0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.10.5914–5922.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Variant Upstream Regulatory Region Sequences Differentially
Regulate Human Papillomavirus Type 16 DNA Replication
throughout the Viral Life Cycle
Walter G. Hubert*
Department of Dermatology, University of Arkansas for Medical Sciences, 4301 W. Markham St., Little Rock, Arkansas 72205
Received 11 October 2004/Accepted 8 January 2005
While the central role of the viral upstream regulatory region (URR) in the human papillomavirus (HPV) life cycle has been well established, its effects on viral replication factor expression and plasmid replication of HPV type 16 (HPV16) remain unclear. Some nonprototypic variants of HPV16 contain altered URR sequences and are considered to increase the oncogenic risk of infections. To determine the relationship between viral replication and variant URRs, hybrid viral genomes were constructed with the replication-competent HPV16 prototype W12 and analyzed in assays which recapitulate the different phases of normal viral replication. The establishment efficiencies of hybrid HPV16 genomes differed about 20-fold among European prototypes and variants from Africa and America. Generally, European and African genomes exhibited the lowest replication efficiencies. The high replication levels observed with American variants were primarily attributable to their efficient expression of the replication factors E1 and E2. The maintenance levels of these viral genomes varied
about fivefold, which correlated with their respective establishment phenotypes and published P97activities.
Vegetative DNA amplification could also be observed with replicating HPV16 genomes. These results indicate that efficient E1/E2 expression and elevated plasmid replication levels during the persistent stage of infection may comprise a risk factor in HPV16-mediated oncogenesis.
Epidemiology. Infection with human papillomaviruses
(HPVs) leads to hyperproliferative lesions in humans at spe-cific anatomical sites which are determined by the tropism of individual viral genotypes. Among the more than 100 types of HPVs identified to date, a subset of mucosotropic viruses, which infect the epithelial lining of the anogenital tract and oral cavity, are causally associated with the great majority of cervical cancers worldwide. This group of high-risk HPVs in-cludes HPV type 16 (HPV16), HPV18, HPV31, HPV33, and HPV35, with HPV16 being the most prevalent. In malignant anogenital lesions, high-risk HPV DNA is present in infected cells and the viral oncogenes are expressed detectably. While type 16 is also found in over 80% of HPV-positive head and neck cancers (39, 40, 54), and less frequently in HPV-positive cutaneous squamous cell carcinomas (64), its etiology in these diseases is still unclear. Over the past decade, numerous HPV16 variants have been identified which exhibit one or more single nucleotide changes compared to the European prototype E (19, 48, 60). Typically, L1, E6, and also upstream regulatory region (URR) sequences of HPV16 DNA from cervical cancers were analyzed (69, 70, 73) and variant classes established for different continents (73). Recent epidemiologic studies indicate that non-European variants of HPV16 exhibit increased oncogenicity (71, 72), specifically members of the Asian-American class AA (6). The underlying causes for the pathogenic differences of HPV16 variants, particularly those with altered URR sequences, are not well understood.
Viral life cycle, DNA replication, and oncogenesis.The life
cycle of papillomaviruses is closely linked to the differentiation program of their host cells. HPVs infect dividing basal kera-tinocytes in the stratified epithelium and replicate as nuclear plasmids at low copy number. During this persistent stage, only a subset of viral genes is expressed to ensure stable viral plas-mid replication in dividing cells without generating progeny virions (43). The productive stage of infection ensues as HPV-positive cells differentiate and migrate to the upper layers of the epithelium. At this time, viral genomes are amplified to a high copy number per cell, capsid genes are expressed, and progeny virions are assembled (30). Persistent infections with high-risk HPVs, such as types 16, 18, 31, and 33, may last for years, during which the continued expression of the viral on-cogenes induces genetic instability in host cells and initiates malignant conversion (18, 75). High-risk HPV-mediated onco-genesis is commonly associated with integration of the viral DNA into the host genome (20). In tumor cells with integrated HPV DNA, high levels of the viral oncoproteins have been observed. As a consequence, the proliferative capacity of such cells is increased, which leads to a preferential expansion dur-ing cell culture (1, 21, 34, 35, 57). Viral DNA integration, however, is not required for HPV16-induced oncogenesis, as cervical cancers which harbor extrachromosomal HPV16 ge-nomes have been identified. In such biopsies, analysis of the URR showed that YY1 factor binding sites were altered or deleted, which increased the transcriptional activity of the ma-jor viral promoter P97(17). At present, the molecular
param-eters which abolish normal viral plasmid replication and lead to integration of HPV16 DNA into the host chromosome are not known. However, the behavior of YY1 mutants of HPV16 indicates that long-term viral plasmid stability may depend on * Mailing address: Department of Dermatology, MS576, University
of Arkansas for Medical Sciences, 4301 W. Markham St., Little Rock, AR 72205. Phone: (501) 686-5110. Fax: (501) 686-7264. E-mail: [email protected].
5914
on November 8, 2019 by guest
http://jvi.asm.org/
higher transcriptional activities than those provided by the HPV16 prototype URR. More recently, elevated P97activities
have also been observed with biopsy-derived variant URRs which contain multiple single-nucleotide changes (36, 68).
URR-dependent replication of HPV16.Earlier efforts to
ge-netically analyze the regulation of HPV16 DNA replication were hampered by the lack of a cell culture system which could support normal viral plasmid replication. Following the recent isolation of the HPV16 prototype W12 (22), however, stable replication of these viral genomes was demonstrated by differ-ent laboratories (22, 62). Using this replication-competdiffer-ent HPV16 prototype, the hypothesis was tested in this study that variant URR sequences, which alter P97activity, also modulate
viral DNA replication. To determine the viral establishment, maintenance, and vegetative replication phenotypes of HPV16, URR sequences from the European prototype (E) and five African (Af1aB2, Af1aD1, Af2aE1, Af2aF1, and Af2aF3) and two American (AAc and NA1) variants (36) were analyzed in the background of an HPV16W12 genome, adapt-ing methodologies initially developed for the related HPV types 18 and 31 (23–25, 47). Comparison of the replication behavior of the HPV16 genomes with their published tran-scriptional phenotypes (36) indicates that the low replication levels observed with European and African hybrids correlate with inefficient expression of the viral replication factors E1 and E2. In contrast, American URR variants were found to replicate at much higher levels, which may be related to their elevated oncogenicity (8).
MATERIALS AND METHODS
Plasmids.The parental HPV16 genome pBRmin3-HPV16W12 (542.203) was derived from pEFHPV16W12E (provided by P. Lambert, Madison, WI) (22), which had been isolated from a clonal population (34, 35) of the W12 biopsy cell line (63). The viral genome was excised from pEFHPV16W12E by BamHI digestion and cloned into pBRmin3, a minimal vector with bacterial origin, -lactamase, andropgenes, which was obtained from pBRmin-HPV31 (31) and adapted by inserting a BamHI linker at its unique EcoRI site. Nucleotide num-bering for the W12 genome is based on its published sequence (22). Numnum-bering for the non-W12 URRs is based on the revised HPV16R sequence (48) of the European prototype E (19, 60). Hybrid HPV16W12 genomes were prepared by transferring URR fragments (PmlI at nucleotide [nt] 7268 to PpuMI at nt 111) from the European prototype E (provided by L. Turek, Iowa City, IA) and variants Af1aB2, Af1aD1, Af2aE1, Af2aF1, Af2aF3, AAc, and NA1 (36) (pALuc reporter constructs provided by C. Wheeler, Albuquerque, NM) into pBRmin3-HPV16W12, which was opened at PmlI (nt 7266) and PpuMI (nt 111). Two control plasmids were also created in pBRmin3-HPV16W12. (i) E1TTL (543.301) contains a six-frame translational termination linker (52), 5⬘-d(gcgcC TAACCTAGGTTAG), with KasI-compatible ends at KasI (nt 1310), which terminates translation of E1 after codon 150. (ii) E2FS (808.103) was prepared by partial digestion of pBRmin3-HPV16W12 with BstXI, blunt ending with T4 DNA polymerase, and religation. The deletion of nt 2896 to 2899 results in a frameshift mutation in E2 which terminates translation after codon 75. pSK-E116 and pSK-E216(58), provided by P. Howley, Boston, MA, are expression vectors which contain a human cytomegalovirus promoter and intact E1 or E2 coding sequences, respectively, derived from the European HPV16 prototype plasmid p1203 (55). Both vectors have been shown to support replication of HPV16 origin plasmids (58).
Short-term replication.Viral establishment efficiency was analyzed in tran-sient replication assays essentially as previously described for HPV31 (31). Briefly, viral genomes were released from bacterial vector sequences and uni-molecularly religated at a low DNA concentration. HPV-negative human squa-mous cell carcinoma 13 (SCC13) (53) cells (5⫻106) were transfected by elec-troporation either with ligated HPV genomes (5g of viral DNA) by themselves or together with equimolar amounts of pSK-E116 and pSK-E216 each. After expanding the transfected SCC13 cells on J2-3T3 fibroblast feeder layers for 5 days, low-molecular-weight DNA was extracted by a modified Hirt DNA
proce-dure. Prior to gel electrophoresis, sample DNAs were digested to completion with NcoI to linearize HPV16 genomes and DpnI to remove bacterially meth-ylated input DNA.
Long-term replication.Viral maintenance efficiency was analyzed in stable replication assays as published for HPV31 (31). Briefly, ligated viral genomes were prepared as described for the transient assay. Subconfluent, low-passage, primary human foreskin keratinocytes (HFKs; isolated from anonymous neona-tal donors) were transfected with HPV16 genomes (5g of viral DNA) and a 0.5 molar equivalent of the marker plasmid pSV2neo using FuGENE 6 (Roche) or Lipofectamine (Invitrogen). Transfected HFKs were transferred to J2-3T3 fibro-blast feeder layers and selected briefly with G418. Drug-resistant colonies from each transfection were pooled within 3 weeks and expanded as mass cultures. After removal of fibroblast feeders, total cellular DNA was isolated from HFKs 4 to 6 weeks posttransfection using proteinase K digestion and differential salt precipitation to minimize shearing (31). Gel electrophoresis was performed with DpnI-digested total cellular DNA samples (5g) which were either sheared or digested with the single-cut enzyme NcoI.
Vegetative replication.Viral DNA amplification efficiency was analyzed by in vitro differentiation of stably transfected cells as previously described for HPV31 (31, 56). Briefly, fibroblast feeders were removed from the expanded, HPV16-positive mass cultures (see above), and HFKs trypsinized. After collection, cell pellets were divided equally into two samples and suspended separately in meth-ylcellulose (1.6%, wt/vol)-containing E medium. Cells were rinsed out from methylcellulose cultures immediately or after 24 h of incubation at 37°C. Total cellular DNA was isolated for the two time points, and sheared samples analyzed as in the stable replication assay (see above).
Southern hybridization and analysis.DNA samples from all replication assays were resolved on agarose gels (0.8%, wt/vol) and transferred to positively charged nylon membranes (Magna Probe; Osmonics) under alkali conditions as previously described for HPV31 (31). HPV16W12-derived DNA standards of 500, 25, 2.5, and 0.5 pg of genome size viral DNA were included on each blot. Prehybridization and hybridization were performed with dextran sulfate- and formamide-containing solutions at 42°C (31). HPV16-specific probe was pre-pared by labeling genome length viral DNA (W12 prototype) with [␣-32
P]deoxy-cytosine triphosphate by random priming (Prime-It-RmT; Stratagene). After stringent washing, viral DNA was visualized by storage phosphor-mediated au-toradiography (Molecular Dynamics). Autoradiograms were quantified with Im-ageQuant software (Molecular Dynamics) or IPLab gel (Signal Analytics), and figures assembled with Photoshop (Adobe) and Freehand (Macromedia), per-forming only linear adjustments.
Sequence analysis.The published HPV16R (48) sequence was used as a base for variant URRs Af1aB2, Af1aD1, Af2aE1, Af2aF1, Af2aF3, AAc, and NA1 (36). The approximate positions of variant nucleotides within the URR are shown in Fig. 1B. The search for potential factor binding sites in the URR was performed with DNA Strider (Christian Marck, CEA, France) as described in the legend to Fig. 1.
Research compliance.All experiments with human cells and human papillo-mavirus DNAs were in compliance with federal, state, and institutional regula-tions and were approved by the chairman of the Institutional Review Board at the University of Arkansas for Medical Sciences.
RESULTS
Establishment efficiencies of URR variants are strongly
modulated by E1 and E2 expression.The viral establishment
phase is the first phase of the HPV life cycle during which a stable low plasmid copy number is attained within an infected cell population. In this phase, viral plasmids replicate more frequently than the host genome for multiple cell division cycles. For a transient replication assay which mimics the viral establishment phase, permissive human epithelial cells are transfected with HPV genomes and their replication levels are quantified after 5 days. In this assay, the expression efficiency and functionality of the viral replication factors E1 and E2 can be analyzed, as well as the function of the replication origin on the viral genome. Under physiological conditions, papilloma-virus replication requires sufficient levels of both E1 and E2 in
transand an intact origin incis(67). Since both the
transcrip-tional control elements for HPV gene expression and the
on November 8, 2019 by guest
http://jvi.asm.org/
lication origin are located within the URR, this domain plays a central role in viral regulation. Depending on transfection efficiency and other assay variations, the replication levels of HPV16 prototype genomes (E and W12) are 5- to 50-fold lower than that of HPV31 (W. G. Hubert, unpublished data). This low replication phenotype may be responsible for the earlier assessment that HPV16 genomes did not replicate when permissive cells were transfected transiently (14). Suffi-cient signal can now be obtained, however, and replication of HPV16 determined reliably with assay protocols adapted from studies with HPV31 (31).
To test the hypothesis that the transcriptional phenotypes exhibited by a panel of variant HPV16 URRs (36) could dif-ferentially modulate transient viral replication, hybrid HPV16
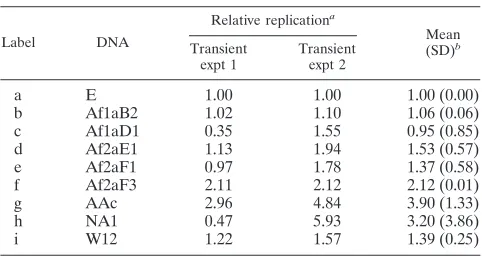
[image:3.585.49.534.69.454.2]DNAs were constructed which contain variant URR sequences in a common background of the replication-competent HPV16 W12 genome. This strategy focuses the experimental analysis only on URR-mediated effects, excluding the potential contri-butions of sequence variations in other parts of the viral ge-nome. The URR fragments of HPV16 variants Af1aB2, Af1aD1, Af2aE1, Af2aF1, Af2aF3, AAc, and NA1 (36), which were originally collected for an epidemiologic study (73), and the URR of the European (E) prototype were transferred to the HPV16W12 genome as described in Materials and Meth-ods. Transient replication assays were then performed by transfecting HPV-negative SCC13 cells (53) with recircular-ized viral genomes and analyzing de novo replication. As shown in Fig. 2 A, the replication levels of the European FIG. 1. HPV16 genome (prototype E). (A) Circular map depicting the early (E genes) and late (L genes) regulatory regions and URR of the viral genome. It shows the open reading frames (shaded arcs), protein coding sequences (solid arrows within arcs), and promoters (solid arrows at periphery). The positions of the E2 DNA binding sites in the URR are indicated by flags. (B) Linear map of the entire URR. Potential sites for cellular regulatory proteins are indicated by position (gray boxes at center line) and name (flags above) and were identified by searching for the following degenerate motifs: AP1 (TKWNTMA) (9), AP2/TEF2 (SCACMY) (11), NF1 (TTGGC) (2, 28), Oct1 (AANWGYAB) (49), Sp1 (GGGMGK) (3, 65), TEF1 (YRCATDBYDB) (33), and YY1 (MCATNKT) (16, 17, 46, 50). The DNA binding sites for the viral replication factors are shown by position (black and gray boxes at center line) and name (flags below) as identified for E1 (by homology with its counterpart in HPV31 [Hubert, unpublished]) and E2 (HPV16R sequence (48). The positions of URR alterations versus prototype (data from reference 36) are shown schematically under the factor binding sites for variants Af1aB2 (b), Af1aD1 (c), Af2aE1 (d), Af2aF1 (e), Af2aF3 (f), AAc (g), and NA1 (h). Single nucleotide changes (solid circles) and a single nucleotide deletion (open circle) are indicated.
on November 8, 2019 by guest
http://jvi.asm.org/
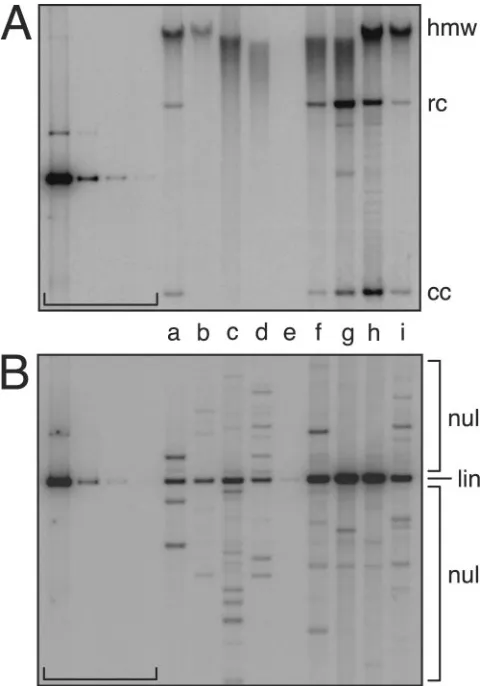
prototypes (hybrid W12-URR-E and parental W12, lanes a and i) and most African URR variants (W12 hybrids b through e) were relatively low. In contrast, both American variants exhibited high replication levels (Fig. 2 A, lanes g and h) and were about 20-fold more efficient than prototype E (Table 1, mean relative replication). HPV16 DNAs defective in either E1 or E2 protein translation (E1TTL, E2FS; Materials and Methods) were replication defective in control transfections (Table 1), which is consistent with the physiological require-ment for both replication factors (67).
HPV DNA replication is regulated in a dose-dependent manner by the abundance of the viral replication factors (10, 31, 41, 44, 45, 51, 74). Since the short-term replication assay with entire HPV genomes analyzes the efficiency of replication
[image:4.585.44.285.69.170.2]factor expression, as well as origin function, the differences in autonomous transient replication of the HPV16 DNAs could have been the result of bothcis- andtrans-acting changes. To more specifically test how E1 and E2 expression modulates replication, viral genomes were cotransfected with expression vectors for both viral replication factors (Materials and Meth-ods), which had previously been shown to support replication of HPV16 origin plasmids (58). In the presence of heterolo-gous E1 and E2, prototype (hybrid W12-URR-E and parental W12, a and i) and most African variant (W12 hybrids b through e) genomes exhibited similar replication levels (up to 1.53-fold, Fig. 2 B and Table 2, mean relative replication). Replication of variants Af2aF3, AAc, and NA1 exceeded that of prototype E by as much as 3.9-fold, possibly due to a combination of high levels of endogenous and exogenous E1 and E2. These data indicate (i) that HPV16 origin efficiency is not strongly affected by variant URR sequences and (ii) that, consistent with the behavior of HPV31 mutants with defects in replication factor expression (31), the low-establishment phenotypes of Euro-pean prototypes and African URR variants of HPV16 are primarily the consequence of inefficient E1 and E2 expression. FIG. 2. Transient replication of HPV16 URR variants. These
[image:4.585.301.542.87.215.2]au-toradiograms show the replication of HPV16 genomes during the establishment phase of the viral life cycle. After removal of bacterial vector sequences and recircularization, HPV16 genomes were trans-fected into HPV-negative human SCC13 cells. Five days posttransfec-tion, low-molecular-weight DNA was isolated, digested, resolved on agarose gels, and visualized by Southern blotting and hybridization (Materials and Methods). The autoradiograms show DpnI-resistant (replicated) and linearized HPV DNAs detected with an HPV16W12 genome-specific probe. The brackets indicate standard lanes of 500, 25, 2.5, and 0.5 pg of genome size HPV16W12 DNA. Hybrid HPV16W12 genomes contained the following URRs (see Materials and Methods): European prototype (E, a); variants Af1aB2 (b), Af1aD1 (c), Af2aE1 (d), Af2aF1 (e), Af2aF3 (f), AAc (g), and NA1 (h); and parental prototype HPV16W12 (i). (A) Autonomous HPV16 replication in the absence of any expression vectors. (B) HPV16 rep-lication in the presence of HPV16-derived E1 and E2 expression vec-tors (Materials and Methods).
TABLE 1. Autonomous, transient replication of HPV16 genomes
Label DNA
Absolute replicationa Relative replicationb
Mean (SD)c
Transient expt 1
Transient expt 2
Transient expt 1
Transient expt 2
a E 1.31 1.62 1.00 1.00 1.00 (0.00)
b Af1aB2 1.88 4.01 1.44 2.48 1.96 (0.74)
c Af1aD1 1.14 3.36 0.87 2.08 1.48 (0.85)
d Af2aE1 1.31 1.81 1.00 1.12 1.06 (0.08)
e Af2aF1 0.79 1.86 0.61 1.15 0.88 (0.39)
f Af2aF3 4.67 1.89 3.58 1.17 2.37 (1.70)
g AAc 25.43 23.72 19.48 14.69 17.09 (3.39)
h NA1 16.31 45.87 12.50 28.40 20.45 (11.24)
i W12 2.26 1.67 1.74 1.04 1.39 (0.49)
W12-E1TTL NDd 0.15 ND 0.09 ND
W12-E2FS ND 0.12 ND 0.07 ND
aData are from transient experiments 1 (shown in Fig. 2A) and 2; absolute replication levels in pg were calculated with blot-specific DNA standards. bRelative ratios versus E, calculated from absolute data.
cOf relative transient levels 1 and 2. dND, not determined.
TABLE 2. Transient replication of HPV16 genomes with exogenous E1 and E2
Label DNA
Relative replicationa
Mean (SD)b
Transient expt 1
Transient expt 2
a E 1.00 1.00 1.00 (0.00)
b Af1aB2 1.02 1.10 1.06 (0.06)
c Af1aD1 0.35 1.55 0.95 (0.85)
d Af2aE1 1.13 1.94 1.53 (0.57)
e Af2aF1 0.97 1.78 1.37 (0.58)
f Af2aF3 2.11 2.12 2.12 (0.01)
g AAc 2.96 4.84 3.90 (1.33)
h NA1 0.47 5.93 3.20 (3.86)
i W12 1.22 1.57 1.39 (0.25)
a
Data are from transient experiments 1 and 2 (Fig. 2B); relative ratios versus E were calculated from raw densitometry data.
b
Of relative transient levels 1 and 2.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.585.48.540.547.690.2]Maintenance levels of URR variants are similar to their
establishment efficiencies. Maintenance phase is the second
phase of the viral life cycle, during which HPV plasmids rep-licate as frequently as the cellular genome on average (26), resulting in a stable copy number in an HPV-positive cell population. In the stable replication assay, which mimics the viral maintenance phase, primary HFKs are transfected with HPV genomes. It is performed over a 4- to 6-week time course, and the major viral parameters which determine replication behavior are (i) expression of the viral replication factors, (ii) viral origin function, and (iii) expression of the viral oncogenes (32, 66).
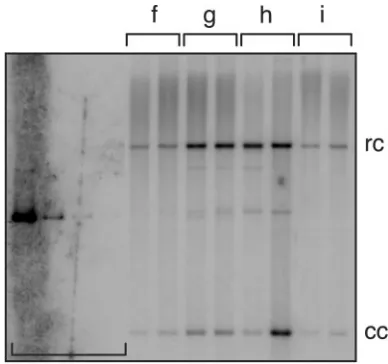
To test the hypothesis that the transcriptional phenotypes exhibited by variant HPV16 URRs (36) could differentially modulate viral maintenance levels, the panel of hybrid HPV16 genomes was analyzed in stable replication assays (Materials and Methods). HFKs were transfected with recircularized viral genomes and a selectable marker. After selection and expan-sion of drug-resistant colonies as mass culture cell lines, total cellular DNA was isolated and analyzed by Southern hybrid-ization. Normal viral plasmid replication yields characteristic autoradiographic bands which indicate the presence of closed circular (cc) and relaxed circular (rc) monomers. As shown in Fig. 3 A, the replication levels of the European prototypes (hybrid W12-URR-E and parental W12, lanes a and i) and the African URR variant (W12 hybrid, f) were relatively low. In contrast, both American variants consistently replicated at high levels (Fig. 3A, lanes g and h, and Table 3, mean absolute replication). Interestingly, strong variations in the relative lev-els of the inefficient replicators were observed in multiple ex-periments which were performed with different primary cell isolates and transfection agents (Table 3, rows i and a through e, mean relative replication). These findings suggest that inef-ficient viral DNA replication is also modulated by other factors (see Discussion). Consistent with their behavior in transient assays (Fig. 2A. and Table 1), the E1 and E2 mutants of HPV16W12 were also found to be defective for stable repli-cation (Table 3).
The additional HPV topologies present in total DNA sam-ples are referred to as high molecular weight (hmw, Fig. 3A). hmw signals generally indicate (i) integrated viral DNA, which migrates as part of the randomly sheared cellular DNA, (ii) viral plasmid concatemers, or (iii) trapped monomeric or mul-timeric HPV plasmids. To resolve this potential mixture, un-sheared total cell DNA was digested with a restriction enzyme which linearizes HPV plasmid monomers (lin). As shown in Fig. 3B, all analyzed cell populations harbor detectable amounts of unit length viral DNA. Additional bands of larger or smaller size than linear HPV16 DNA (non-unit length, nul) are also detected. nul bands indicate the presence of junction fragments containing both cellular and viral DNA but could also result from replicating or integrated HPV genomes which are rearranged. However, in light of the fact that no replica-tion-competent multimeric plasmid species, including rear-ranged partial multimers, were detected in earlier experiments with HPV31 (31) or HPV16 (Fig. 3A), nul hybridization signals likely indicate only rearranged, integrated viral DNA. Such HPV16 DNA species have also been observed in cloned cell populations from the W12 biopsy cell line (34). As hmw hy-bridization signals (Fig. 3A) and nul-specific bands (Fig. 3B)
are present in most sample lanes, integration of viral DNA appears to occur with both prototypes and URR variants of HPV16.
URR variant and prototype genomes replicate vegetatively.
Vegetative HPV replication is the final phase of the viral life cycle. Typically, HPV plasmid DNA is amplified from a low, stable copy number to several thousand in competent, supra-basal keratinocytes. Viral DNA amplification is required for HPV capsid gene expression (25), which precedes virion as-FIG. 3. Stable DNA replication of HPV16 URR variants. These autoradiograms show replication of HPV16 genomes during the main-tenance phase of the viral life cycle. DNA standards (horizontal brack-et): 500, 25, 2.5, and 0.5 pg, equivalent to 100, 5, 0.5, and 0.1 viral copies per cell (31). Hybrid HPV16W12 DNAs: prototype E (a); vari-ants Af1aB2 (b), Af1aD1 (c), Af2aE1 (d), Af2aF1 (e), Af2aF3 (f), AAc (g), and NA1 (h); and parental prototype W12 (i). Primary HFKs were stably transfected with recircularized HPV genomes (Materials and Methods). Total cellular DNA samples were isolated from expanded mass cultures and either sheared and digested with DpnI (for panel A) or digested with both NcoI and DpnI (for panel B) prior to gel elec-trophoresis. Southern blot analysis was performed as described above. (A) HPV DNA in sheared samples: closed-circular monomer (cc), linearized (lin), relaxed-circular monomer (rc), and high molecular weight (hmw). Normal stable HPV replication is indicated by plasmid-specific cc and rc bands. (B) HPV DNA in digested samples: unit length (linearized [lin]) bands indicate that viral DNAs either replicate as plasmids or are integrated in tandem repeats. Numerous integra-tion-specific small and large junction bands (non-unit length [nul]) appear in all transfected cell populations.
on November 8, 2019 by guest
http://jvi.asm.org/
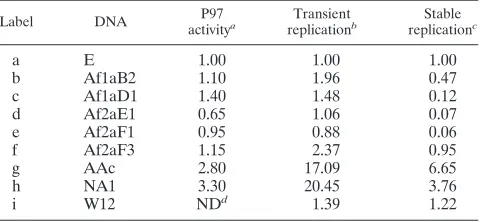
[image:5.585.301.541.69.412.2]sembly and egress. Vegetative replication only occurs in dif-ferentiated keratinocytes, a process which can be induced by suspending epithelial cells in methylcellulose-containing growth medium to prevent their adherence to the substrate (29). During in vitro differentiation, a subset of HPV-positive keratinocytes are competent to amplify viral plasmid DNA in 24 to 48 h, which, in the case of HPV31, increases the plasmid copy number two- to fivefold in a treated cell population (31, 56). To test the capacity of HPV16 URR variants to amplify viral DNA, cell lines with detectable HPV16 replication were analyzed (Materials and Methods). The amounts of replicated DNA were obtained by combining the respective rc and cc signals at each time point, which were then normalized for DNA loading within each sample pair. Comparing viral repli-cation in differentiated (24 h) cells to that in undifferentiated (0 h) cells, moderate DNA amplification ratios could be ob-served with the HPV16 prototype (parental W12, i) and URR variants Af2aF3, AAc, and NA1 (W12 hybrids f, g, and h, Fig. 4 and Table 4). While a relationship between URR variation and DNA amplification efficiency was not apparent in the two independent experiments, these data indicate that HPV16 DNA amplification can be analyzed with the in vitro differen-tiation assay. Since the 24-h amplification ratios of HPV16 appear disproportionally smaller than those observed with HPV31 in 48 h (31, 56), these data may also indicate that vegetative replication of HPV16 is less efficient overall.
DISCUSSION
Viral transcription and replication phenotypes are related.
When the detected transient and stable replication levels of the HPV16 URR variants were normalized to the European pro-totype (hybrid W12-URR-E) and compared to the published P97activities (36), the viral transcription and persistent
repli-cation phenotypes were found to correlate for most of the tested HPV16 DNAs (Table 5). The American variants (Table 5, rows g and h) exhibited the highest levels of replication in
both establishment and maintenance phases, as well as the strongest P97activity. In contrast, both phenotypes were more
moderate with most African and European strains. The repli-cation levels of prototype E (row a) and variants Af1aB2 and Af2aF3 (b and f) also correlated with their P97activity. And the
low P97activities of variants Af2aE1 and Af2aF1 (d and e) are
[image:6.585.42.552.80.224.2]in line with their moderate transient replication and lack of TABLE 3. Stable replication of HPV16 genomes
Label DNA
Absolute replicationd Relative replicatione
Stable expt 1a
Stable expt 2b
Stable
expt 3c Mean (SD)f
Stable expt 1
Stable expt 2
Stable
expt 3 Mean (SD)
g
a E 29.83 5.98 0.84 12.22 (15.47) 0.402 0.074 0.009 0.162 (0.210)
b Af1aB2 15.31 1.29 0.52 5.71 (8.32) 0.206 0.016 0.006 0.076 (0.113)
c Af1aD1 2.43 1.22 0.57 1.41 (0.94) 0.033 0.015 0.006 0.018 (0.013)
d Af2aE1 0.33 1.32 NDh 0.83 (0.70) 0.004 0.016 ND 0.010 (0.008)
e Af2aF1 0.34 1.25 ND 0.79 (0.64) 0.005 0.015 ND 0.010 (0.008)
f Af2aF3 12.53 10.66 ND 11.60 (1.32) 0.169 0.132 ND 0.150 (0.026)
g AAc 74.24 81.00 88.40 81.21 (7.09) 1.000 1.000 1.000 1.000 (0.000)
h NA1 17.12 52.42 68.29 45.95 (26.19) 0.231 0.647 0.773 0.550 (0.284)
i W12 37.48 5.36 2.01 14.95 (19.59) 0.505 0.066 0.023 0.198 (0.267)
W12-E1TTL 0.22 ND 0.26 ND 0.003 ND 0.003 ND
W12-E2FS 0.19 ND 0.36 ND 0.002 ND 0.004 ND
aKeratinocyte isolate 407.1 passage 5, FuGene 6 (Roche) transfection.
bKeratinocyte isolate 407.1 passage 6, Lipofectamine (Invitrogen) transfection (shown in Fig. 3A). cKeratinocyte isolate 503.1 passage 6, Lipofectamine (Invitrogen) transfection.
dData are from stable experiments 1, 2 (shown in Fig. 3A), and 3; absolute replication levels in pg were calculated from the sum of rc and cc signals with blot-specific
DNA standards; 5 pg of HPV DNA in 5g of total cellular DNA is equivalent to 1 viral copy per cell (31).
eRelative ratios versus AAc (row g) calculated from absolute data. fOf absolute levels 1, 2, and 3 or 1 and 2 (for d through f). gOf relative levels 1, 2, and 3 or 1 and 2 (for d through f). hND, not determined.
FIG. 4. DNA amplification of HPV16 URR variants. This autora-diogram shows vegetative DNA replication of HPV16 genomes in HFKs after 24 h of induced differentiation. DNA standards (left brack-et): 500, 25, 2.5, and 0.5 pg. HPV16 DNAs (see Materials and Meth-ods): hybrid HPV16W12 URR variants Af2aF3 (f), AAc (g), and NA1 (h) and parental prototype W12 (i). Paired lanes for each sample contain 5g of total cellular DNA from undifferentiated (left lane in pair) and differentiated (right lane in pair) cells. Southern blot analysis was performed as described for Fig. 2 and 3. HPV DNA topomers: cc monomer (cc) and rc monomer (rc). The high-molecular-weight (hmw) DNA smear is less pronounced in this assay (compared to Fig. 3A), owing to the fact that lower total DNA concentrations qualita-tively affect DNA shearing.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.323.517.423.605.2]stable replication. These data indicate that altered P97
activi-ties are primarily responsible for the strong differences in rep-lication levels observed with these HPV16 URR variants.
Host-specific factors and replication of HPV16.Stable
rep-lication levels of high-risk HPV genomes vary significantly among different isolates of primary cells. Such variability in supporting long-term HPV plasmid replication has been ob-served in other laboratories and are thought to be donor spe-cific (W. G. Hubert, unpublished data; L. A. Laimins, personal communication; P. F. Lambert, personal communication; L. Turek, personal communication). Evidence for this variable property of HFKs is presented in Table 3. A threshold of a minimal replication efficiency appears to be required for at-taining a stable copy number, as the levels of inefficiently replicating genomes (Table 3, rows a through c, stable exper-iment 3; rows d and e, stable experexper-iment 1) are close to the detection limit. The URR variants AAc and NA1 consistently replicated at a high level in three experiments (Table 3, rows g and h, mean absolute replication). While such a threshold may be overcome by increased transfection efficiency in some cases, depending on the agent used (footnotes to Table 3), it may primarily be an intrinsic characteristic of an HFK isolate. Since the outcome of HPV-induced disease is likely affected by such donor-specific factors, future studies are necessary in this area.
Role of the major viral promoter. P97activity, as well as
transient- and stable-replication efficiencies, correlate for most of the tested URR variants (Table 5). These findings indicate that the strong activity of P97, which is known to be involved in
the expression of E1 and E2 (32, 39), as well as E6 and E7 (13), during the persistent stage of infection, is also associated with high-establishment and -maintenance phenotypes. Restoring the replication signals of intrinsically low prototypes (hybrid W12-URR-E and parental W12, a and i) and African URR variants (c through e) to similar levels (Table 2) by overex-pressing E1 and E2 from heterologous vectors confirms previ-ous observations about the central role of P97activity in the
pathogenesis of HPV16. Similar to the intrinsic efficiencies of URR variants for expressing the viral replication factors, their capacity for viral oncogene expression is also expected to cor-relate with P97activity. Since significant levels of E7 protein
have already been observed with replicating HPV16 at high copy number in clonal W12 cell populations (34, 35), the se-verely oncogenic potential of the HPV16 variant AAc (6, 8) may be related to its strong transcription and replication phe-notypes.
Transcription factor-specific regulation by the URR. The
importance of the URR in the HPV life cycle was recognized quite early. Numerous studies have characterized the function of common factor binding sites in P97-mediated transcription
of HPV16, such as AP1 (9), AP2/TEF2 (11), NF1 (2, 28), Oct1 (49), Sp1 (3, 65), TEF1 (33), and YY1 (16, 17, 46, 50). Our understanding of viral regulation can now be expanded by testing the importance of these sites for viral replication throughout the life cycle. Preliminary analysis of the variant changes in the URR has already shown that the low replication phenotypes of African variants may correlate with alterations of the P97⫺proximal Sp1 binding site. And the high-replication
phenotype of the American variants may be aided by the ac-quisition of an additional AP1 site. Both Sp1 (3, 7, 15, 27, 59, 61) and AP1 (11, 12, 38, 42) are known to play complex acti-vating roles in viral transcription and DNA replication. In addition, several URR variants also have a reduced number of YY1 binding sites which, depending on their sequence context, may increase transcriptional activation (5, 37) or repression (4, 46, 50). The effects of selected factor binding sites on viral replication will be analyzed in future studies.
ACKNOWLEDGMENTS
Thanks to Peter Howley, Lou Laimins, Paul Lambert, Lubos Turek, and Cosette Wheeler for providing essential plasmids for this study and to Paul Lambert and Lou Laimins for critically reading the manu-script.
This research was funded by grants from the American Cancer Society, Murphy Oil Corporation, and the University of Arkansas for Medical Sciences. W.G.H. is supported by a career development award from the Dermatology Foundation.
REFERENCES
1.Alazawi, W., M. Pett, B. Arch, L. Scott, T. Freeman, M. A. Stanley, and N. Coleman.2002. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res.62:6959–6965. 2.Apt, D., T. Chong, Y. Liu, and H. U. Bernard.1993. Nuclear factor I and
epithelial cell-specific transcription of human papillomavirus type 16. J. Vi-rol.67:4455–4463.
3.Apt, D., R. M. Watts, G. Suske, and H. U. Bernard.1996. High Sp1/Sp3 ratios in epithelial cells during epithelial differentiation and cellular trans-formation correlate with the activation of the HPV-16 promoter. Virology
224:281–291.
4.Bauknecht, T., P. Angel, H. D. Royer, and H. zur Hausen.1992. Identifica-tion of a negative regulatory domain in the human papillomavirus type 18 promoter: interaction with the transcriptional repressor YY1. EMBO J.
11:4607–4617.
[image:7.585.44.285.82.161.2]5.Bauknecht, T., F. Jundt, I. Herr, T. Oehler, H. Delius, Y. Shi, P. Angel, and
TABLE 4. Vegetative replication of HPV16 genomes
Label DNA
Relative replicationa
Mean (SD)b
Vegetative expt 1
Vegetative expt 2
f Af2aF3 1.31 NDc ND
g AAc 1.05 1.21 1.13 (0.12)
h NA1 1.84 1.28 1.56 (0.39)
i W12 1.63 ND ND
a
Data are from vegetative experiments 1 (shown in Fig. 4) and 2; relative DNA amplification ratios (24 h versus 0 h level⫽100%) were calculated from the sum of rc and cc signals, which were normalized to DNA loading.
b
Of relative levels 1 and 2.
c
ND, not determined.
TABLE 5. Transcription and replication phenotypes of HPV16
Label DNA P97
activitya Transient
replicationb Stable
replicationc
a E 1.00 1.00 1.00
b Af1aB2 1.10 1.96 0.47
c Af1aD1 1.40 1.48 0.12
d Af2aE1 0.65 1.06 0.07
e Af2aF1 0.95 0.88 0.06
f Af2aF3 1.15 2.37 0.95
g AAc 2.80 17.09 6.65
h NA1 3.30 20.45 3.76
i W12 NDd 1.39 1.22
aRelative versus E; data are from reference 36. bRelative versus E; mean relative ratios are from Table 1.
cRelative to E; mean relative ratios were calculated from the absolute means
in Table 3.
dND, not determined.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.585.44.284.573.684.2]H. zur Hausen. 1995. A switch region determines the cell type-specific positive or negative action of YY1 on the activity of the human papilloma-virus type 18 promoter. J. Virol.69:1–12.
6.Berumen, J., R. M. Ordonez, E. Lazcano, J. Salmeron, S. C. Galvan, R. A. Estrada, E. Yunes, A. Garcia-Carranca, G. Gonzalez-Lira, and A. Madri-gal-de la Campa.2001. Asian-American variants of human papillomavirus 16 and risk for cervical cancer: a case-control study. J. Natl. Cancer Inst.93:
1325–1330.
7.Butz, K., and F. Hoppe-Seyler.1993. Transcriptional control of human pap-illomavirus (HPV) oncogene expression: composition of the HPV type 18 upstream regulatory region. J. Virol.67:6476–6486.
8.Calleja-Macias, I. E., M. Kalantari, J. Huh, R. Ortiz-Lopez, A. Rojas-Mar-tinez, J. F. Gonzalez-Guerrero, A. L. Williamson, B. Hagmar, D. J. Wiley, L. Villarreal, H. U. Bernard, and H. A. Barrera-Saldana.2004. Genomic di-versity of human papillomavirus-16, 18, 31, and 35 isolates in a Mexican population and relationship to European, African, and Native American variants. Virology319:315–323.
9.Chan, W. K., T. Chong, H. U. Bernard, and G. Klock.1990. Transcription of the transforming genes of the oncogenic human papillomavirus-16 is stimu-lated by tumor promotors through AP1 binding sites. Nucleic Acids Res.
18:763–769.
10.Chiang, C. M., G. Dong, T. R. Broker, and L. T. Chow.1992. Control of human papillomavirus type 11 origin of replication by the E2 family of transcription regulatory proteins. J. Virol.66:5224–5231.
11.Chong, T., D. Apt, B. Gloss, M. Isa, and H. U. Bernard.1991. The enhancer of human papillomavirus type 16: binding sites for the ubiquitous transcrip-tion factors oct-1, NFA, TEF-2, NF1, and AP-1 participate in epithelial cell-specific transcription. J. Virol.65:5933–5943.
12.Cripe, T. P., A. Alderborn, R. D. Anderson, S. Parkkinen, P. Bergman, T. H. Haugen, U. Pettersson, and L. P. Turek.1990. Transcriptional activation of the human papillomavirus-16 P97 promoter by an 88-nucleotide enhancer containing distinct cell-dependent and AP-1-responsive modules. New Biol.
2:450–463.
13.Cripe, T. P., T. H. Haugen, J. P. Turk, F. Tabatabai, P. G. D. Schmid, M. Durst, L. Gissmann, A. Roman, and L. P. Turek.1987. Transcriptional regulation of the human papillomavirus-16 E6-E7 promoter by a keratino-cyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: implications for cervical carcinogenesis. EMBO J.6:3745–3753. 14.Del Vecchio, A. M., H. Romanczuk, P. M. Howley, and C. C. Baker.1992.
Transient replication of human papillomavirus DNAs. J. Virol.66:5949– 5958.
15.Demeret, C., M. Le Moal, M. Yaniv, and F. Thierry.1995. Control of HPV 18 DNA replication by cellular and viral transcription factors. Nucleic Acids Res.23:4777–4784.
16.Dong, X. P., and H. Pfister.1999. Overlapping YY1- and aberrant SP1-binding sites proximal to the early promoter of human papillomavirus type 16. J. Gen. Virol.80:2097–2101.
17.Dong, X. P., F. Stubenrauch, E. Beyer-Finkler, and H. Pfister.1994. Preva-lence of deletions of YY1-binding sites in episomal HPV 16 DNA from cervical cancers. Int. J. Cancer58:803–808.
18.Duensing, S., and K. Munger.2004. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncopro-teins. Int. J. Cancer109:157–162.
19.Durst, M., L. Gissmann, H. Ikenberg, and H. zur Hausen.1983. A papillo-mavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA80:
3812–3815.
20.Durst, M., A. Kleinheinz, M. Hotz, and L. Gissman.1985. The physical state of human papillomavirus type 16 DNA in benign and malignant genital tumours. J. Gen. Virol.66:1515–1522.
21.Durst, M., S. Seagon, S. Wanschura, H. zur Hausen, and J. Bullerdiek.1995. Malignant progression of an HPV16-immortalized human keratinocyte cell line (HPKIA) in vitro. Cancer Genet. Cytogenet.85:105–112.
22.Flores, E. R., B. L. Allen-Hoffmann, D. Lee, C. A. Sattler, and P. F. Lambert.
1999. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology
262:344–354.
23.Frattini, M. G., and L. A. Laimins.1994. The role of the E1 and E2 proteins in the replication of human papillomavirus type 31b. Virology204:799–804. 24.Frattini, M. G., H. B. Lim, J. Doorbar, and L. A. Laimins.1997. Induction of human papillomavirus type 18 late gene expression and genomic ampli-fication in organotypic cultures from transfected DNA templates. J. Virol.
71:7068–7072.
25.Frattini, M. G., H. B. Lim, and L. A. Laimins.1996. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differen-tiation-dependent late expression. Proc. Natl. Acad. Sci. USA93:3062–3067. 26.Gilbert, D. M., and S. N. Cohen.1987. Bovine papilloma virus plasmids replicate randomly in mouse fibroblasts throughout S phase of the cell cycle. Cell50:59–68.
27.Gloss, B., and H. U. Bernard.1990. The E6/E7 promoter of human papil-lomavirus type 16 is activated in the absence of E2 proteins by a sequence-aberrant Sp1 distal element. J. Virol.64:5577–5584.
28.Gloss, B., M. Yeo-Gloss, M. Meisterenst, L. Rogge, E. L. Winnacker, and H. U. Bernard. 1989. Clusters of nuclear factor I binding sites identify enhancers of several papillomaviruses but alone are not sufficient for en-hancer function. Nucleic Acids Res.17:3519–3533.
29.Green, H.1977. Terminal differentiation of cultured human epidermal cells. Cell11:405–416.
30.Howley, P. M.1996. Papillomavirinae: the viruses and their replication, p. 2045–2076.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa. 31.Hubert, W. G., T. Kanaya, and L. A. Laimins.1999. DNA replication of
human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5⬘of the minimal origin. J. Virol.73:1835–1845. 32.Hubert, W. G., and L. A. Laimins.2002. Human papillomavirus type 31
replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of E1 and E2 expression. J. Virol.76:2263–2273.
33.Ishiji, T., M. J. Lace, S. Parkkinen, R. D. Anderson, T. H. Haugen, T. P. Cripe, J. H. Xiao, I. Davidson, P. Chambon, and L. P. Turek.1992. Tran-scriptional enhancer factor (TEF)-1 and its cell-specific co-activator activate human papillomavirus-16 E6 and E7 oncogene transcription in keratinocytes and cervical carcinoma cells. EMBO J.11:2271–2281.
34.Jeon, S., B. L. Allen-Hoffmann, and P. F. Lambert.1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol.69:2989–2997.
35.Jeon, S., and P. F. Lambert.1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA
92:1654–1658.
36.Kammer, C., U. Warthorst, N. Torrez-Martinez, C. M. Wheeler, and H. Pfister.2000. Sequence analysis of the long control region of human papil-lomavirus type 16 variants and functional consequences for P97 promoter activity. J. Gen. Virol.81:1975–1981.
37.Kanaya, T., S. Kyo, and L. A. Laimins.1997. The 5⬘region of the human papillomavirus type 31 upstream regulatory region acts as an enhancer which augments viral early expression through the action of YY1. Virology237:
159–169.
38.Kikuchi, K., A. Taniguchi, and S. Yasumoto.1996. Induction of the HPV16 enhancer activity by Jun-B and c-Fos through cooperation of the promoter-proximal AP-1 site and the epithelial cell type-specific regulatory element in fibroblasts. Virus Genes13:45–52.
39.Klumpp, D. J., and L. A. Laimins.1999. Differentiation-induced changes in promoter usage for transcripts encoding the human papillomavirus type 31 replication protein E1. Virology257:239–246.
40.Koskinen, W. J., R. W. Chen, I. Leivo, A. Makitie, L. Back, R. Kontio, R. Suuronen, C. Lindqvist, E. Auvinen, A. Molijn, W. G. Quint, A. Vaheri, and L. M. Aaltonen.2003. Prevalence and physical status of human papilloma-virus in squamous cell carcinomas of the head and neck. Int. J. Cancer
107:401–406.
41.Kuo, S. R., J. S. Liu, T. R. Broker, and L. T. Chow.1994. Cell-free replication of the human papillomavirus DNA with homologous viral E1 and E2 pro-teins and human cell extracts. J. Biol. Chem.269:24058–24065.
42.Kyo, S., A. Tam, and L. A. Laimins.1995. Transcriptional activity of human papillomavirus type 31b enhancer is regulated through synergistic interaction of AP1 with two novel cellular factors. Virology211:184–197.
43.Laimins, L. A.1998. Regulation of transcription and replication by human papillomaviruses, p. 201–223.InD. J. McCance (ed.), Human tumor viruses. ASM Press, Washington, D.C.
44.Lambert, P. F., B. C. Monk, and P. M. Howley.1990. Phenotypic analysis of bovine papillomavirus type 1 E2 repressor mutants. J. Virol.64:950–956. 45.Liu, J. S., S. R. Kuo, T. R. Broker, and L. T. Chow.1995. The functions of
human papillomavirus type 11 E1, E2, and E2C proteins in cell-free DNA replication. J. Biol. Chem.270:27283–27291.
46.May, M., X. P. Dong, E. Beyer-Finkler, F. Stubenrauch, P. G. Fuchs, and H. Pfister.1994. The E6/E7 promoter of extrachromosomal HPV16 DNA in cervical cancers escapes from cellular repression by mutation of target se-quences for YY1. EMBO J.13:1460–1466.
47.Meyers, C., and L. A. Laimins.1994. In vitro systems for the study and propagation of human papillomaviruses. Curr. Top. Microbiol. Immunol.
186:199–215.
48.Myers, G.1995. Human papillomaviruses. Los Alamos National Laboratory, Los Alamos, N.Mex.
49.O’Connor, M., and H. U. Bernard.1995. Oct-1 activates the epithelial-specific enhancer of human papillomavirus type 16 via a synergistic interac-tion with NFI at a conserved composite regulatory element. Virology207:
77–88.
50.O’Connor, M. J., S. H. Tan, C. H. Tan, and H. U. Bernard.1996. YY1 represses human papillomavirus type 16 transcription by quenching AP-1 activity. J. Virol.70:6529–6539.
51.Plumpton, M., N. A. Sharp, L. H. Liddicoat, M. Remm, D. O. Tucker, F. J. Hughes, S. M. Russell, and M. A. Romanos.1995. A high capacity assay for inhibitors of human papillomavirus DNA replication. BioTechnology 13:
1210–1214.
on November 8, 2019 by guest
http://jvi.asm.org/
52.Rabson, M. S., C. Yee, Y. C. Yang, and P. M. Howley.1986. Bovine papil-lomavirus type 1 3⬘early region transformation and plasmid maintenance functions. J. Virol.60:626–634.
53.Rheinwald, J. G., and M. A. Beckett.1981. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res.41:1657–1663.
54.Ritchie, J. M., E. M. Smith, K. F. Summersgill, H. T. Hoffman, D. Wang, J. P. Klussmann, L. P. Turek, and T. H. Haugen.2003. Human papilloma-virus infection as a prognostic factor in carcinomas of the oral cavity and oropharynx. Int. J. Cancer104:336–344.
55.Romanczuk, H., and P. M. Howley.1992. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immor-talization capacity. Proc. Natl. Acad. Sci. USA89:3159–3163.
56.Ruesch, M. N., and L. A. Laimins.1998. Human papillomavirus oncopro-teins alter differentiation-dependent cell cycle exit on suspension in semi-solid medium. Virology250:19–29.
57.Ruutu, M., P. Peitsaro, B. Johansson, and S. Syrjanen.2002. Transcriptional profiling of a human papillomavirus 33-positive squamous epithelial cell line which acquired a selective growth advantage after viral integration. Int. J. Cancer100:318–326.
58.Sakai, H., T. Yasugi, J. D. Benson, J. J. Dowhanick, and P. M. Howley.1996. Targeted mutagenesis of the human papillomavirus type 16 E2 transactiva-tion domain reveals separable transcriptransactiva-tional activatransactiva-tion and DNA replica-tion funcreplica-tions. J. Virol.70:1602–1611.
59.Sandler, A. B., C. C. Baker, and B. A. Spalholz.1996. Sp1 is critical for basal and E2-transactivated transcription from the bovine papillomavirus type 1 P89 promoter. J. Gen. Virol.77:189–198.
60.Seedorf, K., G. Krammer, M. Durst, S. Suhai, and W. G. Rowekamp.1985. Human papillomavirus type 16 DNA sequence. Virology145:181–185. 61.Spalholz, B. A., S. B. Vande Pol, and P. M. Howley.1991. Characterization
of theciselements involved in basal and E2-transactivated expression of the bovine papillomavirus P2443 promoter. J. Virol.65:743–753.
62.Sprague, D. L., S. L. Phillips, C. J. Mitchell, K. L. Berger, M. Lace, L. P. Turek, and A. J. Klingelhutz.2002. Telomerase activation in cervical kera-tinocytes containing stably replicating human papillomavirus type 16 epi-somes. Virology301:247–254.
63.Stanley, M. A., H. M. Browne, M. Appleby, and A. C. Minson.1989. Prop-erties of a non-tumorigenic human cervical keratinocyte cell line. Int. J. Cancer43:672–676.
64.Stark, L. A., M. J. Arends, K. M. McLaren, E. C. Benton, H. Shahidullah,
J. A. Hunter, and C. C. Bird.1994. Prevalence of human papillomavirus DNA in cutaneous neoplasms from renal allograft recipients supports a possible viral role in tumour promotion. Br. J. Cancer69:222–229. 65.Tan, S. H., B. Gloss, and H. U. Bernard.1992. During negative regulation of
the human papillomavirus-16 E6 promoter, the viral E2 protein can displace Sp1 from a proximal promoter element. Nucleic Acids Res.20:251–256. 66.Thomas, J. T., W. G. Hubert, M. N. Ruesch, and L. A. Laimins.1999. Human
papillomavirus type 31 oncoproteins E6 and E7 are required for the main-tenance of episomes during the viral life cycle in normal human keratino-cytes. Proc. Natl. Acad. Sci. USA96:8449–8454.
67.Ustav, M., and A. Stenlund.1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J.10:449–457.
68.Veress, G., K. Szarka, X. P. Dong, L. Gergely, and H. Pfister.1999. Func-tional significance of sequence variation in the E2 gene and the long control region of human papillomavirus type 16. J. Gen. Virol.80:1035–1043. 69.Villa, L. L., L. Sichero, P. Rahal, O. Caballero, A. Ferenczy, T. Rohan, and
E. L. Franco.2000. Molecular variants of human papillomavirus types 16 and 18 preferentially associated with cervical neoplasia. J. Gen. Virol.81:2959– 2968.
70.Wheeler, C. M., T. Yamada, A. Hildesheim, and S. A. Jenison.1997. Human papillomavirus type 16 sequence variants: identification by E6 and L1 lin-eage-specific hybridization. J. Clin. Microbiol.35:11–19.
71.Xi, L. F., J. J. Carter, D. A. Galloway, J. Kuypers, J. P. Hughes, S. K. Lee, D. E. Adam, N. B. Kiviat, and L. A. Koutsky.2002. Acquisition and natural history of human papillomavirus type 16 variant infection among a cohort of female university students. Cancer Epidemiol. Biomarkers Prev.11:343–351. 72.Xi, L. F., L. A. Koutsky, D. A. Galloway, J. Kuypers, J. P. Hughes, C. M. Wheeler, K. K. Holmes, and N. B. Kiviat.1997. Genomic variation of human papillomavirus type 16 and risk for high grade cervical intraepithelial neo-plasia. J. Natl. Cancer Inst.89:796–802.
73.Yamada, T., M. M. Manos, J. Peto, C. E. Greer, N. Munoz, F. X. Bosch, and C. M. Wheeler.1997. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J. Virol.71:2463–2472. 74.Yang, L., R. Li, I. J. Mohr, R. Clark, and M. R. Botchan.1991. Activation of
BPV-1 replication in vitro by the transcription factor E2. Nature353:628– 632.
75.zur Hausen, H.1999. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin. Cancer Biol.9:405–411.