Analysis of the cleavage site of the human immunodeficiency virus type 1 glycoprotein: requirement of precursor cleavage for glycoprotein incorporation.
Full text
Figure
Related documents
According to the standard DHS definition, “the unmet need group includes all fecund women who are married or living in union, and thus presumed to be sexually active, who either
fail the test, Auditory Brain Stem evoked response audiometry can..
In summary, these analyses indicate that (i) introduction of sim- ple BLV producer cells or proviral DNA induces production of anti-BLV antibodies; (ii) injection of simple BLV
To study the role of CD44 in HIV-1 infection and tropism, we have used cells of the human T-lymphoblast line Jurkat, a CD4-positive, CD44-negative cell line which can be infected
Most of the mutant viruses also had normal Western blot (immunoblot) profiles, although three of the mutations resulted in reduced signals for IN relative to the wild type on
(43, 44) reported the role of Ca2+ in the in vitro assembly of murine polyomavirus recombinant VP1 capsomeres into capsid-like particles, and now we have reported a similar mechanism
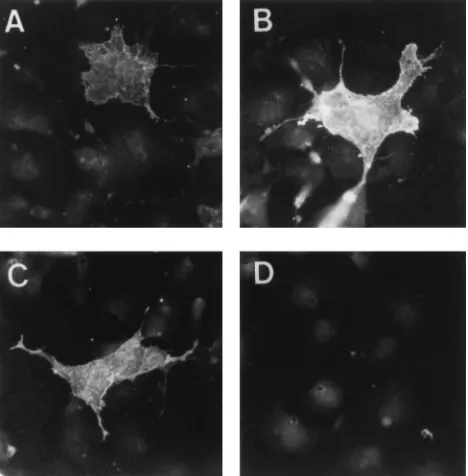
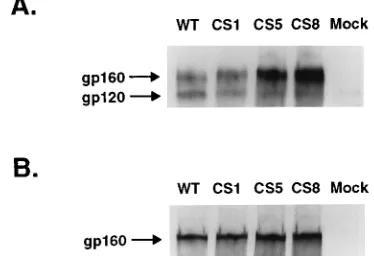
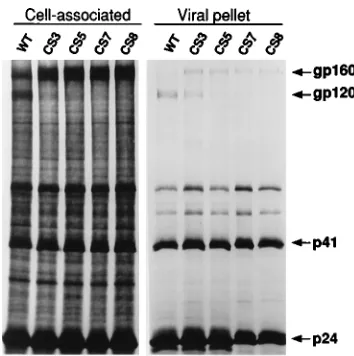
AIDS patient sera precipitated both precursor and processed forms of the wild-type and AV1/2, AV3, and AV1/2/3 glyco- proteins from transfected COS-1 cells lysates..
The positions (bases) of full-length donor template-directed DNA synthesis products (F) and products produced by strand transfer events (T) are indicated. The region where the donor
