0022-538X/86/020563-09$02.00/0
Copyright © 1986, American Society for Microbiology
Functional
Mapping
of
a
trans-Activating
Gene
Required for
Expression of
a
Baculovirus Delayed-Early Gene
LINDA A.GUARINO* AND MAX D. SUMMERS
Departmentof Entomology, Texas A&M University, and TexasAgricultural Experiment Station, College Station, Texas 77843
Received 25July 1985/Accepted 9 October 1985
The temporal regulation of an early gene of the baculovirus Autographa californica nuclear polyhedrosis virus wasexamined. We constructed a plasmid (plasmid 39CAT) in which the bacterial gene for chloram-phenicolacetyltransferase was placed under the control of the promoter for the gene for a A.californicanuclear polyhedrosisvirus39,000-dalton protein(39K).Atransientexpressionassay ofplasmid39CATrevealed that the 39Kgene wasexpressedininfectedcellsbut not inuninfectedcels, indicatingthat the39K gene should be classifiedas adelayed-earlygene. The 39Kpromoter also efficiently directed the synthesis of chloramphenicol acetyltransferasewhen theplasmidwascotransfected withviral DNA which hadbeenrestricted with several restrictionenzymes. To map the location of the gene(s) required for the synthesis of39K,plasmid39CAT was cotransfected withpurifiedrestrictionfragments of A. californica nuclear polyhedrosis virus DNA. Fragments which mapped between 90.7 and 100.8 map units induced plasmid 39CAT. Plasmid pEcoRI-B, containing EcoRIfragment B (90 to 100 map units), activatedplasmid39CAT.Functionalmapping of
plasmid
pEcoRI-B indicatedthat the essential region was located between 95.0 and 97.5 map units. The 5' end of this gene was mapped, and the chloramphenicol acetyltransferase gene was inserted under the control of its promoter. Transient assayexperiments indicated that the trans-acting regulatory genewasexpressed in uninfected cells and istherefore animmediate-early gene. This gene was named IE-1.The genome of Autographa californica nuclear poly-hedrosis virus (AcNPV) is adouble-stranded,
supercoiled,
circularDNAmoleculeofapproximately 128kilobasepairs. Thetemporal regulation ofAcNPV geneexpression is sim-ilar to that ofmost large DNA viruses. The expression of immediate-early (ca) genes does not require prior viral pro-tein synthesis, while delayed-early (,B) genes apparently require an ageneproductfor expression (25). Synthesis of late
(y)
genesisconcomitant with, butmay notbe dependentupon,,
the onset of viral DNA synthesis (12, 25). Bac-uloviruses are unique in having a fourth temporal class of genes,theverylate(6)
genes,whichareassociatedwithviral occlusion.The
8genes arehighly expressed after the release of extracellularvirus, whilethesynthesis ofa, ,B,andfy gene products isrepressed (9, 31, 47). Some genes areexpressed inmorethanonetemporal phase. Forexample,thegenefor aprotein
having a molecularweight of 39,000 (39K) is the mostabundantlyexpressed earlygeneand is also expressed late ininfection(37, 41).Synthesis of39K can be detectedby 3 to 6 h postinfection, but it has not been determined whetherthe39K gene is an a gene ora 13gene.AcNPVearlytranscriptsmap to afewdispersed regions of the genome, while late in infection most ofthe genome is transcribed (13, 14, 35). Overlapping nests of transcripts with coterminal 3' or 5' ends havebeen mapped in several regions of the genome (15, 27). Onetranscription unit was composed of
five
early and late RNAs with a common 3' end. The smallest RNAsappeared early and were replaced in time by successively larger transcripts, suggesting that temporal regulation involves sequential activation of up-stream promoters along with coordinate deactivation of downstream promoters (15). In contrast, the very late (b)*Correspondingauthor.
transcription unitsarecomposed ofoverlapping RNAs with coterminal 5' ends.
Splicing
has not been detected in baculovirus transcripts (26).Nothing is known about the mechanisms of regulation of temporal expression in baculoviruses. Two temperature-sensitive mutants blocked at an early stage of replication havebeen describedpreviously(16,33).Themutationswere mapped to two different regions of the genome, but the identity and normal function of the mutated genes are unknown.
We identified and functionally mapped a gene which regulates
temporal expression
inAcNPV.Thebacterialgene forchloramphenicol acetyltransferase
(CAT) was cloned underthe control ofthe 39K gene. The resulting construc-tion, plasmid 39CAT,was active ininfected cellsbut notin uninfected cells, indicatingthat the 39K gene is a delayed-early gene. By using cotransfection of plasmid 39CAT and purified restriction fragments of viral DNA or cloned vital DNA fragments, we identified a viral immediate-early gene which trans-activates 39K gene expression.MATERIALS ANDMETHODS
Cells and virus.Spodoptera
frugiperda
IPL-21AEIIIcells obtained from S. Weiss (Cetus Corp., Emeryville, Calif.) were used in transfection experiments. These cells were propagated in IPL-41 monolayer medium (46) containing10% fetal bovine serum (KC Biologicals, Lenexa, Kans.). For preparation of viral DNA and infected cell RNA, S. frugiperda clone 9spinner culturesweregrown in TNMFH medium (20) supplemented with 10% fetal calfserum. The cellswereconcentrated bycentrifugationat1,000 x g for 5 min,suspendedat adensityof 107cellsperml,andinoculated with extracellular virus at amultiplicity of 20 PFU percell. After a 1-hadsorption periodatroomtemperature, the cells 563on November 10, 2019 by guest
http://jvi.asm.org/
564 GUARINO AND SUMMERS
were centrifuged as describedabove andsuspendedin fresh TNMFH medium at a density of106 cells per ml. After 48 h, the cells were centrifuged at 1,000 x g. Thesupernatant was adjusted to 10%polyethylene glycol and 0.1 MNaCl, andthe preparation was gently stirred at 4°C for 1 h. The virus precipitate was collected bycentrifugation at10,000 x g for 10min. The pellet was gentlysuspendedin5 mlof 10 mMTris (pH
7.5)-i
mM EDTA and then mixedwith anequal volume of20%polyethyleneglycolto 0.2 MNaCl.After 1 h at 4°C, the precipitated virus was centrifuged and suspended as described above. The virus suspension was adjusted to 1% sodium dodecyl sulfate and extracted with phenol.RNA isolation. Cells wereinfected asdescribed above. At 6 or 18 h postinfection the cells were centrifuged, washed once in phosphate-buffered saline, and suspended in five times the packed cell volume of 6 M guanidinium isothio-cyanate-5 mM sodium citrate-0.1 M
P-mercaptoetha-nol-0.5% Sarkosyl. Then 1 g ofCsCl per ml ofsupernatant was added, the mixture was layered onto 1.2 ml of5.7 M CsCl, and thepreparationwascentrifuged at30,000rpm in a type SW60 rotor for 16 h. The RNA pellet was suspended and extracted asdescribed previously (30).
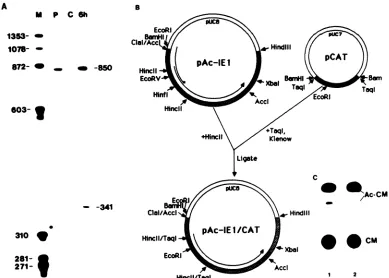
Recombinant plasmids. The 39CAT plasmids were con-structed in several steps. First, the termination and polyadenylation sequences for the polyhedrin gene were excised from plasmid pAc373 (G. E. Smith, G. Ju, B. L. Erickson, J. Moschera, H.-W. Lahm, R. Chizzonite, and M. D. Summers, Proc. Natl. Acad. Sci. USA, in press) with BamHI and EcoRI. The resulting 3-kilobase (kb) fragment was cloned into the BamHI and EcoRI sites of pUC8. The coding sequences for the bacterial CAT gene were derived fromplasmid pCAT (28). The CAT gene was excised with BamHI and cloned into the BamHI site adjacent to the polyhedrin sequences. The proper orientation of the CAT gene was determined byEcoRI digestion. This plasmid had only oneunique cloning siteupstreamfrom the CAT gene. A second unique site wascreated by removingthe SalI site in the polyhedrin sequences by successively performing the following steps: partial Sall digestion, purification of full-length DNA, Klenow repair, blunt end ligation, and trans-formation of Escherichia coli strain JM83 cells. Plasmid pAc3075, which had a singleSalI site in themultiple cloning region, was selected. The 5' end of the 39K message has been mapped to a site ca. 220base pairs (bp)upstream from a PvuII site inPstI fragment K of the AcNPV genome (J. Vlak, no. 23, Abstr. Soc. Invertebr. Pathol. 1985). To remove thecoding sequences betweenthe promoter atdthe PvuII site, plasmidpPstI-K (20 ,ug) wasdigested with PvuII and incubated with 0.5 U of Bal 31 buffer at 30°C (30). At 5-min intervals,
10-p.l
portionswereremovedandadjustedto 50 mM EGTA [ethylene glycol-bis(,-aminoethyl ether)-N,N,N',N'-tetraacetic acid], and the digested ends were made blunt byincubationwith theKlenowfragmentof DNA polymerase I.The digested DNAs wereextracted with phenol, precipi-tated with ethanol, suspended, and digested withPstI. After electrophoresisin 1%agarose,fragmentsca. 750 to 1,050bp long were eluted andclonedinto the
PstI
andSalI (repaired) sites ofpAc3075. Seven plasmids containing different-length promoter sequences were tested by transfecting AcNPV-infected S.frugiperda cells and assaying for CAT activity. Theplasmid yielding thehighest activity (plasmid 39CAT-4) waschosen forfurther study.The HindIII and EcoRI plasmids were constructed by cloningthe appropriate restriction fragments in the HindIII and EcoRI sites of
PUC8.
Recombinant plasmids werepropagated in E. coli strains JM83 and TB1. Plasmid DNA wasisolated
by
the methodof Holmes andQuigley(21)andpurified by centrifugation
in cesium chloride-ethidium bro-midegradients.TransfectionsandCAT assays. Cellsweretransfected bya modification of the procedure of Graham and van der Eb (18). Subconfluent monolayers of S. frugiperda IPL-21 AEIIIcellswereharvestedby scraping witharubber
police-man and suspended by gentle pipetting. The cells were seeded at a
density
of 106 cells per 60-mm dish. Thecells
were allowed to attach for 1 h, and then the mediumwas
replaced
with 0.5 ml of fresh medium. DNAs to be transfected were diluted in 0.5 ml of HEPES(N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic
acid)-buffered
saline
(25
mMHEPES, pH
7.1, 140 mMNaCl)
containing
125 mM
CaCI2.
The unprecipitated DNAs were addeddropwise
tothecellmonolayers.
Calciumphosphateprecip-itates formedduetotheCaCl2 intheHEPES-bufferedsaline and thephosphateinthemedium. Each tratisfection mixture contained2
,ug
of 39CAT-4and 2,g of viralorplasmidDNA unless otherwise noted. For transfections of purified gelfragments,
10,ug
of viral DNA was digested with a restric-tion enzyme, and fragments were separated by electropho-resis through 0.6% low-melting-temperature agarose. Thepurified fragments
wereelutedby heating the preparation at650C
for 15min,
followed by phenol extractionand ethanol precipitation. The amountofaDNAfragment purifiedfrom 10jig
of digested DNA was added to each dish. For transfections inwhich the total amount oftransfected DNA waslessthan 4,g,calf thymus DNA was added to bringthe amount up to 4,ug.
Cells were harvested 24 h after transfection, and CAT assays were performed asdescribed previously (18), except that cells were broken by three cycles of freezing and
thawing
instead of sonication and each assay mixture con-tained only0.2 ,uCiof[14C]chloramphenicol.
DNAsequencing.The39CATplasmidsweredigestedwith PstI and BamHI, and the promoterfragments were cloned into the PstI and BamHI sites of M13mp9 (32). Single-stranded recombinantphage was purifiedand sequenced by thedideoxy chain terminationmethod (36). The sequence of the oppositestrandwasdeterminedby cloningand sequenc-ingthe appropriateHpaII restriction fragments of PstI-K in the AccI site ofmp9.
Si nuclease mapping of AcNPV DNA. The 5' ends of the 39K and IE-1transcripts were mapped by the S1procedure ofBerkandSharp (6), asmodifiedby WeaverandWeissman (45); 5'-end-labeled probes were generated by digesting pPstI-K and pIE-1 with ClaIl and Hinfl,respectively.The5' endsweredephosphorylatedwith calf intestinalphosphatase (Boehringer Mannheim Biochemicals, Indianapolis, Ind.) and radiolabeled with T4 polynucleotide kinase (Bethesda Research Laboratories, Inc., Gaithersburg, Md.) and
[y-32P]ATP (4,000 Ci/mmol). The DNAs were then digested with PstI (pPstI-K) or BamHI (IE-1), and the appropriate restriction fragments were purified by agarose gel electro-phoresis. DNAprobes (0.1 ,ug) were hybridized to 10 ,ug of total cellular RNA in a solution containing 80%formamide, 40 mM PIPES [piperazine-N,N'-bis(2-ethansulfonic acid)] (pH 6.4), 0.4 M NaCl, and 1 mM EDTA for 4 h at 42°C. Samples were diluted 10-fold into Si buffer (30 mM sodium acetate, pH 4.6, 4.5 mM ZnSo4, 0.25 M NaCI, 20,ug of single-stranded DNA perml) containing 100 U of Si nucle-ase per ml and incubated at 37°C for 30 min. Digestion was terminated by adding 75 ,ul of S1 stop buffer (2.5 M ammonium acetate, 0.1 M EDTA). The Si hybrids were J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
M P C 6h 18h
* -1080
} -653
B
HHrUUrILJlUHI U IdHLH1THHtHUU UI UULUH,UHrI U TTGGACATCGTGTCGTTTGAGCGTRTRRRRGRRTRTRTR AGAGCTARTTTAGGCCRTTTCRCAGTRRTCACCORCARR
6
CTGTTCAAGCCTRAGGTGCCTGTCTTCATCRCRRRCGRA
TVTGCCAGGTTGTTGGGCRTTAARAAATATRTCATCRRGR
4
RTACRRACGGGTTGTTTCRARGGTTTRCRRGRAGCRRRCA
9
ATGAGTA
ARC GTG CCG GRG CAR CAR TCT CCG GRG ACT GCG GCC GTG TOC RAR ART GRA ARG CTG TTG ART AAR CTG GARARC ARA TCC ARC RTG GRC
TCG RCC TCT TRC CAG
A
C
C -260
-244
-240
_i _ -237
.*e
*
CMcontrol 901 3904 3906 3908 3909 3916 3920
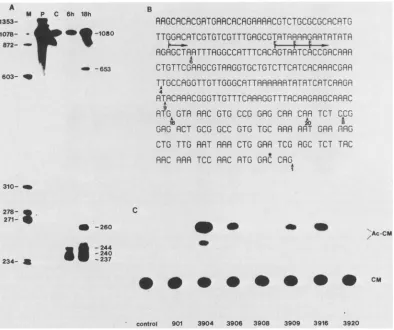
FIG. 1. Si nucleasemappingof the 5' ends ofthe 39Kgene(A), the nucleotide sequenceof the 5' end of the gene(B), and transient
expressionofchimeric 39CATplasmids (C). (A) Mappingof the5' endsofviraltranscriptsisolated 6 and 18 hpostinfectionorfromcontrol, uninfected cells (lane C). Total cellularRNA(10 ,ug)washybridizedwith 0.1 Figof the1.07-kbPstI-ClaIIfragment(the positionofthe label intheClaIIsite is indicatedbyanasterisk inpanel B).The size of the Si-resistantfragment wasdetermined by electrophoresison a7 M
urea-5%polyacrylamide gel.Anautoradiograph of thegelis shown. The sizes ofthe molecularweight markers(+X174HaeIIIfragments)
areindicatedontheleft,and the sizes of the Si-resistant DNAsareindicatedontheright. LanePcontained the5'-end-labeled DNAprobe beforedigestion. Lane M contained the molecularweightmarkers.(B) Sequenceof the5' endof the 39Kgene.Thenumbered arrowheads
indicate the extents of the Bal 31 deletions for the plasmids indicated. The arrows marked with E and L indicate the early and late
transcriptional start sites, respectively. The positionof the 5' end label used for S1 mapping is indicated by an asterisk. (C) Transient expression of 39CATplasmids. S.frugiperdacellswereinfected with AcNPV 1 hpriortotransfection with2-,ug portionsoftheplasmids indicated atthe bottom. At 24 h posttransfection, cell extracts were prepared andanalyzedforCAT activity. Anautoradiograph ofthe
thin-layerchromatogramis shown.Thepositionsofinput unacetylatedchloramphenicol (CM)andacetylated products(Ac-CM)areindicated.
precipitated withethanol, denatured, andanalyzed by elec-trophoresis on 5% polyacrylamide-7 M urea-89 mM Tris hydrochloride (pH 8.3)-89mMboricacid-2 mM EDTAgels. Size standards were derived from 3'-end-labeled HaeIII digests of XX174DNA(30).
RESULTS
Temporal regulation of 39K. The 39K gene has been mappedto EcoRIfragment J of AcNPV DNA (14, 41) (see Fig.3 forthelocation ofthe39K gene). The 5' end is located approximately220bpupstreamfromaPvuII site in AcNPV
DNA PstIfragmentK (Vlak,no. 23,Abstr. Soc. Invertebr.
Pathol.1985). Becausebaculovirusgenesapparentlyarenot spliced (26), we used high-resolution S1 mapping to deter-mine theprecise location of the5'end of the 39Kgene.The
1,080-bp PstI-ClaII fragment of PstI-K was labeled at the ClaII site andhybridized tototal cellular RNA isolatedat6 and 18 h postinfection (Fig. 1A). The following four
S1-resistant DNA fragments were resolved when the DNA
probe was hybridized to 6-h RNA: a major band at 237 nucleotides, two minor bands at 240 and 244 nucleotides, and a very faint band at 653 nucleotides. When the probe
was hybridized to 18-h RNA, the same four S1-resistant
bandswereobserved. Inaddition,anothermajorbandat260 bpwas present.
In orderto clone the CATgene underthe control ofthe
39K promoter, Bal 31 was used to remove the coding
sequenceslocated between thepromoterandthe PvuIIsite. The approximate extent of the Bal 31 deletions was
deter-minedby restrictiondigestion andagarose gel
electrophore-sis. Six plasmids with promoter fragments of different lengths were sequenced to determine the precise point at
which the CAT gene was inserted (Fig. 1B). Plasmid
pAc3901 contained the complete 1,080-bp PstI-PvuII frag-ment. Assuming that the first AUG sequence downstream from the transcriptional start sites was the initiating AUG
sequence, pAc3901 would be expected to code for an in-phasefusionprotein. In threeofthedeletions,theCATgene
A
1353-
1078-
872-603- *
310- _
278-
11
271-
234-"Ac-CM
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.107.501.66.396.2]566 GUARINO AND SUMMERS
A B
-A -B
-CD *
-E -FG
-HI
-J
-K I
-L
-M
XhoI
Es
AB CD E FG H I J K
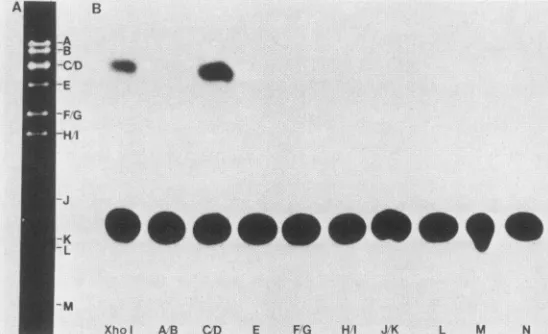
FIG. 2. XhoIrestriction digest ofAcNPV DNA (A)and transient expressionassayofcellextractscotransfected with 39CAT-4andpurified fragments(B). AcNPV DNA (10,ug)wasdigested withXhol, and restriction fragmentswereseparated electrophoreticallyon0.6% agarose
gels. Purified fragmentswere eluted and testedfor the abilitytoinduce 39CAT expression. Each transfectionmixture contained the total
amountoffragmentpurified from 10 ,ugof viral DNA.
was inserted into the 39K coding region. Two plasmids, pAc3908 and pAc3920, were expected to code for
out-of-phase fusion proteins. Plasmid pAc3916 was expected to
code foranin-phase fusionprotein. In plasmids pAc3904 and
pAc3909, the CAT gene was inserted 50 or 90 bp
down-stream from the major early transcription start site. In pAc3906, the early transcriptionstartsitesweredeleted, and
the CATgenewasinserted 4 bp downstream from the major
late start site. Because there were noAUG codons located
between the early and latestart sites, both transcripts were
expected tocode for thesame protein.
To examine thetemporal regulation of the 39Kpromoter, S.frugiperda cells were infected with AcNPV and 1 h later were transfected with
2-pxg
portions of the various 39CATplasmids. After 24 h, cellular extracts were prepared, and
CATassays were performed (Fig. 1C). As expected, there wasno CAT activity detected in thetwoplasmids in which the CATgene wasinsertedoutof phase. The in-phase fusion protein produced by pAc3916 was active. These results
indicate that the assignment of the initiating AUG codonwas
correct. Plasmid pAc3901, which encoded a larger fusion
protein, had little activity. The two plasmids in which the CAT gene was inserted in the 5' leader of the early tran-scripts were active in directing the synthesis of CAT. The
latepromoteralso efficiently directed the synthesis of CAT,
asevidenced by the activity observed in cells cotransfected
withpAc3906.
Thesevenplasmids whichwe constructedwerealso used
to transfect S. frugiperda cells that had not been infected priorto transfection. No CAT activity was observed with
anyofthe plasmids in extractsof uninfected cells(datanot shown), indicating that the 39Kpromoterwas not transcrip-tionally active in uninfected cells. These resultsare
consist-entwith aclassification ofdelayed early for the 39K gene,
because a viral function is apparently required for
expres-sionof the 39Kgene.PlasmidpAc3904 (=plasmid 39CAT-4)
waschosen for further study.
Functional mapping ofthe gene required for 39K
expres-sion. The 39K promoter was efficiently induced when
uninfected cells were cotransfected with 2 ,ug of 39CAT-4 and 2,ug of viral DNA.Thepromoterwasalsostimulatedby
viral DNA thatwasdigested with several different restriction
enzymes priortotransfection(datanot shown). Tomapthe
location of the gene(s) required for expression of the 39K
gene, AcNPV DNA was restricted with either KpnI, SstI,
SstII, XhoI, or BamHI. It was necessary to use several
restriction enzymes because many restriction fragments
comigrated on agarose gels. For example, the restriction
digestpatternfor XhoI is shown inFig. 2A. Fragments A and B, C and D, FandG, and H and I comigrated andso were
testedaspairs. The results of the CATassaysafter cotrans-fection of gel-purified XhoI restriction fragments and 39CAT-4 are shown in Fig. 2B. Cells cotransfected with
39CAT-4 and XhoI fragments C and D synthesized CAT, indicating that the gene(s) neededto activate the 39K gene are located on either one or both of these fragments. The other fragments which induced CAT activity were KpnI
fragments A, B, and C, SacII fragments C and D, SacI fragments A and B, and BamHI fragment B (data not shown). By constructing a consensus map from several digests,itwaspossibletoeliminate all butonefragment from
each digest (Fig. 3). Our combined data indicated that the region from 90.7to0.8map units(m.u.) contained thegene
required for expression of the 39K gene. The consensus
region wasdefined by the left end of XhoI fragment C and
the right end of SstII fragment D. The circular AcNPV
genome is presented as linear by convention, with the 0 position of themapatthe leftend ofEcoRIfragment I(40). EcoRI fragment B (90 to 100 m.u.) covered most ofthe
consensus region indicated in Fig. 3. This fragment was
cloned into the EcoRI site of pUC8 and tested in the transient assay system for its ability to induce 39CAT expression. CAT was efficiently expressed in cells
transfected with 39CAT-4 andpEcoRI-B (data notshown). To further define the location of the gene required for expression of the 39K gene, a detailed restriction map of pEcoRI-B wasgenerated (Fig. 4A). A seriesof deletions in
EcoRI fragmentB was constructedby digestion and
religa-tion with Sall, ClaI, EcoRV, XbaI, or BglII (Fig. 4B). In addition, plasmids containing HindIII fragments G and F
were cloned and tested. All of the activity was located in
HindlIlfragmentG. Deletion of the XbaIfragmentbetween 96.7 and97.5m.u.didnoteliminate CATactivity, indicating that the gene was located to the left of 96.7 m.u. on the physicalmap.Deletion oftheregion between the SalIsite in
themultiple cloningsite and theSall siteat92.2m.u.hadno
L M N
J. VIROL.
11
14 ol
04
OtTo4b
4b
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.171.445.70.237.2]w uKp I A J KI1111N F *~C I S 0 ALI I I ai
_~~~~~~~~~~
MMl
J IKtlHl
"I,
I L c"D ^L s1 tJFI
C A , ,LMi K
jP~ ~~~ III A I U I0,1 IJII
ECAfl
stIff
BAasI
I I I
0 10 20
I I I I I I
s 0 0710
40
XACNPVU
FIG. 3. Consensusmapof theDNAfragmentswhich activate 39CAT-4. Fragmentswhichwerepositivein thetransientexpressionassay
areindicated by heavy lines. Theconsensusregion is shownas ashaded line. The circular AcNPVgenomewaslinearizedatEcoRIfragment Iby convention. The genomic location of the 39Kgeneis indicated.
effect, indicating that the gene was located to the right of 92.2m.u.
Two of the deletions (the ClaI and EcoRV deletions) did not activate 39CAT expression. The ClaI fragment (95 to 99.8m.u.) and the EcoRV fragment (95.3to97.8m.u.)were
cloned into theAccI orHinclI sites ofpUC8and tested for
their ability to activate 39CAT-4 in the transient assay
system. Theplasmid containing the ClaI fragment induced CAT, but the plasmid containing the EcoRV fragment did not (data notshown). This result indicated that oneend of
thegenerequired for expression of the 39Kgenewaslocated
between the leftmost ClaI and EcoRV sites and the other end
waslocatedtotheleft of the XbaI siteat97.5m.u. Because
mostAcNPVgenesaretranscribedfrom lefttoright (15, 27)
on the linear map (clockwise on the circular map), we
expected that the end located between the ClaI and EcoRV siteswasthe 5' end.
A
HyIIIEcORI KhoI Sol I Sell
I I I
90.0 90.7 912 90.6
922
Plasmid pEcoRI-B was cotransfected with pAc3916,
pAc3909, and pAc3906 to determine whether these CAT plasmidswereactivatedby the samegeneas39CAT-4(data
not shown). CAT was expressed in cells transfected with
pAc3916, the plasmid producinganin-phase fusion protein,
and in cells transfected with pAc3909, in which the CAT
genewas insertedin the leader downstreamfrom 39CAT-4. No CAT activity was detected in cells transfected with pAc3906, in which the 5' end of the early transcript was
deleted, indicating that the early and late transcripts were
differentially regulated.
Temporal regulation of IE-1. The ClaIfragment of EcoRI-Bwascloned intothe AccIsite ofpUC8 in both orientations. Inoneorientation, digestionwithHindIII followed by religa-tion removed the sequences to the right of 96.9 m.u. The
resulting plasmid, which contained 3 kb of viral DNA, was
designated pIE-1 (for immediate-early gene 1). To confirm
HelEIIIIII
11
Clol EcoRV Xbol I XbolEed
Pe
I Clot';co*l
95.0 95.3 %.7 97.0 975 97.8 9 99.6 100
96.9 98Z3
B
cUT kfAtli
C,.,
EcoeN
Xhal
*iii/
MAVdVl-G
A- Awn-r
FIG. 4. Detailed restrictionmapof EcoRI-B (A) and constructionof EcoRI fragment B subclones (B). Five subcloneswereconstructed by restriction withSall, ClaI, EcoRV, XbaI, and BglII, followed by religation. The regions of EcoRI fragment B deleted by digestionare
indicatedby diagonal lines. pHindIII-G and pHindIII-F contained the restriction fragments indicated. The results ofatransient expression
assayfor each cloneareshown byaplus sign (induced 39CAT)or aminus sign(noCAT activity).
m0 T VU
m
L T- I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.107.502.71.278.2] [image:5.612.58.550.507.680.2]568 GUARINO AND SUMMERS
A
M P CSh B
1353- _ 1078-~
872- * _ _ -850
603- I
_ -341
0
31094 281-
271-Hincil/Taql 1 2
FIG. 5. S1mapping of the 5' end of the IE-1 gene (A), construction of anIE-1-CAT plasmid (B), and expression of the plasmid in infected anduninfected cells (c). (A)Sinuclease mapping of the 5' end of the IE-1 gene. Total cell RNAs (10 ,ug) from cells infected for 6 h orcontrol, uninfected (c) cellswerehybridized with 100-pg portions of an 850-bp BamHI-Hinfl probe (lane P), which was specifically radiolabeled at the Hinfl site.Sl-resistant hybrids were denatured and analyzed on 7 M urea-5% polyacrylamide gels. The sizes of the XX174 molecular weight standards(lane M)are indicated on the left, and the sizes of theSi-resistant fragments are indicated on the right. (B) Plasmid IE-1 was digested withHincII,and a770-bpTaqIfragment of pCAT which was made blunt ended by Klenow repairtreatment was cloned into the HinclI site. The location and direction of transcription of the IE-1 transcript are indicated by an arrow. The location of the probe forS1 mapping is indicated byanasterisk. (C) AcNPV-infected cells (lane 2) and uninfected cells (lane 1) were transfected with2,ug ofIE-1-CAT. After 24h, the CAT activity in cellularextracts wasassayed.CM, Input unacetylatedchloramphenicol; Ac-Cm, acetylated chloramphenicol products.
tht thetemporalregulation of thisgene wascharacteristic of animmediate-early gene, achimeric IE-1-CAT plasmidwas constructed. The5' end ofthe IE-1 genewas locatedbySi mapping, using an 850-bp BamHI-Hinfl probe exclusively labeledattheHinfl site. Figure SB shows the maplocation oftheprobe. The5'endofthe IE-1 genewaslocated341bp upstream from the Hinfl site. A HincII site was mapped approximately20bpdownstreamfromthetranscriptionstart site. The CAT gene was cloned into this HincIl site after TaqIdigestion of pCAT, followed by Klenow repairofthe ends
(Fig.
SB). Theresulting plasmid, pIE-1/CAT,wasused totransfectS.frugiperda cells.AsshowninFig.5,CATwas expressed in uninfected cells, as well as in infected cells,indicating
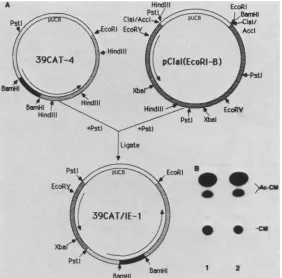
that the gene which trans-activated 39K gene expression was an immediate-early gene by definition be-cause prior viral protein synthesis was not required for expression.Construction ofa plasmid containing 39CAT and IE-1. A plasmid containing 38CAT and the gene required for its expressionwasconstructed. AClaI subclone of pEcoRI-B, cloned in the opposite orientation as pIE-1 relative to multiple cloning site,was digested with PstI. PstI cut in the multiple cloning site and at 97.0 m.u. The resulting 3-kb fragmentwascloned into the PstI site of 39CAT-4 (Fig. 6A). TheCATactivity inducedby the 39CAT-IE-1 plasmid was equivalent to the activity induced by cotransfection of the twoseparateplasmids (Fig. 6B).
DISCUSSION
Weexamined the temporalregulation of anearly gene of the baculovirus AcNPV. A protein having a molecular weight of 39,000 (39K) is the most abundantly expressed early protein and is also synthesized late in infection. To studythetemporal regulationofthisprotein, wecloned the bacterial CAT gene under the control of the 39K promoter andassayedfor theinductionof the CAT genetomonitorthe expressionof the 39K gene.
Becausebaculovirusgenesapparentlyarenotspliced (26), we used
Si
nucleaseto mapthe 5' ends of39Ktranscripts isolatedat 6and 18 h postinfection. With RNA isolated 6 hpostinfection,
amajor
transcriptional startsitewas mapped 124 nucleotides upstream from theinitiatingAUG codon of the 39K gene. Two minor start siteswere detected 3 and 7 nucleotides upstream from the major site. With RNA iso-lated 18 hpostinfection,amajorlatetranscriptionalstartsite was mapped 23 nucleotides upstream from the majorearly site. The sequence of the 39K promoter indicates that the early and latetranscripts should codefor thesameprotein,
because therearenoinitiatingAUG codons located between thetwosites. Expressionof the CAT gene under the control of both promoters indicates that early and late transcripts direct thesynthesis of 39K. The use of dual promoters for earlyand late genes has beendescribedpreviouslyfor genes of vaccinia virus (10), and simian virus 40 (43), and for a J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.120.509.68.346.2],EcoRIl
[image:7.612.164.446.68.346.2]Pstl
FIG. 6. Construction ofasingle clone containing 39CAT and the generequired for itsexpression (A) and 39CATexpressionintransfected cells(B). (A)Aplasmidcontaining theClaIfragment ofEcoRIfragment B(oppositeorientation of theplasmidmentionedabove)wasdigested withPstI,which cutinthemultiple cloning site and in the insertion. Theresulting3.0-kbfragmentwascloned into thePstIsite of39CAT-4. (B) Results ofaCAT assay done with cells cotransfected with 39CAT-4 and
pCIaI(EcoRI-B)
(lane 1)or cells transfected with plasmid 39CAT-4/IE-1 alone (lane 2).CM, Input unacetylated chloramphenicol; Ac-Cm, acetylatedchloramphenicol products.developmentally regulatedgeneofDrosophilamelanogaster
(4).
The 5' endofanotherminor early transcriptwasdetected approximately 400 bp upstream from the 39K gene. This transcriptisprobably notinvolved in39Kgeneexpression, because therearenumerousAUG codonsupstreamfromthe 39K gene. Overlapping unspliced transcripts have been describedpreviouslyinseveral otherregions ofthe AcNPV genome (15, 27), as well as in vaccinia virus (3, 29) and herpes simplex virus (1, 42). Friesen and Miller (15) de-scribed a 3' AcNPV coterminal nest of overlapping tran-scripts in which the smaller RNAs were synthesized early andthelargerRNAs were synthesized later. These authors proposedamodelin whichupstream promoters are sequen-tially activated, accompanied by deactivation of downstream promoters.
We transfected S.
frugiperda
cells with seven plasmids which had the CATgene inserted at various positions in the 39Kgene 5' nontranslated leader andcodingsequences. To confirm thattheassignment oftheinitiating AUG codonwas correct, we testedin-phaseandout-of-phase fusion proteins. CATactivity
wasdetected inAcNPV-infected cellsbut not inuninfected cells, indicating thatthe 39Kgene requires a viralfunction and is therefore not animmediate-earlygene. Becauseexpressionof the 39K gene can bedetected as early as 3 to 6hpostinfection and afterDNA synthesis (37), the 39K geneshould be classified as adelayed-early/late gene.The genewhich trans-activates 39K gene expression was functionally mapped by cotransfecting cells with 39CAT-4 andfragments of viral DNA,followed by anassayfor CAT activity. The gene required for 39K gene expression was
designated IE-1 (for immediate-early gene 1). IE-1 was demonstratedtobe an immediate-earlygene by cloningthe CATgene under thecontrolof the IE-1promoter. IE-1-CAT was efficiently expressed in uninfected cells as well as in infected cells. IE-1 didnot stimulatepAC3906, theplasmid in which the earlypromoter wasdeletedand the CATgene wasapparentlyunder thecontrol of the latepromoter.This result indicates that IE-1 trans-activates the delayed early halfofthe tandem promoter and that thelate half is differ-entially regulated. Itwillbeof interesttodetermine whether IE-1trans-activates alldelayed-earlygenes orwhether other viral factorsare required.
Functionalmapping with CATwasusedpreviouslytomap the trans-activating gene of human T-cell lymphtropic vi-ruses (2, 38, 39). A similar procedure with the thymidine kinase gene was used to map the structural protein which activatesherpessimplexvirus ICP4(8). Traditionally, regu-latory genes have been identified by using temperature-sensitivemutants.Cotransfection with CATplasmids should be useful for functional
mapping
of viral genomes when libraries oftemperature-sensitive mutants are notavailable. The mutation of an early temperature-sensitive mutant of AcNPV, blocked at an early stage of replication, was mapped to 90.7 to 1.9 m.u. (33), the same region of the genome as IE-1,indicating
that the two procedures are complimentary. We intend to use the cotransfection map-ping procedure to identify the gene(s) requiredfor expres-sionofpAc3906.The AcNPV IE-1 gene producthas a trans-acting regula-tory role in 39K gene expression. trans-Acting regulatory genes have been described in adenoviruses (5, 23), simian
on November 10, 2019 by guest
http://jvi.asm.org/
570 GUARINO AND SUMMERS
virus 40 (7, 24), herpes simplex virus (11, 44), and human T-cell lymphotrophic viruses (2, 38, 39). While these genes are functionally similar, their mechanisms of action may differ. Simian virus 40 T-antigen acts by direct binding to specific DNA sequences (7, 24), and the nucleotide sequence of the trans-acting gene of human T-cell lymphotrophic viruses is consistent with a DNA-binding activity (39). However, adenovirus type 5 and herpes simplex virus ap-pear to actindirectly(19, 22) and mayinactivate a host cell factor which inhibits viral transcription (34). It will be interesting to determine whether IE-1 acts at the level of transcription, the nucleotide sequence of IE-1, and the cellular location of the gene product.
ACKNOWLEDGMENTS
We thank Pat Chen and Mark Gonzalez for expert technical assistance. We thank J. Brusca and G. Smith for helpful discussions andcritical review of this manuscript.
This investigation was supported by Public Health Service grant 2RO1 Al 14755 from the National Institutes of Health and Texas Agricultural Experiment Station Project6316.
LITERATURE CITED
1. Anderson, K.P.,R.J. Frink, G.B.Devi,B. H.Gaylord,R. H. Costa,and E. K.Wagner. 1981.Detailed characterization of the mRNAmapping in theHindlll fragmentKregion of the herpes simplex virus type1genome. J. Virol. 37:1011-1027.
2. Arya,S. K., C. Guo, S. F. Josephs,and F. Wong-Staal. 1985. Trans-activator gene of human T-lymphotrophic virus typeIII (HTLV-III). Science229:69-73.
3. Bajszar,G.,R.Wittek, J.P.Weir,and B. Moss.1983. Vaccinia virus thymidine kinase and neighboring genes: mRNAs and polypeptides of wild-type virus and putativenonsense mutants. J. Virol. 45:62-72.
4. Benyajati, C., N. Spoerel, H. Haymerle, and M. Ashburner. 1983. The messenger RNA for alcohol dehydrogenase in Drosophila melanogaster differs in its5'end indifferent stages ofdevelopment. Cell 33:125-133.
5. Berk, A. J., F. Lee, T. Harrison, J. Williams, and P. Sharp. 1979.Pre-early adenovirus5geneproduct regulatessynthesis of early viral mRNA's. Cell 17:935-944.
6. Berk,A.J.,and P. A.Sharp. 1977.Sizing and mapping of early adenovirus mRNAs by gelelectrophoresis of S1 endonuclease-digestedhybrids. Cell 12:721-730.
7. Brady, J., J. Bolen,M.Radonovich,N.Salzman,andG.Koury. 1985. Stimulation of SV40 late geneexpression by SV40tumor antigen.Proc. Natl.Acad. Sci. USA 81:2040-2044.
8. Campbell, M. E. M., J. W. Palfreyman, and C. M. Preston. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimu-lation of immediateearly transcription. J. Mol. Biol. 180:1-19. 9. Carstens,E.B.,S. T. Tjia,and W. Doerfler. 1979. Infection of
Spodopterafrugiperda cells with Autographa californica
nu-clearpolyhedrosis virus. Virology 99:386-398.
10. Cochran, M. A., C. Puckett, and M. Moss. 1985. In vitro mutagenesis of the promoter region fora vaccinia virus gene: evidence for tandemearly and lateregulatory signals.J. Virol. 54:30-37.
11. Dixon,R. A.F.,and P.Schaffer. 1980. Finestructuremapping and functionalanalysis oftsmutantsinthegenesencodingthe herpes simplex virus type1 immediateearly protein VP175.J. Virol. 36:189-203.
12. Dobos, P.,and M. A.Cochran. 1980. Proteinsynthesis in cells infectedby Autographacalifornica nuclear polyhedrosis virus: the effect ofcytosine arabinoside. Virology 103:446-464. 13. Erlandson,M. A., and E. Carstens. 1983.Mapping early
tran-scription products of Autographa californica nuclear poly-hedrosis virus. Virology 126:398-402.
14. Esche, H.,H.Lubbert,B.Siegman,and W. Doerfler. 1982.The translation map of the Autographa californica nuclear polyhedrosis virus (AcNPV) genome. EMBO J. 1:1629-1633.
15. Friesen, P. D.,and L. K. Miller. 1985. Temporal regulationof baculovirus RNA: overlapping early and late transcripts. J. Virol. 54:392-400.
16. Gordon, J. D.,and E. B.Carstens. 1984.Phenotypic character-ization andphysical mapping ofatemperaturesensitivemutant ofAutographa californica nuclear polyhedrosis virus defective inDNAsynthesis. Virology 138:69-81.
17. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinantgenomes whichexpresschloramphenicol acetyl-transferase in mammalian cells. Mol. Cell. Biol. 2:1044-1051. 18. Graham,F. L., and A.vander Eb. 1973. Anewtechnique for
theassayofinfectivity ofhumanadenovirus5 DNA.Virology 52:456-460.
19. Green, M.J., R. Triesman, and T. Maniatis. 1983. Transcrip-tional activation of cloned human ,-genes by viral immediate-earlygeneproducts.Cell35:137-148.
20. Hink, W. F. 1970. Estblished insect cell line for the cabbage looper, Trichoplusiani. Nature(London)226:466-467. 21. Holmes,D. S.,and M. Quigley. 1981. Arapid methodfor the
preparationof bacterialplasmids.Anal.Biochem. 114:193-198. 22. Imperiale, M. J., L. T. Feldman, and J. R. Nevins. 1983. Activation ofgeneexpression by adenovirus and herpesvirus regulatorygenesactingintransandby acis-acting adenovirus enhancer element. Cell35:127-136.
23. Jones, N., and T. Shenk. 1979. An adenovirus 5 early gene product function which regulates expression of other early genes. Proc.Natl. Acad. Sci. USA76:3665-3669.
24. Keller, J. M.,andJ. C. Alwine. 1984. Activationofthe SV40 latepromoter:directeffects ofT-antigenin the absenceof viral DNAreplication. Cell36:381-389.
25. Kelley, D. C., and T. Lescott. 1980. Baculovirus replication: protein synthesis in Spodoptera frugiperda cells infected with Trichoplusia ni nuclear polyhedrosis virus. Microbiologica 4:35-57.
26. Lubbert, H., and W.Doerfier. 1984. Mapping of early and late transcripts encoded by the Autographa californica nuclear polyhedrosis virus genome: is viral RNA spliced? J. Virol. 50:497-506.
27. Lubbert, H., and W.Doerfier. 1984. Transcription of overlap-pingsetsofRNAsfrom thegenomeofAutographacalifornica nuclearpolyhedrosis virus:anovel methodformappingRNAs. J.Virol. 52:255-265.
28. Mackett, M.,G. L.Smith,and B. Moss. 1984.Generalmethod forproductionand selection ofinfectious vaccinia virus
recom-binants. J. Virol. 49:857-864.
29. Mahr, A.,and B. E. Roberts. 1984.Arrangementof late RNAs transcribed from a 7.1-kilobase EcoRI vaccinia virus DNA fragment. J. Virol.50:510-520.
30. Maniatis, T.,E. F. Fritsch, andJ. Sambrook. 1982. Molecular cloning. Cold SpringHarborLaboratory,ColdSpringHarbor, N.Y.
31. Maruniak, J. E., and M. D. Summers. 1981. Autographa californica nuclear polyhedrosis virus phosphoproteins and synthesisof intracellularproteinsafter virus infection.Virology 109:25-34.
32. Messing, J. 1983. New M13 vectors for cloning. Methods Enzymol. 101:20-78.
33. Miller,L.K.,R. E.Trimarchi,D.Browne,andG. D.Pennock. 1983. A temperature sensitive mutant of the baculovirus Autographa californica nuclearpolyhedrosisvirus inan early function required for further gene expression. Virology 126:376-380.
34. Nevins, J. R. 1981. Mechanism of activation of early viral transcription by the adenovirus ElA gene product. Cell 26:213-220.
35. Rohel,D.Z.,and P. Faulkner.1984. Timecourseanalysisand mapping ofAutographa californicanuclearpolyhedrosisvirus transcripts. J.Virol. 50:739-747.
36. Sanger,F.,S.Nicklen,andA.R.Coulson. 1977. DNA sequenc-ing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA74:5463-5468.
37. Smith, G. E., J. M. Vlak, and M. D. Summers. 1982. Invitro translation ofAutographa californica nuclearpolyhedrosis vi-J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
rusearly and late mRNAs. J. Virol. 44:199-208.
38. Sodrowski, J., R. Patarca, C. Rosen, F. Wong-Staal, W.
Haseltine. 1985. Location of the trans-activating region on the genomeof human T-celllymphotrophic virustype Ill. Science 229:74-77.
39. Sodrowski, J., C. Rosen, W. C. Goh, andW.Haseltine. 1985. A transcriptional activator protein encoded by the x-lorregionof
the human T-cell lymphotrophic virus. Science
228:1430-1434.
40. Vlak, J. M., and G. E. Smith. 1982. Orientation of thegenome
ofAutographa californica nuclear polyhedrosis virus: a
pro-posal. J. Virol. 41:1118-1121.
41. VIak, J. M., G. E. Smith, and M. D. Summers. 1981. Hybrid-ization selection and in vitro translation of Autographa californica nuclear polyhedrosis virus mRNA. J. Virol. 40:762-771.
42. Wagner, E. K. 1983. TranscriptionalpatternsinHSVinfections.
Adv. Viral Oncol. 3:239-270.
43. Wasylyk, B., C. Wasylyk, H. Matthes, M. Winterith, and P. Chambon. 1983. Transcription from the SV40 early-early and late-earlyoverlapping promotersin the absence of DNA
repli-cation. EMBO J. 2:1605-1611.
44. Watson, R. J., and J. B.Clements.1982. Aherpes simplex virus
type I function required for early and late gene expression.
Nature(London) 285:329-330.
45. Weaver, R. F., and C. Weissman. 1979. Mapping of RNA bya
modification of theBerk-Sharp procedure: the 5' termini of 15S 3-globin mRNAprecursorandmature10S fI-globin mRNA have identical mapcoordinates. Nucleic Acids Res.7:1175-1193.
46. Weiss,S. A.,G. C.Smith,S. S.Kalter, and J. L. Vaughn.1981. Improved method for the production of insect cell cultures in large volume. In Vitro 17:495-502.
47. Wood, H.A.1980. Aiutographa californica nuclear polyhedrosis virus-induced proteins intissueculture. Virology 102:21-27.