Sendai Virus C Proteins Counteract the Interferon-Mediated Induction of an Antiviral State
Full text
Figure


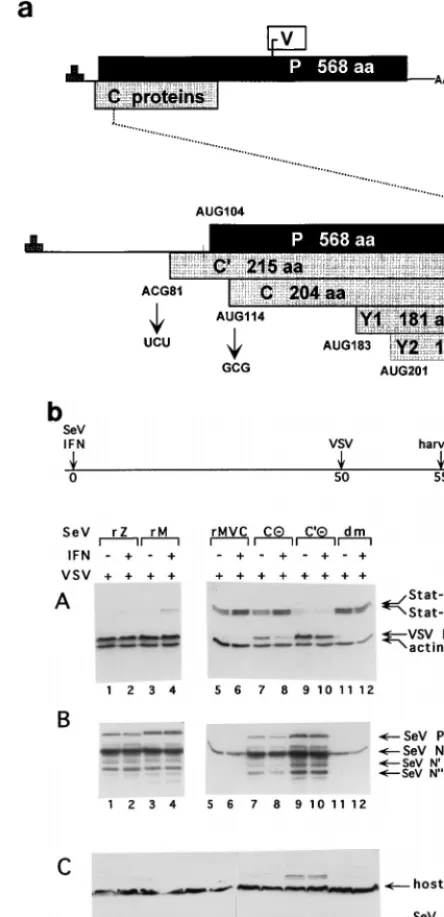

Related documents
To determine the capacity of the 5=pppRNA RIG-I agonist to induce a protective antiviral response to DENV infection, A549 cells were challenged with DENV at different multiplicities
To approximate virus infection whereby both NS proteins are produced, we expressed each deletion mutant form of one NS protein in combination with the wild- type protein of the other
Using HeLa cell lines expressing truncated or mutated SeV C proteins, we found that the C-terminal half has anti-IFN capacity, and that K 151.. A,
Using two strains (Cantell and 52) of the murine respiratory Sendai virus (SeV) with differential abilities to induce type I IFN production from infected cells, together with type I
Previously, we created by reverse genetics the 4C( ⫺ ) virus that expresses none of the four C proteins and concluded that the SeV C proteins are categorically nonessential gene
This rescue could be clearly attributed to a specific inhibition of the antiviral action of IFN, since no increase in EMCV titer was observed in UHCV cells as a result of the
Gunduz et al Virology Journal 2012, 9 143 http //www virologyj com/content/9/1/143 RESEARCH Open Access Free fatty acids induce ER stress and block antiviral activity of interferon
We also show that the Tat proteins specifically associate with TAK in vivo, since wild-type Tat-1 and Tat-2 proteins expressed in mammalian cells, but not mutant Tat proteins
