C and three times for 10 min each in 13SSC at 60°C. Slides were incubated with 0.3% Triton X-100 in PBS for 5 min and blocked for 30 min with 10% fetal bovine serum (Gibco BRL Life Technologies) in PBS containing 0.3% Triton X-100. Similar techniques with modifications were used for the detection of the viral antigen (26). Briefly, slides were incubated for 1 h at room temperature with rabbit polyclonal anti-Hz-1 virus antibody diluted at 1:1,000 in PBS containing 0.3% Triton X-100. After three washes with PBS–0.3% Triton X-100, Cy5-conjugated goat anti-rabbit immunoglobulin G (Jackson Im-munoResearch Laboratories) was added at a dilution of 1:500 in PBS containing 0.3% Triton X-100 and incubated for 1 h. Subsequently, the slides were incu-bated with 0.4 mg of antidigoxigenin fluorescein-conjugated antibodies per ml diluted in 0.3% Triton X-100 in PBS at room temperature for 1 h and then were washed in three changes of 0.3% Triton X-100 in PBS. Finally, slides were incubated in PBS containing 0.5 mg of propidium iodide (Molecular Probes) per ml for 5 min, washed briefly with PBS, and visualized with a Laser Scanning confocal microscope (Zeiss LSM 310).
Nucleotide sequence accession number.The sequence reported in this paper has been deposited in the GenBank database (accession no. U03488).
RESULTS
No significant ORFs except clustered repeated sequences
were found in the PAT1 coding region.
Previously we showed
that the PAT1 sequence resides in the EcoRI-M fragment of
the viral genome (10). This fragment was further subdivided
into A, B, C, D, and E fragments by using the restriction
enzyme KpnI (Fig. 1A, EcoRI-M fragment). The coding region
of PAT1 and the putative promoter region were sequenced
(Fig. 1A and B).
The initiation and termination sites of PAT1, which were
roughly analyzed previously by using RNase protection assays
in agarose gels, were found to localize in fragments B and C,
respectively (Fig. 1A) (10). To position both the 5
9
and 3
9
ends
of PAT1 precisely, primer extensions and RNase protection
anal-yses in a high-resolution polyacrylamide gel were undertaken.
[image:5.612.68.528.73.197.2]A 78-base extension product (Fig. 1A and C) was observed
by using a 35-base primer beginning at position 1143 (Fig. 1A),
suggesting that the transcript was initiated from nucleotide A
at position 1066 (Fig. 1B). This position is very close to a
conserved transcription start sequence, CAGT, of baculovirus
early transcripts (Fig. 1B, boxed). For the transcription
termi-nation site of PAT1, the RNase protection experiment
indi-cated the protection of a major 82-base band (Fig. 1A and D),
which can be mapped to nucleotide C at position 4002 (Fig.
1B), and several closely associated weaker bands (Fig. 1D).
FIG. 3. Amplification of PAT1 cDNA fragments by RNA PCR. Overlapping fragments covering the entire 2.8-kb cDNA were amplified by using paired primers. Regions of the amplified fragments H, M, M1, M2, and T were sequenced. The experiments, from RNA PCR to sequencing, were repeated twice. The P fragment, which resides in the promoter region, was subjected to RNA PCR amplification to serve as a negative control.on November 9, 2019 by guest
http://jvi.asm.org/
Analysis with the GCG Sequence Analysis Software Package
showed, interestingly, that the coding region of PAT1 contains
no significant ORF in any forward or reverse translation
frames (Fig. 2A). GCG Dotplot analysis showed that pag1
contains several clustered direct repeats. These repeats were
organized primarily into three clusters within nucleotides in
the regions of positions 1400 to 1550, 1800 to 2000, and 2100 to
2200 (Fig. 2B and C). The lack of a significant ORF together
with unusually clustered repeats suggested that PAT1 may not
encode a protein.
The cDNA sequence of PAT1 is identical to that of genomic
DNA.
Overlapped cDNAs of PAT1 were amplified and cloned
from persistently infected TNP3 cells (10) by using an RNA
PCR technique. Multiple primers were used to amplify H (251
bp), M (1,638 bp), M1 (870 bp), M2 (809 bp), and T (1,320 bp)
cDNA fragments which run across the entire PAT1 coding
region (Fig. 3). Since it was previously found that no transcript
other than PAT1 was detectable during persistent viral
infec-tion, a pair of primers was also used to amplify the promoter
region to serve as a negative control. The sequence of PAT1
cDNAs was determined and was found to be identical to the
sequence of genomic DNA, indicating that RNA editing or
splicing does not occur after transcription.
Viral factors are not essential for pag1 transcription.
It has
been shown previously that PAT1 can be detected very early
during productive viral infection and that it is the only
virus-specific transcript detectable during persistent viral infection
(10). To test whether the expression of PAT1 is independent
from the expression of other viral genes, a plasmid, pHzE-M,
which contains only the putative promoter and the PAT1
cod-ing region (10) was transfected into Sf9 cells. At 4 and 8 h after
transfection, total RNA was extracted and analyzed by
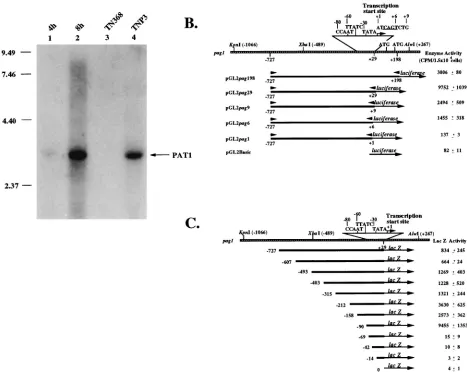
North-FIG. 4. Promoter activity analysis of pag1. (A) Transfection of pag1 into host cells, indicating that viral factors are not essential for PAT1 transcription by the pag1 promoter. Plasmid pHzE-M (10), which contains the entire pag1 gene, was transfected into Sf9 cells. Total RNAs were harvested and analyzed by Northern hybridization. PAT1 signals can be found at 4 and 8 h after transfection. Total RNAs harvested from parental TN368 cells and persistently infected TNP3 cells were also used as negative and positive controls, respectively. The probe used for this analysis was a gel-purified subfragment E derived from the viral genomic EcoRI-M fragment (see Fig. 1A). Numbers on the left are in kilobases. (B) Progressive deletion analysis at the 39end of the upstream regulatory region of pag1. These fragments were ligated with the protein-coding region of the luciferase gene, and the activity of this enzyme was determined after the transfection of the constructs into Sf9 cells. All of the constructs were cotransfected with another construct containing a Drosophila actin promoter-driven CAT gene as an internal control. Data (means6standard deviations) were collected from triplicate assays in three independent experiments. (C) Progressive deletion analysis at the 59end of the upstream regulatory region of pag1. All the fragments ended at129 bp and were ligated with the protein-coding region of the lacZ gene. CAAT, GATA (TTATC), and TATA motifs are shown. Following transfection of the constructs into Sf9 cells, LacZ activities were determined. All of the constructs were cotransfected with a construct containing a Drosophila actin promoter-driven CAT gene to serve as negative controls. One unit of LacZ activity is equal to the intensity emitted by 0.1 nM 4-methylumbelliferone. Data (means6standard deviations) were collected from triplicate assays in three independent experiments.on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.77.544.68.440.2]ern hybridization. Figure 4A shows that PAT1 was detected 4 h
after transfection and that the intensity of the signal had
in-creased greatly by 8 h after transfection. These results
indi-cated that host factors alone were sufficient for the
transcrip-tion of PAT1, although it was still possible that some viral
factors further modulate the expression of pag1 upon infection
with Hz-1 virus.
The pag1 promoter is close to the transcriptional start site
of PAT1.
To further identify and characterize the promoter of
pag1, progressive deletions from both upstream and
stream sequences were constructed and analyzed. For
down-stream progressive deletions, regions between fixed updown-stream
position
2
727 and various downstream positions, i.e.,
1
1,
1
6,
1
9,
1
29, and
1
198, were each cloned and fused to a
lucif-erase-coding sequence (Fig. 4B). Luciferase activity was
ana-lyzed after transfection of these constructs into Sf9 cells. The
results showed that upstream sequences between nucleotide
2
727 and the start site at nucleotide
1
1 gave rise to weak
luciferase activity. The luciferase activity gradually increased as
the 3
9
end of the promoter region was extended to nucleotide
1
29. However, the promoter activity dropped again upon
fur-ther extension to nucleotide
1
198, where two ATG codons are
found between nucleotides
1
29 and
1
198 (Fig. 4B). The
re-sults showed that nucleotides
1
1 to
1
29 downstream of the
transcription start site were required for better expression of
the ligated luciferase sequence (Fig. 4B).
To analyze the upstream sequence required for promoter
activity, the region between nucleotides
2
727 and
1
29 was
first ligated to a LacZ-coding sequence (Fig. 4C). Plasmids
containing progressive deletions from the upstream region
were transfected separately into Sf9 cells, and the intensity of
lacZ expression from each promoter deletion construct was
analyzed. Rather similar levels of promoter activity were
ob-served in the constructs containing nucleotides
2
727/
1
29 to
2
315/
1
29. Activity gradually increased when the construct was
deleted up to nucleotide
2
158. The best promoter activity was
observed when the construct was further deleted to nucleotide
2
90, which still retained the putative CAAT and TATA boxes
and a GATA motif. Further deletion into the nucleotide
2
90/
1
29 region abolished the activity of the promoter, indicating
that the closely associated TATA box and CAAT and GATA
motifs were crucial for PAT1 expression (Fig. 4C).
[image:7.612.53.547.69.404.2]PAT1 is not associated with ribosomes and is localized
pre-dominantly in the nucleus.
Although the GCG computer
pro-gram predicted that PAT1 lacks protein-coding potential (Fig.
2A), it was necessary to test whether PAT1 is associated with
the cellular protein synthesis machinery. We therefore
exam-ined polysome profiles in postmitochondrial fractions obtaexam-ined
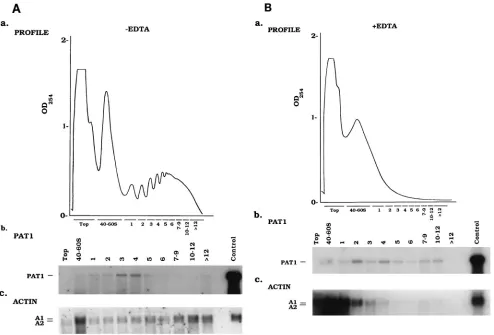
FIG. 5. Polysome fractionation of persistently infected TNP3 cells. (A) Postmitochondrial lysates collected from 107persistently infected TNP3 cells were subjected to sucrose gradient centrifugation. (a) Profile of optical density at 254 nm of postmitochondrial lysates. Each fraction was then collected and analyzed by Northern hybridization with either a pag1 (b) or actin (c) probe. One-tenth of the total RNA extracted from 107TNP3 cells was loaded into the control lanes prior to fractionation to serve as a control. (B) (a) Ribosome knockout by EDTA treatment in the polysome fractionation experiment. A profile of the optical density at 254 nm of the gradient isolated from 107cells (a) and Northern analysis of each fraction with either a pag1 (b) or an actin (c) probe are shown. One-tenth of the total RNA extracted from 107TNP3 cells was loaded into the control lanes prior to fractionation to serve as a control. The probe used for pag1 hybridization was the same as in Fig. 4A, and the actin probe used was the 0.6-kb BglII/SalI subfragment of the original 1.8-kb Bombyx mori actin clone in pGem (32).on November 9, 2019 by guest
http://jvi.asm.org/
from persistently infected TNP3 cells. As a control, polysomes
were dissociated from mRNA by adding EDTA. Gradient
frac-tions were then assayed for PAT1 RNA by Northern blot
analysis. A low level of PAT1 signal was detected throughout
gradient fractions either without (Fig. 5A) or with (Fig. 5B)
EDTA treatment. These results indicated that PAT1 was not
associated with ribosomes. In contrast, actin mRNA was
de-tected mainly in the heavy polysome regions when EDTA was
omitted (panel c in Fig. 5A). However, in the presence of
EDTA, the majority of actin mRNA shifted dramatically to the
free ribosome fractions (panel c in Fig. 5B).
Total signal intensities of PAT1 from all postmitochondrial
fractions were found to be much lower than PAT1 signals in
the control lanes which contained unfractionated total cellular
RNAs (panel b in Fig. 5). These results are contradictory to
those for the control actin signals, in that the intensities of all
postmitochondrial fractions should be 10 times that of the
control lanes (panel c in Fig. 5), thus suggesting that the
ma-jority of PAT1 may not be in the postmitochondrial fractions.
To determine the distribution of PAT1 in the cell, RNAs from
both isolated nuclei and pooled cytoplasmic fractions of TNP3
cells were analyzed separately in slot blots. The results
indi-FIG. 6. PAT1 is localized in the nucleus. (A) Slot blots of nuclear (Nu) and cytoplasmic (Cyt) RNAs from persistently infected TNP3 cells. Slot blots with a series of 103dilutions of nuclear and cytoplasmic RNAs start from 5mg per slot. The blots were hybridized with a pag1 (a) or actin (b) probe. The pag1 and actin probes used for this analysis were the same as for Fig. 5. (B) Fluorescent in situ hybridization showing that PAT1 is localized in the nucleus. (a1, b1, and c1) PAT1 fluorescent in situ hybridization of persistently infected SfP2 cells, stably pag1-transfected SfPAG1-1 cells, and parental Sf9 cells, respectively. (a2, b2, and c2) DAPI-stained nuclei of the cells in panels a1, b1, and c1, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
cated that PAT1 was present almost exclusively in the nuclear
fraction, with less than 1% of the PAT1 signal detected in the
cytoplasm (Fig. 6A, panel a). In contrast, over 90% of actin RNA
was found in the cytoplasmic fraction (Fig. 6A, panel b). Results
[image:9.612.108.488.64.598.2]of fluorescent in situ hybridization provided additional proof that
PAT1 is indeed localized in the nucleus regardless of whether it is
expressed by the genome of the virus (Fig. 6B, panel a) or by the
stably transfected pag1 gene in the cells (Fig. 6B, panel b).
FIG. 7. The pag1 gene functions in the establishment of persistent viral infection. (A) Map of the plasmids as it appeared in this experiment. Arrows indicate the regions of the promoters and their directions of gene expression. Phsp70, hsp70 promoter; Ppag1, pag1 promoter; neo, neomycin resistance gene; pag1, PAT1 coding region; pKSMII, bacterial plasmid vector (Stratagene). (B) Generation of persistently infected clones by Hz-1 virus infection in the parental and stably pag1-transfected cells. Sf9, untransfected host cells. SfPKN3H and SfPKN4H are two cell lines stably transfected with only the neomycin resistance gene. The stably pag1-transfected clones, SfPAG1-1, SfPAG1-2, SfPAG2-1, and SfPAG2-2, were established from different transfection experiments. Sf9 cells transiently transfected with plasmids containing pag1 (pPAGN) were also tested. In these experiments, cells (43104) were challenged with Hz-1 virus, and the numbers of surviving persistently infected cell clones were calculated. Data (means6standard deviations) were collected from triplicate assays of three independent viral infection experiments. (C) Colonies which were established by the infection of Hz-1 virus in the parental Sf9 cells (a) and the stably pag1-transfected SfPAG1-1 cells (b).on November 9, 2019 by guest
http://jvi.asm.org/
on November 9, 2019 by guest
http://jvi.asm.org/
of Hz-1 virus with the green fluorescent protein (GFP) gene of
the jellyfish Aequorea victoria (7, 8). After cotransfection of the
GFP gene-containing transfer plasmid and the viral DNA, the
supernatants containing viruses were harvested. Emission of
green fluorescence in individual cells could be detected when
cells were infected at a high multiplicity of viruses from the
supernatant. This observation suggested that recombinant
vi-ruses containing the GFP gene were formed. However, green
fluorescence was no longer detectable in any cells when the
supernatants were highly diluted, suggesting that pag1 is likely
to be an essential gene in the life cycle of the virus. In addition,
we reported previously that there are at least two other large
viral transcripts which traverse the coding region of PAT1
during productive viral infection (10). If one or both of these
transcripts are essential for the completion of productive viral
infection, then the removal of pag1 from the viral genome will
also be fatal to viral replication. This observation may explain
why the pag1-deleted Hz-1 virus could not be constructed even
in cells stably transfected with pag1.
Because pag1-deleted Hz-1 virus could not be constructed in
cells stably transfected with pag1, another approach was taken.
pag1 was ligated with a neomycin resistance gene (Fig. 7A) and
transfected into Sf9 cells which were free of infection with Hz-1
virus. Sf9 cell clones stably transfected with pag1 were isolated
and propagated in monolayers before infection with the Hz-1
virus. Fluorescent in situ hybridization showed that PAT1 was
properly expressed and retained in the nuclei of these stably
pag1-transfected cells (Fig. 6B, panel b). Most of the Sf9 cells
died when infected with Hz-1 virus, leaving only a small
per-centage of the cells that became persistently infected (Fig. 7B,
Sf9). The percentage of persistently infected cells did not
in-crease in cells stably transfected with the neomycin resistance
gene alone (Fig. 7B, SfPKN3H and SfPKN4H). However, the
number of persistently infected cell clones increased drastically
when Sf9 cell clones containing a stably integrated pag1 gene
were infected with Hz-1 virus (Fig. 7B, SfPAG1-1, SfPAG1-2,
SfPAG2-1, and SfPAG2-2).
Cloning and propagation of G418-resistant cells that were
stably transfected with the pag1 gene required a significant
amount of time, and many of the cell clones died during clonal
expansion. We were concerned that the death of part of the
cells might bias the response of the remaining clones to
infec-tion by Hz-1 virus, so the pag1 funcinfec-tion was also tested by
transient pag1 gene transfection. The results showed that
per-sistently infected cell clones again increased significantly in Sf9
cells transiently transfected with pag1 but not in Sf9 cells
tran-siently transfected with the neomycin resistance gene or in
untransfected cells (Fig. 7B and data not shown).
We found that the increase in persistently infected colonies
in pag1-containing cells was not merely due to a pag1 function
of increasing either colony formation or viral resistance,
be-cause our results showed that the colony formation rates were
the same between a plasmid containing only a neomycin
resis-tance gene or another plasmid, with further ligation of the pag1
virus infection according to assays of yields of viral progeny
(data not shown).
To determine if all or only some of the persistently infected
cells contain viral DNA and if the clones established by Hz-1
virus infection of pag1-containing cells were persistently virus
infected, these cells were analyzed by fluorescent in situ
hy-bridization (Fig. 8). The results showed that in cells
produc-tively infected with Hz-1 virus, viral DNA was detectable in
essentially all of the cells, and viral antigens were also
detect-able in many of the cells having undergone productive viral
infection (Fig. 8B). In the persistently infected SfP2 cells, viral
DNA was detectable in all of the cells (Fig. 8C), whereas viral
antigens were not visible (Fig. 8C, inset). Persistently infected
clones established by the infection of SfPAG1-1 cells were
analyzed after they were mixed and cultured for four passages.
Similar to the case for the persistently infected SfP2 cells
established by using non-pag1-containing cells, viral DNA (Fig.
8D) but no viral antigen (Fig. 8D, inset) was again detectable
in all persistently infected cells established from SfPAG1-1
cells. These results suggest that although upon viral infection
the number of clones was increased in cells stably transfected
with pag1, all of these cells was persistently virus infected, as
were the persistently infected clones derived from the parental
Sf cells.
DISCUSSION
The pag1 gene of Hz-1 virus was found to transcribe a
unique PAT1 RNA. Its lack of significant ORFs, lack of
poly-some binding, and localization in the nucleus argue that PAT1
may not encode a peptide. So far, PAT1 or related sequences
have not been reported to exist in any other viruses, including
the type baculovirus Autographa californica nuclear
polyhedro-sis virus. A computer gene bank search also did not find any
other DNA or RNA species which have significant sequence
homology to pag1.
[image:11.612.308.547.624.708.2]Although the PAT1 transcript initiates from position
1
1,
progressive deletions of the downstream promoter region
showed that the promoter activity increased drastically when
the CAGT motif, a conserved motif found in baculovirus early
TABLE 1. Comparisons between PAT1 and XIST RNA
Property PAT1 XIST RNA
Localization
Nucleus
Nucleus
Significant ORFs
No
No
Clustered repeats
Yes
Yes
Expression upon genome inactivation
Yes
Yes
Functions linked to genome inactivation
Yes
Yes
Sequence homology
a(%)
49
49
Size (kb)
2.9
15–17
aSequence homology in a window of 20 bases through a 2.9-kb region between PAT1 and XIST.
on November 9, 2019 by guest
http://jvi.asm.org/
genes (1, 35), was included (Fig. 4B, positions
2
727 to
1
1
versus
2
727 to
1
6). This observation suggested that this
CAGT motif may play a crucial role in the proper expression
of pag1 promoter. Progressive deletion of the upstream
pro-moter region showed that a 90-base region containing putative
CAAT, GATA (TTATC), and TATA motifs (Fig. 4C,
posi-tions
2
90 to
1
29) is also important for promoter activity.
Although high levels of LacZ expression were observed for the
progressively deleted pag1 promoter constructs containing
up-stream regions from positions
2
493 to
2
158 (Fig. 4C,
con-structs
2
493 to
1
29,
2
403 to
1
29,
2
315 to
1
29,
2
212 to
1
29,
and
2
158 to
1
29), further increases were observed with a
short
2
90 promoter construct in which all upstream regions
were removed (Fig. 4C,
2
90 to
1
29). These expression
pat-terns suggest that a repressor and/or activator from host cells
(or virus) may play a role in the expression of the pag1
pro-moter.
Collectively, the involvement of pag1 transcription with the
TATA box- and CAGT motif-containing regions and its
ter-mination at dinucleotide CA, which is 25 bases downstream
from the AATAAA motif, revealed that although pag1 does
not have a peptide-coding potential, it is still likely to be
tran-scribed by RNA polymerase II. Furthermore, peptides can be
properly synthesized if protein-coding genes, e.g., the
lucif-erase gene and lacZ, are ligated to the pag1 promoter (Fig. 4B
and C).
PAT1 is the only detectable viral transcript during persistent
Hz-1 virus infection (10). Similar global viral gene shutoff is
also observed on latent infection of herpesviruses in mammals,
where viral gene expression is limited to the transcription of
only one latency-associated gene which gives rise to three
nu-clear-localized RNAs, the LATs (39, 42). Mutational analysis
has demonstrated that LATs are not responsible for the
initi-ation of latent infection (19, 40). Rather, they could be
in-volved in herpes simplex virus type 1 reactivation (13, 29, 40),
although contradictory results have also been reported (3, 19,
30). The promoter predicted for LATs is over 660 bases
up-stream from their 5
9
ends, suggesting that LATs may be introns
of a larger unstable 8.3-kb RNA which is transcribed only 28
bases downstream from the promoter (14, 48). This suggestion
is also supported by the observation that some LAT RNA
species contain lariat structures (47).
The pag1 gene is located in a heavily transcribed region, and
many other transcripts traverse the PAT1 coding region in the
same orientation (10). However, PAT1 is unlikely to be an
intron of another longer transcript for the following reasons.
(i) Only PAT-1, and no other overlapping long transcripts, is
detectable during persistent viral infection (10). (ii) Unlike the
case for LATs, the TATA box of the pag1 promoter is only 25
bases upstream from the 5
9
end of PAT1. (iii) PAT-1 is readily
detectable when a viral EcoRI-M fragment which contains only
the promoter and coding region of pag1 is transiently
trans-fected (Fig. 4A) or stably transtrans-fected (Fig. 6B) into virus-free
cells. Fragment EcoRI-M contains the pag1 promoter but is
not long enough to contain the promoters or the transcription
start sites of the other transcripts initiating upstream from the
PAT1 coding region (10). (iv) The 90-base region directly
upstream from the transcription start site, which contains the
TATA, CAAT, and GATA motifs, is essential for promoter
activity. (v) The cDNA sequence combined from
PCR-gener-ated subfragments of PAT1 is the same as that of the genomic
DNA (Fig. 3 and data not shown). These results and
observa-tions show that the sequence of PAT1 is most likely to be
identical to that of pag1, although we can not exclude the
possibility that spliced minor PAT1-related RNA species exist.
PAT1 has some similarities with the human X-inactive
spe-cific transcript (XIST) (5) and its mouse counterpart (Xist) (4),
two recently described genes which map to the X-chromosome
inactivation center of mammals. This gene is expressed only
from the inactive X chromosome in which the majority of
X-linked genes are inactivated (4, 5, 23) and is required for
X-chromosome inactivation (27, 33; for a review, see reference
38). The 17-kb human XIST and 15-kb mouse Xist RNAs lack
significant ORFs. XIST/Xist RNA is not associated with the
translational machinery and is located almost exclusively in the
nucleus (4, 5). A unique feature of the XIST/Xist sequence is
the presence of several regions comprised of direct tandem
repeats. These repeats are conserved in both mice and humans,
suggesting that they may have functional significance (5).
It was suggested that the clustered repeats of XIST may
serve either as binding sites for nuclear attachment or as a
factor for X-chromosome inactivation to occur (5). Xlsirts, a
family of interspersed repeat RNAs that contain from 3 to 13
repeat units, is the Xenopus laevis homolog to the mammalian
XIST transcript. The Xlsirt RNA repeat sequences were found
to be required for translocation of RNAs to the vegetal cortex
(25). Currently, we lack information regarding the function of
clustered repeats of PAT1. They may function as signals for
nuclear localization or retention. Another possibility is that
they may serve as domains for PAT1 to bind to the activator
protein or to the viral genome. Alternatively, they may be
functional domains in the genome of the virus for interaction
with PAT1 or may serve as origins for genomic inactivation.
Experiments for further elucidating all of these possibilities are
in progress.
Although XIST RNA and PAT1 differ in sequence and it is
likely that their modes of function are different, similarities
between PAT1 and XIST RNA are still evident (Table 1). The
sequence homology between XIST RNA and PAT1 was
esti-mated to be 49% by computer analysis. However, the
homol-ogy is mainly due to the AT-rich nature of these two
tran-scripts, and highly homologous stretches were not found. At
present, it is not known whether PAT1 is directly responsible
for the establishment of persistent viral infection or only
en-hances this process. Even if the latter is true, the multifold
enhancement of persistent viral infection is significant and
warrants further investigation of its mechanisms. In addition,
further experiments are necessary to determine whether pag1
functions only in the establishment of persistent viral infection
and viral gene shutoff is a later consequence or, alternatively,
whether pag1 functions directly in the shutoff of viral gene
expression, which then results in persistent viral infection.
ACKNOWLEDGMENTS
We thank D. Chamberlin, D. Platt, and Eli Libas for editing the
manuscript; C. C. Wang, Karla Kirkegaard, and H. J. Kung for useful
revisions and discussions; and Chi-Wu Chen, Cherng-Yui Chang, and
Mi-I Hu-Tsai for technical assistance.
This work was supported by grants NSC86-2316-B-001-014 from the
National Science Council and BT-86-02 from Academia Sinica,
Tai-wan, Republic of China.
REFERENCES
1. Blissard, G. W., P. H. Kogan, R. Wei, and G. F. Rohrmann. 1992. A synthetic early promoter from a baculovirus: roles of the TATA box and conserved start site CAGT sequence in basal levels of transcription. Virology 190:783–793. 2. Blissard, G. W., and G. F. Rohrmann. 1990. Baculovirus diversity and
mo-lecular biology. Annu. Rev. Entomol. 35:127–155.
3. Block, T. M., J. G. Spivack, I. Steiner, S. Deshmane, M. T. McIntosh, R. P.
Lirette, and N. W. Fraser. 1990. A herpes simplex virus type 1 latency-associated transcript mutant reactivates with normal kinetics from latent infection. J. Virol. 64:3417–3426.
4. Brockdorff, N., A. Ashworth, F. K. Graham, V. M. McCabe, D. P. Norris,
P. J. Cooper, S. Swift, and S. Rastan.1992. The product of the mouse XIST
on November 9, 2019 by guest
http://jvi.asm.org/
Virol. 71:1265–1270.
10. Chao, Y. C., H. A. Wood, C. Y. Chang, H. T. Lee, and H. R. Lee. 1992. Differential gene expressions of Hz-1 baculovirus during viral productive and persistent infections. J. Virol. 66:1442–1448.
11. Chao, Y. C., S. Y. Young, K. S. Kim, and H. A. Scott. 1985. A newly isolated densonucleosis virus from Pseudoplusia includens (Lepidoptera:Noctuidae). J. Invertebr. Pathol. 46:70–82.
12. Chen, E. Y., and P. H. Seeburg. 1985. Supercoil sequencing: a fast and simple method for sequencing plasmid DNA. DNA 4:165–170.
13. Dobson, A. T., F. Sederati, G. Devi-Rao, W. M. Flanagan, M. J. Farrell, J. G.
Stevens, E. K. Wagner, and L. T. Feldman. 1989. Identification of the latency-associated transcript promoter by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J. Virol. 63:3844–3851.
14. Farrell, M. J., A. T. Dobson, and L. T. Feldman. 1991. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA
88:790–794.
15. Granados, R. R., T. Nguyen, and B. Cato. 1978. An insect cell line persis-tently infected with a baculovirus-like particle. Intervirology 10:309–317. 16. Han, K., M. S. Levine, and J. L. Manley. 1989. Synergistic activation and
repression of transcription by Drosophila homeobox proteins. Cell 56:573–583. 17. Henikoff, S. 1984. Unidirectional digestion with exonuclease III creates
tar-geted breakpoints for DNA sequencing. Gene 28:351–359.
18. Hink, W. F. 1970. Established insect cell line from the cabbage looper, Trichoplusia ni. Nature 226:466–467.
19. Ho, D. Y., and E. S. Mocarski. 1989. Herpes simplex virus latent RNA (LAT) is not required for latent infection in the mouse. Proc. Natl. Acad. Sci. USA
86:7596–7600.
20. Huang, Y. S., M. Hedberg, and C. Y. Kawanishi. 1982. Characterization of the DNA of a nonoccluded baculovirus, Hz-1 V. J. Virol. 43:174–181. 21. Hughes, D. S., R. D. Possee, and L. A. King. 1993. Activation and detection
of a latent baculovirus resembling Mamestra brassicae nuclear polyhedrosis virus in M. brassicae insects. Virology 194:608–615.
22. Jurkovicoba, M. 1979. Activation of latent infections in larvae of Adoxophyes orana (Lepidoptera: Torticidae) and Barathra brassicae (Lepidoptera: Noc-tuidae) by foreign polyhedra. J. Invertebr. Pathol. 34:213–215.
23. Kay, G. F., G. D. Penny, D. Patel, A. Ashworth, N. Brockdorff, and S. Rastan. 1993. Expression of XIST during mouse development suggests a role in the initiation of X chromosome inactivation. Cell 72:171–182.
24. Klein, G. 1989. Viral latency and transformation: the strategy of Epstein-Barr virus. Cell 58:5–8.
25. Kloc, M., G. Spohr, and L. D. Etkin. 1993. Translocation of repetitive RNA sequences with the germ plasm in Xenopus oocytes. Science 262: 1712–1714.
26. Lee, J. C., H. H. Chen, H. L. Wei, and Y. C. Chao. 1993. Superinfection-induced apoptosis and its correlation with the reduction of viral progeny in cells persistently infected with Hz-1 baculovirus. J. Virol. 67:6989–6994. 27. Lee, J. T., W. M. Strauss, J. A. Dausman, and R. Jaenisch. 1996. A 450 kb
transgene displays properties of the mammalian X inactivation center. Cell
86:83–94.
28. Lee, S. T., S. M. Yu, E. L. Hsu, and Y. C. Chao. 1995. Identification of a very
of the coding region of two actin genes in Bombyx mori. Nucleic Acids Res.
15:2781.
33. Penny, G. D., G. F. Kay, S. A. Sheardown, S. Rastan, and N. Brockdorff. 1996. Requirement for XIST in X chromosome inactivation. Nature 379: 131–137.
34. Podgwaite, J. D., and H. M. Mazzone. 1986. Latency of insect viruses. Adv. Virus Res. 31:293–320.
35. Pullen, S. S., and P. D. Friesen. 1995. The CAGT motif functions as an initiator element during early transcription of the baculovirus transregulator ie-1. J. Virol. 69:3575–3583.
36. Ralston, A. L., Y. Huang, and C. Y. Kawanishi. 1981. Cell culture studies with the IMC-Hz-1 nonoccluded virus. Virology 115:33–44.
37. Schmidt, E. E., and G. F. Merrill. 1991. Changes in dihydrofolate reductase (DHFR) mRNA levels can account fully for changes in DHFR synthesis rates during terminal differentiation in a highly amplified myogenic cell line. Mol. Cell. Biol. 11:3726–3734.
38. Solter, D., and G. Wei. 1997. Ends XIST, but where are the beginnings? Genes Dev. 11:153–155.
39. Spivack, J., and N. W. Fraser. 1987. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol. 61:3841–3847. 40. Steiner, I., J. G. Spivack, R. P. Lirette, S. M. Brown, A. R. McLean, J. H.
Subak-Sharpe, and N. W. Fraser.1989. Herpes simplex virus type 1 latency associated transcripts are evidently not essential for latent infection. EMBO J. 8:505–511.
41. Steller, H., and V. Pirrotta. 1985. A transposable P vector that confers selectable G418 resistance to Drosophila larvae. EMBO J. 4:167–171. 42. Stevens, J. G., E. K. Wagner, G. B. Devi-Rao, M. L. Cook, and L. T. Feldman.
1987. RNA complementary to a herpesvirus gene mRNA is prominent in latently infected neurons. Science 235:1056–1059.
43. Summers, M. D., and G. E. Smith. 1988. A manual of methods for baculo-virus vectors and insect cell culture procedures. Texas Agricultural Experi-ment Station Bulletin no. 1555.
44. Volkman, L. E. 1995. Virus taxonomy: the classification and nomenclature of viruses, p. 104–113. In F. A. Murphy, C. M. Fauquet, D. H. L. Bishop, S. A. Ghabrial, A. W. Javis, G. P. Martelli, M. A. Mayo, and M. D. Summers (ed.), The sixth report of the ICTV. Springer-Verlag Wien, Inc., New York, N.Y. 45. Wilson, M. 1991. The family and groups of Baculoviridae, p. 117–123. In R. I. B. Francki, C. M. Fauquet, D. L. Knudson, and F. Brown (ed.), Classifi-cation and nomenclature of viruses. Fifth report of the International Committee on Taxonomy of Viruses. Springer-Verlag Wien, Inc., New York, N.Y. 46. Wood, H. A., and J. P. Burand. 1986. Persistent and productive infections
with the Hz-1 baculovirus. Curr. Top. Microbiol. Immunol. 131:119–134. 47. Wu, T. T., Y. H. Su, T. M. Block, and J. M. Taylor. 1996. Evidence that two
latency-associated transcripts of herpes simplex virus type 1 are nonlinear. J. Virol. 70:5962–5967.
48. Zwaagstra, J. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, S. C. Wheatley, K.
Lillycrop, J. Wood, D. S. Latchman, K. Patel, and S. L. Wechsler.1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J. Virol. 64:5019–5028.