Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Full-Length GB Virus C (Hepatitis G Virus) RNA Transcripts
Are Infectious in Primary CD4-Positive T Cells
JINHUA XIANG,1SABINA WU¨ NSCHMANN,1WARREN SCHMIDT,1JIANQIANG SHAO,2
ANDJACK T. STAPLETON1*
Departments of Internal Medicine and Research1and the University of Iowa Central Microscopy Research Facility,2
Iowa City Veterans Administration Medical Center and The University of Iowa College of Medicine, Iowa City, Iowa 52242
Received 24 February 2000/Accepted 21 June 2000
GB virus C (GBV-C or hepatitis G virus) is a recently described flavivirus which frequently leads to chronic viremia in humans. Although GBV-C is associated with acute posttransfusion hepatitis, it is not clear if the virus is pathogenic for humans. We constructed a full-length cDNA from the plasma of a person with chronic GBV-C viremia. Peripheral blood mononuclear cells (PBMCs) transfected with full-length RNA transcripts from this GBV-C clone resulted in viral replication. This was demonstrated by serial passage of virus from cell culture supernatants, detection of increasing concentrations of positive- and negative-sense GBV-C RNA over time, and the detection of the GBV-C E2 antigen by confocal microscopy. In addition, two types of GBV-C particles were identified in cell lysates; these particles had buoyant densities of 1.06 and 1.12 to 1.17 g/ml in sucrose gradients. PBMCs sorted for expression of CD4 contained 100-fold-more GBV-C RNA than CD4-negative cells. Taken together, these data demonstrate that RNA transcripts from GBV-C full-length cDNA are infectious in primary CD4-positive T cells. In contrast, RNA transcripts from an infectious hepatitis C virus clone did not replicate in the same cell culture system. Infectious RNA transcripts from GBV-C cDNA should prove useful for studying viral replication and may allow identification of differences between GBV-C and hepatitis C virus cultivation in vitro.
GB virus type C (GBV-C, also called hepatitis G virus) is a recently described virus whose genomic organization and nu-cleotide sequence place it in theFlaviviridae. It is the most closely related human virus to hepatitis C virus (HCV) (25, 27, 41). Although GBV-C was originally associated with posttrans-fusion hepatitis in humans (27), subsequent epidemiological studies indicated that it does not cause acute or chronic hep-atitis (4, 5). In addition, experimental GBV-C infection of chimpanzees was not associated with acute hepatitis (8). None-theless, persistent GBV-C viremia (as detected by reverse tran-scriptase PCR [RT-PCR]) is common, with 0.9 to 3% of healthy U.S. blood donors and approximately 20 to 30% of patients with HCV infection persistently infected with GBV-C (11, 15, 17, 41, 42, 45). Following infection, about 80% of people clear their viremia, concomitantly developing antibod-ies to the GBV-C E2 protein (15, 17, 47). Thus, it is estimated that approximately 20% of infected people remain viremic for long periods of time. GBV-C appears to be transmitted pri-marily by parenteral exposure (41), although there are data suggesting that sexual and/or household transmission of GBV-C infection may occur (2, 12, 31, 46, 49).
Like other members of theFlaviviridae, GBV-C is a positive-strand RNA virus which encodes a single long open reading frame (ORF) (25). The GBV-C polyprotein is predicted to be cleaved into two envelope proteins (E1 and E2), an RNA helicase, a trypsin-like serine protease, and an RNA-depen-dent RNA polymerase. A major difference between GBV-C and HCV is in the amino terminus of the polyprotein. In many of the reported viral sequences, this region is truncated and no core (or nucleocapsid) coding region is present (41, 51). In
vitro translation experiments suggest that the AUG immedi-ately upstream of the coding sequence for the putative E1 protein is preferentially used to initiate translation, although there may be as many as four AUGs in frame with the polypro-tein coding sequence upstream of this AUG (40). In addition, the mutation frequency in codon positions 1 and 2 of the region upstream of this AUG suggests that it is a noncoding region (32). These observations have led to speculation that GBV-C may not have a core protein or nucleocapsid (13, 40). However, we and others have shown that the sedimentation profiles of GBV-C particles are consistent with the presence of a nucleocapsid (30, 52), and electron microscopy of plasma-derived GBV-C demonstrated enveloped particles with a nu-cleocapsid structure (51). Although the amino acid composi-tion of the nucleocapsid remains undefined, some infected individuals have antibodies to a peptide representing amino acids upstream of the predicted E1 protein in frame with the polyprotein (52). Thus, this region may encode the nucleocap-sid.
Full-length cDNAs or RNA transcripts of several RNA vi-ruses including hepatitis A virus, GBV-B, and HCV are infec-tious in cell culture or animal inoculation studies (6, 7, 9, 14, 18, 22, 53, 54). These infectious clones are useful for genetic studies and allow a precise method for evaluating the evolution of viruses that normally exist in molecular quasispecies. The site of GBV-C replication has not been clearly identified, but it appears that replication in the hepatocyte, if it occurs, is not the primary source of virus in infected individuals (24, 33, 37). Recently, there were reports that human peripheral blood mononuclear cells (PBMCs) and interferon-resistant Daudi cells are permissive for GBV-C replication (16, 38). In addi-tion, transient replication of GBV-C in MT-2 cells (a human T-cell line) (19) and PH5CH cells (a human hepatocyte line immortalized with simian virus 40 large T antigen) (37) was described. In this study, we describe the construction of a * Corresponding author. Mailing address: Department of Internal
Medicine, SW 54, GH UIHC, 200 Hawkins Dr., The University of Iowa, Iowa City, IA 52242. Phone: (319) 3168. Fax: (319) 356-4600. E-mail: [email protected].
9125
on November 9, 2019 by guest
http://jvi.asm.org/
synthesized in vitro were transfected into PBMCs and a T-cell line and proved to be infectious in PBMCs. By comparison, RNA transcripts of a full-length cDNA infectious HCV clone (22) were not infectious under identical conditions.
MATERIALS AND METHODS
Isolation and preparation of cells.PBMCs from healthy blood donors (HCV RNA and antibody negative, GBV-C RNA negative, and hepatitis B virus sur-face antigen negative) were isolated from heparinized blood by centrifugation on Ficoll-Hypaque gradients, washed twice with phosphate-buffered saline (PBS), and suspended in RPMI 1640 medium (Sigma, St. Louis, Mo.) supplemented with 10% fetal calf serum (FCS) and antibiotics as previously described (10). PBMC cultures (2⫻106cells/ml) were maintained in RPMI 1640 containing
phytohemagglutinin (PHA; 10g/ml; Difco, Detroit, Mich.) and 5% interleu-kin-2 (IL-2) (Cellular Products Inc., Buffalo, N.Y.) at 36°C in 5% CO2. PBMC
cultures used for transfection were maintained in the same medium, except that
Escherichia colilipopolysaccharide (10g/ml; Sigma) was added to the medium for 48 h prior to transfection. Following transfection, cells were maintained in RPMI 1640 supplemented with PHA (5g/ml) and IL-2 only. MOLT-4 cells were maintained in RPMI 1640 containing 10% FCS and antibiotics as previ-ously described (50a). All blood donors volunteered to participate in these studies, and informed consent was obtained. These studies were approved by the University of Iowa Institution Review Board.
GBV-C RNA preparation and RT-PCR.A previously described GBV-C RNA-positive patient with a diagnosis of chronic liver disease was selected for this study (52). This patient did not have detectable HCV antibodies (EIA 2.0; Abbott Laboratories, North Chicago, Ill.) or RNA. RNA was prepared from plasma using a previously described guanidinium isothiocyanate RNA extraction method (36). GBV-C RNA was detected using nested oligonucleotide primers from the 5⬘nontranslated region (NTR) as previously described (52). Primers used for producing the full-length clone are described below. All RT-PCRs utilized Moloney murine leukemia virus (MMLV) RT (40 U) as previously described (43); the addition of MMLV RT was followed by 35 cycles of PCR (94°C for 30 s, 55°C for 30 s, and 72°C for 45 s). Three microliters of the first-round PCR mixture served as the template for 35 cycles of second-round PCR using nested primers and the same time and temperature settings (36). To ensure that our RT-PCR methods were specific for GBV-C and did not amplify bovine diarrhea virus (BVDV) potentially present in FCS, we utilized BVDV primers which were previously shown to amplify most strains of BVDV (34). RT-PCR was performed with the sense (5⬘-CATGCCCATAGTAGGAC-3⬘) and antisense (5⬘-CCATGTGCCATGTACAG-3⬘) primers (34). BVDV and BVDV-negative cells (for a BVDV-negative control) were kindly provided by Julia Ridpath, USDA Agricultural Research Laboratory, Ames, Iowa.
Cloning and sequencing of PCR products.PCR products were separated by agarose gel electrophoresis, visualized by ethidium bromide staining, excised, and purified using a DNA purification system kit (Promega Co., Madison, Wis.). Amplicons were ligated into pCR 2.1 (Original TA cloning kit; Invitrogen, Carlsbad, Calif.), and plasmid DNA was sequenced in both directions using primers complementary to the T7 polymerase or the M13 universal primer sequences present in the vector as previously described (43). Automated fluo-rescent-dye terminator cycle sequencing was performed by the University of Iowa DNA Core Facility (automated DNA sequencer 373A; Applied Biosys-tems, Foster City, Calif.).
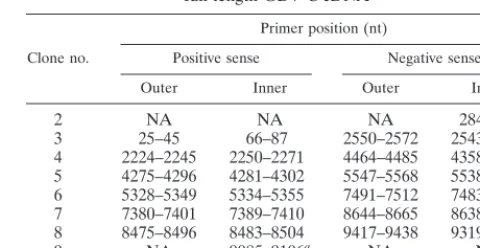
Construction of full-length GBV-C cDNA.Based on conserved sequences throughout the GBV-C genome, a series of primers which contained suitable restriction sites in their overlapping sequences were designed. Primer sets that generated products were used to prepare the full-length clone. Table 1 and Fig. 1 demonstrate the six primer sets and fragments generated in this study. The rapid amplification of cDNA ends (RACE) method was used to prepare the 5⬘ -and 3⬘-terminal RNA (GIBCO BRL, Rockville, Md.). The eight DNA fragments were ligated into a full-length clone by using the restriction enzymes in either the overlapping sequences or in the vector DNA.
RNA transcription and transfection.Ten micrograms of GBV-C full-length DNA in pCR 2.1 was digested into linear DNA bySpeI and transcribed using T7 RNA polymerase (Promega) for 1 h at 37°C. An infectious HCV clone (22) was kindly provided by Charles Rice, Washington University, St. Louis, Mo. (Gen-Bank accession no. AF009606). This clone was digested withSpeI, and RNA transcription was carried out in the same fashion. To eliminate plasmid DNA sequences, RQ1 RNase-free DNase (1 U/g) digestion of template DNA was completed for 15 min at 37°C. RNA transcripts were purified by chloroform extraction and ethanol precipitation. RNA from transcription reactions was denatured with formamide and formaldehyde and analyzed on a 1% agarose-formaldehyde gel. Transcribed RNA in DEAE-dextran (1 mg/ml in Hanks bal-anced salt solution) was added to washed PHA-, IL-2-, and lipopolysaccharide-stimulated PBMCs or MOLT-4 cells (106), and the cells were incubated for 30
min at 36°C in 5% CO2. RPMI 1640 supplemented with PHA, IL-2, and 10%
FCS was added, and the cells were incubated at 36°C for 6 h with gentle rocking. After 6 h the medium was removed and the cells were washed twice and incu-bated in RPMI 1640 (10% FCS) at 36°C in 5% CO2. Fresh PHA- and
IL-2-stimulated donor cells (4⫻106) were added each week to the cultured cells.
After 4 weeks, cell culture supernatant from the transfected cells was used to inoculate fresh PBMCs (2⫻106/ml) for at least four consecutive passages.
Negative-strand GBV-C RT-PCR.For detection of GBV-C antisense RNA, cDNA synthesis was performed with an oligonucleotide primer containing a sequence unrelated to GBV-C (5⬘-TCATGGTGGCGAATAAAAGCCCCAGAA ACCGACGCC-3⬘; boldface letters indicate non-GBV-C sequences), as de-scribed by others (24). cDNA synthesis was stopped by heating at 99°C for 1 h, and samples were treated with 50 g of RNase A/ml at 37°C for 30 min. Subsequent amplification of plus-sense RNA byTaqpolymerase used only the tag sequence (5⬘-TCATGGTGGCGAATAA-3⬘) for both the first round of ampli-fication and the nested PCR.
CD4 staining and flow cytometry.Five days postinfection with passage 5 culture supernatant, PBMCs (2⫻107) were pelleted and resuspended in PBS
containing 10% normal goat serum for 30 min at 4°C prior to incubation with a mouse anti-CD4 antibody or a murine isotype control antibody (10g/ml; Mo-lecular Probes, Eugene, Oreg.) for 45 min at 4°C. Anti-CD4 binding was detected using Texas red-conjugated goat anti-mouse antibody (10g/ml; Molecular Probes) for 45 min at 4°C. Between each step, cells were washed two times with PBS. CD4-positive and CD4-negative cells were sorted by flow cytometry (FACScan; Becton Dickinson, San Jose, Calif.), and the two populations were collected for analysis.
Immunofluorescence.Indirect immunofluorescence was performed using a mouse monoclonal antibody against GBV-C E2 protein (Biodesign, Saco, Maine). Two and five days postinfection with passage 5 virus, PBMCs were fixed with 3.7% formaldehyde for 15 min (29) and then permeabilized in acetone at
⫺20°C for 5 min (3). Following blocking with 10% normal goat sera, the cells were incubated for 1 h at room temperature with the anti-GBV-C E2 antibody (10g/ml). After being washed, cells were incubated for 1 h with fluorescein-Texas red-labeled goat anti-mouse immunoglobulin G (5 g/ml; Molecular Probes). Images were recorded using confocal microscopy (519 nm; Zeiss, Jena, Germany) as previously described (50). The⫻100 objective was used to produce these images.
Equilibrium centrifugation in sucrose. Five hundred microliters of either infected-cell culture supernatant fluid or the infected-cell lysates was layered onto 10 ml of a 20 to 60% sucrose gradient, and centrifugation was performed using a Beckman SW41 rotor at 156,000⫻g for 16 h at 4°C as previously described (51). Fourteen fractions (750l each) were collected, RNA was ex-tracted as described above, and GBV-C RNA was detected by RT-PCR.
Nucleotide sequence accession number.The nucleotide sequence determined in this study has been assigned GenBank accession no. AF121950.
RESULTS
To construct full-length GBV-C cDNA, nested RT-PCR was performed on plasma RNA obtained from a GBV-C-infected individual using a variety of oligonucleotide sets spanning the entire genome. Six primer sets, which generated overlapping products containing restriction sites useful for ligation, were identified (Table 1). These six fragments started at nucleotide 25 and ended at nucleotide 9340. To identify the 5⬘and 3⬘ends of the genome, the 5⬘ and 3⬘ RACE methods were used. Primers used for these reactions were located from nucleotide (nt) 284 to 305 (antisense) for 5⬘RACE and from nt 9085 to 9106 (positive sense) for the 3⬘ RACE. Each of these eight
full-length GBV-C cDNA
Clone no.
Primer position (nt)
Positive sense Negative sense Outer Inner Outer Inner
2 NA NA NA 284–305a
3 25–45 66–87 2550–2572 2543–2564
4 2224–2245 2250–2271 4464–4485 4358–4379 5 4275–4296 4281–4302 5547–5568 5538–5559 6 5328–5349 5334–5355 7491–7512 7483–7504 7 7380–7401 7389–7410 8644–8665 8638–8659 8 8475–8496 8483–8504 9417–9438 9319–9340
9 NA 9085–9106a NA NA
aThe 5⬘and 3⬘ends were generated using the RACE method. The numbers
represent the nucleotide sequence numbers based on this isolate (GenBank accession no. AF121950).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.312.552.90.214.2]PCR amplification products was cloned into the pCR 2.1 vec-tor, and the nucleotide sequence was determined (Fig. 1A). Following ligation of the eight fragments shown in Fig. 1A, a clone containing the full-length GBV-C sequence of our GBV-C isolate was obtained. All cloning sites were again se-quenced to exclude the possibility of cloning artifacts. Restric-tion digests of this full-length cDNA in the pCR 2.1 vector were consistent with the sequence data (data not shown).
GBV-C sequence analysis and comparison with GBV-B and HCV. The GBV-C sequence we obtained (AF121950) con-tained an ORF beginning at nt 351 and extending to nt 9080. This ORF is predicted to encode a 2,910-amino-acid polypro-tein with a molecular mass of 314,548 Da. Based on the pre-dicted site of translation initiation at the start of the E1 protein (40), the polyprotein would be 2,677 amino acids (molecular mass, 288,940 Da) if none of the putative “core” protein up-stream of E1 is translated. The complete GBV-C sequence of this isolate was compared with those of 19 full-length human isolates obtained by searching GenBank for complete GBV-C sequences (GenBank accession no. AB320090, AB320091, AB320092, AB003293, AB008335, D90600, D90601, D87255, D87272, D87708, D87709, D87710, D87711, D87712, D87713, D87714, D87715, U44402, and U63715). Nucleotide and pre-dicted amino acid sequences were aligned, and the evolution-ary distances between sequences were determined using the Jukes-Canter method (DNAMAN software; Lynnon BioSoft Inc., Quebec, Canada). Twelve of these sequences contained the complete 5⬘NTR and 3⬘NTR. A comparison of these 12 isolates and our virus revealed 92.79% homology at the nucle-otide level.
A consensus predicted amino acid sequence was generated for these 20 GBV-C polyproteins (initiating translation at nt
554 of AF121950). Our full-length clone and the consensus sequence differed at 115 amino acids (4.3%). Based on the predicted protein-encoding regions of the genome, most of the differences between our sequence and the consensus sequence occurred in the NS5b (polymerase)-coding region (8%), fol-lowed by the E1 region (7%), E2 (3.5%), NS4 (3.2%), NS2 (1.7%), NS3 (1.5%), and NS5a (1.2%). The high rate of dif-ferences in the predicted polymerase region was surprising; however, 40 of the 53 amino acids differing from those of the consensus sequence occurred in the carboxy-terminal 45 amino acids. If only the amino-terminal 518 amino acids (of 563) had been evaluated, the AF121950 sequence would have only var-ied from the consensus by 2.5%.
The complete 3⬘NTRs from 11 of the 21 full-length isolates studied were available. There was 96.46% homology among these sequences, with AF121950 containing no unique nucle-otide sequences. Of the 312 nt present in the 3⬘NTR, there was only a single nucleotide difference between AF121950 and D90600. In both HCV and GBV-B, additional 3⬘-NTR se-quences were found at the 3⬘terminus subsequent to the orig-inal publication of the sequence (7, 23). Because of this, we were concerned that there may be additional sequences on the 3⬘ end of GBV-C. Consequently, we performed 3⬘ RACE experiments eight times and also carried out RT-PCR across 5⬘- to 3⬘-end-ligated viral RNA four times in an attempt to identify additional sequences downstream of the previously published 3⬘terminus. However, no additional nucleotide se-quences were identified.
Not surprisingly, a comparison of the 3⬘ NTR of GBV-C with the 3⬘NTRs of an infectious GBV-B clone and the infec-tious HCV clone we used as a control demonstrated only 45.69% homology among the three sequences. GBV-C was
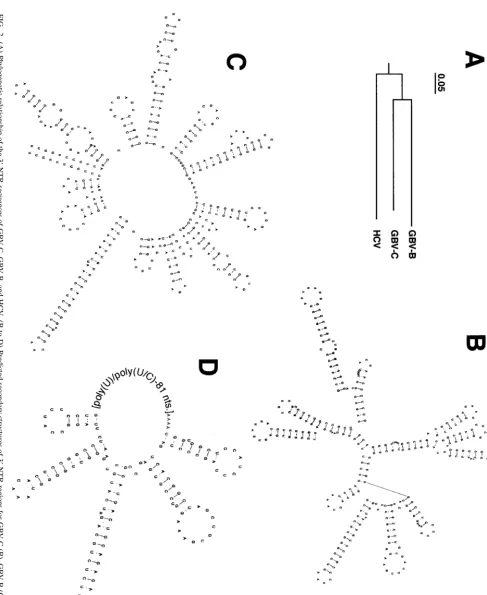
FIG. 1. Cloning strategy for GBV-C full-length clone. (A) Schematic presentation. The full-length GBV-C cDNA sequence is at the top. Each box beneath the full-length sequence represents a cDNA amplified by RT-PCR used to make the full-length clone. The 5⬘and 3⬘ends were generated using the RACE methods. The specific primer sets used for RT-PCR are shown in Table 1. (B) RNA transcripts from GBV-C and HCV full-length cDNA. The RNA size marker is depicted.
on November 9, 2019 by guest
http://jvi.asm.org/
hepatitis agents, GBV-C was more closely related to HCV than was GBV-B (Fig. 2A). Although there was little sequence homology in this region, analysis of the predicted secondary structures demonstrated several similarities (Fig. 2B to D), particularly at the extreme 3⬘ end (RNAdraw, an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows) (28). Although GBV-C does not have a polypyrimidine tract, the 3⬘end has three stem-loop structures, closely resembling the HCV and GBV-B 3⬘ ends (Fig. 2B to D, respectively). In addition, the 5⬘ end of this region bears remarkable structural resemblance to that of GBV-B (Fig. 2B and C). The predicted free energies of the GBV-C, GBV-B, and HCV 3⬘-NTR RNA structures (37°C) were⫺92.98,⫺109.01, and⫺55.08 kcal, respectively (28).
Full-length GBV-C RNA is infectious in cell culture. Full-length GBV-C transcripts were generated using T7 polymer-ase. For comparison, full-length HCV RNA was also tran-scribed from an infectious cDNA clone using T7 polymerase (22). RNA from the transcription reactions was denatured with formamide and analyzed on a 1% agarose-formaldehyde gel (Fig. 1B). The GBV-C transcript was approximately 9.4 kb, whereas the HCV transcript was approximately 9.7 kb. GBV-C and HCV RNA transcripts were transfected into PBMCs and MOLT-4 and HepG2 cell lines. Mock-transfected cells (PBMCs and MOLT-4) were also maintained under identical condi-tions. Following transfection, PBMCs were supplemented with fresh, PHA- and IL2-stimulated PBMCs weekly for 4 weeks. Culture supernatants and cell lysates were evaluated for GBV-C and HCV RNA using 5⬘-NTR primers in RT-PCR experiments described in Materials and Methods. GBV-C RNA was detected in all PBMC lysates and culture supernatants (Table 2). In contrast, HCV RNA was not detected in PBMC cell lysates or culture supernatants after 3 weeks in culture (Table 2). GBV-C and HCV RNA was detected in MOLT-4 cell lysates for 3 or fewer weeks and was detected in the culture supernatants for only the week of transfection (Table 2). GBV-C and HCV RNA was not detected in HepG2 cells within 1 week of transfection (data not shown) or in any of the mock-transfected control cells (Table 2). Cell lysates and cell culture supernatants from the GBV-C-transfected cells were used to infect fresh PHA- and IL2-stimulated PBMC cultures for four passages, and persistent GBV-C infection was dem-onstrated (Table 2). All experiments were performed in dupli-cate or triplidupli-cate, and transfections were repeated twice.
To ensure that the GBV-C RNA detected in PBMC cell lysates and culture supernatants did not represent amplifica-tion of residual plasmid DNA, the cell lysate and supernatant fluids from which GBV-C RNA was detected were amplified in PCR mixtures not containing MMLV RT. These reactions did not produce PCR products (data not shown). In addition, the relative concentrations of GBV-C positive- and negative-sense RNA in culture supernatants and cell lysates were determined by terminal dilution. Figure 3 demonstrates that a low concen-tration of negative-strand RNA was present in culture super-natants following 7 days of infection (passage 5 virus), con-comitantly with an increase in positive-strand RNA. The finding of negative-strand GBV-C RNA in the culture super-natant was not expected. However, negative-strand HCV RNA that is resistant to nucleases has been found in the plasma of infected people, and this RNA is associated with both mem-branes and the HCV glycoprotein E1 (39). Thus, the negative-strand GBV-C RNA in the culture supernatants may represent membrane-associated particles containing minus-strand RNA. Negative-strand RNA was detected 14 days postinfection in the cell lysates and increased on days 21 and 28. Cell culture
not shown).
To ensure that the negative-strand RT-PCR methodology was strand specific, sense and antisense control RNAs were generated by utilizing clones produced by the ligation of am-plicons shown in Fig. 1 into the pCR 2.1 vector (amam-plicons 2 and 3; nt 1 to 2564; specific primers are shown in Table 1) and by ligation in the reverse orientation (nt 2564 to 1). RNA was transcribed from each clone using T7 polymerase to generate a positive- or negative-sense RNA control. Both transcription products were treated with DNase and used as templates in RT-PCRs. Figure 4 demonstrates that the negative-sense-spe-cific primer only generated products with the negative-strand RNA template and was dependent on the presence of RT. Because the standard RT-PCR is not specific for the positive or negative strand (24), this reaction generated products from both the positive-strand and negative-strand templates (Fig. 4). To determine the immunophenotype of the PBMCs that supported GBV-C replication, cells were infected with passage 4 supernatant and 5 days later CD4-positive and CD4-negative cells were sorted by flow cytometry. RNA was extracted from 1.5⫻105CD4-positive cells and from 2⫻105CD4-negative
cells, and GBV-C RNA was evaluated by RT-PCR in each cell population. The relative concentrations of both positive- and negative-strand viral RNA were 100-fold higher in CD4-posi-tive cells than in CD4-negaCD4-posi-tive cells, indicating that 99% of viral replication in PBMCs occurred in the CD4⫹subset (Fig.
5). This experiment was repeated, and cells were sorted for the CD4⫹CD3⫹phenotype to ensure that the CD4⫹cells were of
T-cell origin. In both experiments, GBV-C negative-strand RNA synthesis was only seen in the CD4⫹CD3⫹cells and the
concentration of the positive-strand RNA was 100-fold higher than in the CD4⫺cell population (data not shown).
To further confirm that GBV-C RNA detection in cells represented viral replication, the fifth passage of cells with GBV-C infection was evaluated by indirect immunofluores-cence (IFA). Two and five days postinfection, PBMCs were fixed and GBV-C E2 expression was assessed using a commer-cial anti-E2 monoclonal antibody. Figure 6 demonstrates E2 expression in the cytoplasm of cells from passage 4 (A and B) but not in that of mock-infected cells (D). Figure 6C shows infected cells evaluated as for Fig. 6A; however, an isotype control monoclonal antibody was used in place of the GBV-C E2-specific monoclonal antibody. Five days postinfection, 5% of the PBMCs demonstrated GBV-C E2 expression by IFA. Since 24 to 26% of the PBMCs were CD4⫹in the cell sorting
experiments described above, approximately 20% of the CD4⫹
T cells in our cultures were positive for E2 antigen expression. GBV-C particle characterization.In order to determine the biophysical properties of GBV-C particles generated by the infectious clone, concentrated supernatants and cell lysates from infected PBMCs (fourth passage) were characterized by sucrose gradient centrifugation (Fig. 7A and B, respectively). RNA was extracted from each fraction, and GBV-C RNA was detected by RT-PCR. A very-low-density (1.06 g/ml) particle type, similar to GBV-C particles found in plasma (53), was identified in cell lysates (Fig. 7A). This particle type and an intermediate-density particle were identified in supernatant samples (buoyant densities of 1.06 and 1.12 to 1.17 g/ml, re-spectively) (Fig. 7B).
To ensure that the PCR products observed in Fig. 7 repre-sented GBV-C and not BVDV contamination of the FCS, RT-PCR for BVDV and GBV-C was performed on the con-centrated cell culture supernatant. GBV-C RNA was present in these preparations (6⫻105copies/ml), whereas no BVDV
RNA was identified (data not shown). The BVDV RT-PCR
on November 9, 2019 by guest
http://jvi.asm.org/
FIG.
2.
(A)
Phylogenetic
relationship
of
the
3
⬘
-NTR
sequences
of
GBV-C,
GBV-B,
and
HCV.
(B
to
D)
Predicted
secondary
structures
of
3
⬘
-NTR
regions
for
GBV-C
(B),
GBV-B
(C),
and
HCV
(D).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.58.546.86.681.2]was able to amplify BVDV RNA from the positive-control virus RNA preparation (data not shown).
DISCUSSION
Persistent GBV-C infection is remarkably common in hu-mans, yet the virus does not appear to cause chronic hepatitis (4, 5). In fact, no specific disease has been clearly associated with GBV-C, and the original association with posttransfusion hepatitis has not been supported by subsequent human epide-miological studies (4, 5, 48) or experimental chimpanzee inoc-ulation studies (8). Nevertheless, GBV-C is the most closely related human flavivirus to HCV, a major worldwide pathogen (35). In addition, GBV-C and HCV appear to utilize the low-density lipoprotein receptor for viral entry (1). Thus, compar-ison of GBV-C and HCV may provide insight into the reasons why HCV does not appear to replicate as efficiently in cell culture as GBV-C and why GBV-C is cleared more efficiently by the host immune response than HCV (17, 47, 48). Although several infectious HCV clones have been described, all of these rely on inoculation of transcribed RNA into susceptible pri-mate species, and none were shown to be infectious in vitro (7, 18, 22, 53, 54). Thus, these HCV infectious clones have only limited application (6, 18, 22, 53, 54). The construction of an infectious GBV-C cDNA clone that replicates in vitro provides an important tool for studying the replication of human Flavi-viridae.
Several full-length GBV-C sequences have been submitted to GenBank; however, our data are the first to demonstrate that full-length GBV-C RNA transcripts are infectious in PBMC cultures. Our construction was unlike most previous full-length HCV constructions in that we did not attempt to produce a consensus sequence but rather produced the au-thentic GBV-C amino acid sequence obtained from direct am-plification of viral RNA. The sequence of our virus isolate differs from the consensus sequence by 4.3%, mostly in the carboxyl terminus of NS5b and in the E1 and E2 structural proteins. Viral replication was demonstrated by serial passage of culture supernatants, expression of the envelope
glycopro-tein E2, RNA replication (positive- and negative-strand RNA synthesis), and detection of the viral particles by sucrose gra-dient centrifugation. Characterization of PBMC subsets iden-tified the CD4⫹T cells as the cells supporting GBV-C
repli-cation. Although early studies suggested that GBV-C replicates in the liver, most reported studies indicate that GBV-C is not hepatotropic (20, 24). Our inability to demon-strate infection of HepG2 cells is consistent with this, although we were also unable to demonstrate persistent replication in the CD4⫹ T-cell line (MOLT-4). Thus, host cell factors in
primary cells may be necessary for replication. Studies with primary hepatocyte cultures to test this hypothesis are under way. Nevertheless, several studies have found GBV-C replica-tion in PBMCs, and the concentrareplica-tion of virus in plasma rel-ative to liver tissues suggests that the hepatocyte is not a prominent source of virus (21). Taken together, these data suggest that GBV-C may be lymphotropic.
Simons et al. demonstrated that the AUG codon encoding an amino acid at the amino terminus of the putative E1 protein (AUG-554 in our isolate) was capable of initiating translation, whereas the upstream AUGs were not (40). In many isolates, the amino terminus of the predicted GBV-C polyprotein is truncated or absent (25, 27, 32), and the frequency of poly-morphisms in codon positions 1 and 2 in the upstream ORF suggests that the region is not a coding region (32). Thus, it has been suggested that GBV-C may not have a core protein (13). We previously showed that GBV-C particles have densities and sedimentation characteristics in sucrose and cesium chloride gradients similar to those of HCV (52) and subsequently found particles approximately 65 nm in diameter with 50-nm-diame-ter nucleocapsid structures (51). In this study, two GBV-C particle types were identified by sucrose gradient sedimenta-tion; they had densities of 1.07 and 1.18 g/ml, consistent with virions and nucleocapsids respectively (52). Thus our data sup-port previous work identifying a nucleocapsid for GBV-C. The truncation of the polyprotein upstream of the amino acid en-coded by AUG-554 would be abolished if most isolates did not contain a single-nucleotide deletion at position 381. Given the fact that all sequences have been produced thus far by nested and MOLT-4 cells
Medium and cells Virus Result at wk
a:
0 (0) 1 (Ia) 2 (Ib) 3 (Ic) 4 (Id) 5 (II) 6 (III) 7 (IV) 8 (V)
Cell lysate
PBMC GBV-C T ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹
HCV T ⫹ ⫹ ⫹ ⫺ ⫺ ⫺ ⫺ ⫺
Mockb ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
MOLT-4 GBV-C T ⫹ ⫹ ⫹ ⫺ ⫺ ⫺ ⫺ ⫺
HCV T ⫹ ⫹ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
Mock ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
Culture supernatant
PBMC GBV-C ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹
HCV ⫹ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
Mock ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
MOLT-4 GBV-C ⫹ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
HCV ⫹ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
Mock ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
aT, DEAE transfection of cells with full-length RNA transcripts;⫹, viral RNA detected by RT-PCR;⫺, viral RNA not detected by RT-PCR. The passage generation
for PBMCs is in parentheses. Following transfection, fresh PHA- and IL2-stimulated PBMCs were added to the cells weekly for 4 weeks (generations Ia, Ib, Ic, and Id). Following this, cell culture supernatant was used to infect fresh cells (generations II, III, IV, and V). MOLT-4 cells were passaged weekly.
bMock, mock transfected.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.53.551.92.281.2]RT-PCR, this deletion may represent a polymerase artifact. Nevertheless, propagation of GBV-C in culture should allow the production of sufficient virus for ultimate characterization of the protein content of the GBV-C nucleocapsid. With the exception of the 5⬘-NTR region, the remaining GBV-C se-quences are highly conserved among geographically diverse isolates. Although there is less than 50% sequence homology in the 3⬘-NTR region among GBV-C, GBV-B, and HCV, the predicted secondary structures of these viruses bear striking
similarities. GBV-C does not include a polypyrimidine tract but does have three stem-loop structures at the extreme 3⬘end (Fig. 2). This suggests that the polypyrimidine regions of HCV and GBV-B are not necessarily required for replication.
[image:7.612.343.518.72.288.2]In summary, we have constructed the first GBV-C infectious
[image:7.612.67.280.74.388.2]FIG. 3. Detection of GBV-C RNA in cell culture supernatants (A) and cell lysates (B). Results are expressed as the relative GBV-C RNA end point dilution titers per milliliter of initial culture supernatant fluid. GBV-C RNA was detected 1 day following infection and after 7, 14, 21, and 28 days of culture.
FIG. 4. Validation of strand-specific RT-PCR methods. Strand-specific RNA was transcribed from a plasmid carrying either nt 1 to 2564 or nt 2564 to 1 of GBV-C using T7 polymerase as described in Materials and Methods. The neg-ative-strand-specific RT-PCR with the Tag internal primer generated 298-bp products only when the negative-strand RNA template was used, not with the positive-strand template or the water control (wc). Templates were tested neat (n) or were diluted 1:10. The standard RT-PCR is not specific for either strand, and 220-bp products were generated with both positive- and negative-strand templates. These reactions were dependent on the presence of RT. M, DNA marker.
FIG. 5. CD4-positive and CD4-negative cells were sorted and collected by flow cytometry. RNA was extracted from 1.5⫻105CD4⫹or 2⫻105CD4⫺cells,
and the end point dilution titers (serial log10dilution titer shown above the
[image:7.612.311.552.432.660.2]agarose gels) of sense and antisense GBV-C RNA were measured. FITC, fluo-rescein isothiocyanate.
FIG. 6. GBV-C E2 expression in PBMCs. PBMCs were infected with super-natant from the passage 4 cell culture supersuper-natant fluid, and, 2 (A) and 5 days (B) postinfection, the cells were fixed and processed as described in Materials and Methods. GBV-C E2 expression was detected using a murine monoclonal anti-GBV-C E2 antibody as described in Materials and Methods. The same cells (2 days postinfection) did not show specific cytoplasmic fluorescence when eval-uated with an isotypic control antibody (C), nor did mock-infected PBMCs evaluated with the GBV-C anti-E2 antibody as for panel A (D).
on November 9, 2019 by guest
http://jvi.asm.org/
cDNA clone. We utilized an approach to produce the authen-tic GBV-C sequence, not a consensus sequence, and our syn-thetic GBV-C RNA was capable of initiating replication in vitro. Since GBV-C has much less sequence divergence than HCV, this approach may not be as suitable for producing HCV infectious clones; however, one group has reported construc-tion of an infectious “nonconsensus” HCV clone of genotype 1b (6). Our data indicate that construction of a consensus sequence is not a prerequisite for infectivity among the human
Flaviviridae. This system may serve as a model to compare the replication requirements of GBV-C and HCV and may prove useful for identifying blocks in HCV replication and for ad-dressing questions of pathogenesis. Since GBV-C appears to be a common, nonpathogenic, persistent human virus, it could potentially serve as a gene therapy vector for lifelong expres-sion of foreign genes in humans. In addition, recent data indi-cate that GBV-C viremia occurs in 35 to 40% of human im-munodeficiency virus (HIV)-infected individuals (31, 43), and two clinical reports suggest that GBV-C viremia delays clinical disease progression and mortality in HIV-infected people (26, 44). Since we demonstrated that GBV-C replicates in CD4⫹T
cells in vitro, it is tempting to speculate that GBV-C may cause viral interference with HIV, leading to delayed disease pro-gression. Thus, the study of GBV-C–HIV interactions may lead to new approaches to treat or prevent HIV infection.
ACKNOWLEDGMENTS
We thank Donna Klinzman and Donna Brashear for helpful discus-sions, Lee Ann Allen for suggestions on immunofluorescence and confocal microscopy, Douglas LaBrecque and Mary Jean Perino-Phil-lips for assistance with clinical specimens, Charles Rice (Washington University) for kindly providing the full-length infectious HCV clone, Judith Ridpath and Margaret Walker (USDA Agricultural Research Laboratory) for providing BVDV and BVDV-negative cells, and Donald Smith for helpful discussions regarding sequence and RNA structure analyses. The University of Iowa Flow Cytometry Core Pro-gram was utilized for these studies.
This work was supported in part by a Merit Review and a Career Development Enhancement Award from the Veterans Administration (J.T.S.) and NIH grant RO1 AA12671 (J.T.S.). In addition, W. Schmidt was supported by NIH K08 A101460. The Central Microscopy Research Facility received support from the University of Iowa Office of the Vice President for Research.
REFERENCES
1.Agnello, V., G. Abel, M. Elfahal, G. B. Knight, and X. Zhang.1999. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA96:12766–12771.
K. Tanaka, I. Miyajima, M. Mizokami, and K. Tanikawa.1999. Intraspousal transmission of GB virus C/hepatitis G virus in an hepatitis C virus hyper-endemic area in Japan. Am. J. Gastroenterol.94:1627–1631.
3.Allen, L. H., and A. Aderem.1995. A role for MARCKS, the alpha isozyme of protein kinase C and myosin I in zymosan phagocytosis by macrophages. J. Exp. Med.182:829–840.
4.Alter, H. J., Y. Nakatsuji, J. Melpolder, J. Wages, R. Wesley, J. W.-K. Shih, and J. P. Kim.1997. The incidence of transfusion-associated hepatitis G virus infection and its relation to liver disease. N. Engl. J. Med.336:747–754. 5.Alter, M. J., M. Gallagher, T. T. Morris, L. A. Moyer, E. L. Meeks, K. Krawczynski, J. P. Kim, and H. S. Margolis.1997. Acute non-A-E hepatitis in the United States and the role of hepatitis G virus infection. N. Engl. J. Med.336:741–746.
6.Beard, M. R., G. Abell, M. Honda, A. Carroll, M. Gartland, B. Clarke, K. Suzuki, R. Lanford, D. V. Sangar, and S. M. Lemon.1999. An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology
30:316–324.
7.Bukh, J., C. L. Apgar, and M. Yanagi.1999. Toward a surrogate model for hepatitis C virus: an infectious molecular clone of the GB virus-B hepatitis agent. Virology262:470–478.
8.Bukh, J., J. P. Kim, S. Govindarajan, C. L. Apgar, S. K. Foung, J. Wages, Jr., A. J. Yun, M. Shapiro, and R. H. Purcell.1998. Experimental infection of chimpanzees with hepatitis G virus and genetic analysis of the virus. J. Infect. Dis.177:855–862.
9.Cohen, J. I., J. R. Ticehurst, S. M. Feinstone, B. Rosenblum, and R. H. Purcell.1987. Hepatitis A virus cDNA and its RNA transcripts are infectious in cell culture. J. Virol.61:3035–3039.
10. Cook, R. T., J. T. Stapleton, D. Klinzman, and Z. K. Ballas.1997. Effect of a single ethanol ingestion on lymphocyte subsets and in vitro HIV replica-tion. J. Investig. Med.45:265–271.
11. Dawson, G. J., G. G. Schlauder, T. J. Pilot-Matias, D. Thiele, T. P. Leary, P. Murphy, J. E. Rosenblatt, N. Simons, E. A. Martinson, R. A. Gutierrez, J. R. Lentino, C. Pachucki, A. S. Muerhoff, A. Widell, G. Tegtmeier, S. Desai, and I. K. Mushahwar.1996. Prevalence studies of GB virus-C using reverse-transcriptase-polymerase chain reaction. J. Med. Virol.50:97–103. 12. de Martino, M., C. Azzari, M. Resti, M. Moriondo, M. E. Rossi, L. Galli, and
A. Vierucci.1998. Hepatitis G virus infection in human immunodeficiency virus type 1-infected mothers and their children. J. Infect. Dis.178:862–865. 13. Dickens, T., and S. M. Lemon.1997. GB virus C, hepatitis G virus, or human
orphan flavivirus? Hepatology25:1285–1286.
14. Emerson, S. U., M. Lewis, S. Govindarajan, M. Shapiro, T. Moskal, and R. H. Purcell.1992. cDNA clone of hepatitis A virus encoding a virulent virus: induction of viral hepatitis by direct nucleic acid transfection of mar-mosets. J. Virol.66:6649–6654.
15. Feucht, H. H., B. Zollner, S. Polywka, B. Knodler, M. Schroter, H. Nolte, and R. Laufs.1997. Distribution of hepatitis G viremia and antibody response to recombinant proteins with special regard to risk factors in 709 patients. Hepatology26:491–494.
16. Fogeda, M., S. Navas, J. Martin, M. Casqueiro, E. Rodriguez, C. Arocena, and V. Carrena.1999. In vitro infection of human peripheral blood mono-nuclear cells by GB virus C/hepatitis G virus. J. Virol.73:4052–4061. 17. Gutierrez, R. A., G. J. Dawson, M. F. Knigge, S. I. Melvin, C. A. Heynen,
C. R. Kyrk, C. E. Young, R. J. Carrick, G. G. Schlauder, T. K. Surowy, B. J. Dille, P. F. Coleman, D. L. Thiele, J. R. Lentino, C. Pachucki, and I. K. Mushahwar.1997. Seroprevalence of GB virus C and persistence of RNA and antibody. J. Med. Virol.53:167–173.
18. Hong, Z., M. Beaudet-Miller, R. E. Lanford, B. Guerra, J. Wright-Minogue, A. Skelton, B. M. Baroudy, G. R. Reyes, and Y. N. Lau.1999. Generation of transmissible hepatitis C virions from a molecular clone in chimpanzees. Virology256:36–44.
19. Ikeda, M., K. Sugiyama, T. Mizutani, T. Tanaka, K. Tanaka, D. Shimotohno, and N. Kato.1997. Hepatitis G virus replication in human cultured cells displaying susceptibility to hepatitis C virus infection. Biochem. Biophys. Res. Commun.235:505–508.
20. Kiyosawa, K., and E. Tanaka.1999. GB virus C/hepatitis G virus. Intervi-rology42:185–195.
21. Kobayashi, K., E. Tanaka, J. Nakayama, C. Furuwatari, T. Katsuyama, S. Kawasaki, and K. Kiyosawa.1999. Detection of GB virus-C/hepatitis G virus genome in peripheral blood mononuclear cells and liver tissue. J. Med. Virol.
57:114–121.
22. Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice.1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science277:570–574.
23. Kolykhalov, A. A., S. M. Feinstone, and C. M. Rice.1996. Identification of a highly conserved sequence element at the 3⬘terminus of hepatitis C virus genome RNA. J. Virol.70:3363–3371.
24. Laskus, T., M. Radkowski, L. F. Wang, H. Vargas, and J. Rakela.1998. Detection of hepatitis G virus replication sites by using highly strand-specific Tth-based reverse transcriptase PCR. J. Virol.72:3072–3075.
[image:8.612.62.284.73.195.2]25. Leary, T. P., A. S. Muerhoff, J. N. Simons, T. J. Pilot-Matias, J. C. Erker, M. L. Chalmers, G. G. Schlauder, G. J. Dawson, S. M. Desai, and I. K.
FIG. 7. Cell lysate and concentrated cell culture supernatant from the fourth passage of GBV-C in PBMCs were separated on a 20 to 60% (wt/wt) sucrose equilibrium gradient. The sucrose density of each fraction in grams per milliliter is shown at the top. GBV-C RNA was extracted from each fraction and detected by RT-PCR.
on November 9, 2019 by guest
http://jvi.asm.org/
Mushahwar.1996. Sequence and genomic organization of GBV-C: a novel member of the Flaviviridae associated with human non-A-E hepatitis. J. Med. Virol.48:60–67.
26. Lefre`re, J.-J., S. Guiramand, F. Lefrere, M. Mariotti, P. Aumont, J. Lerable, J.-C. Petit, R. Girot, and L. Morand-Joubert.1997. Full or partial serorever-sion in patients infected by hepatitis C virus. J. Infect. Dis.175:316–322. 27. Linnen, J., J. Wages, Z.-Y. Zhang-Keck, K. E. Fry, K. Z. Krawczynski, H.
Alter, E. Koonin, M. Gallagher, M. Alter, S. Hadziyannis, P. Karayiannis, K. Fung, Y. Nakatsuji, J. W. K. Shih, M. Piatak, C. Hoover, J. Fernandez, S. Chen, J.-C. Zou, T. Morris, K. C. Hyams, S. Ismay, J. D. Lifson, G. Hess, S. K. H. Foung, H. Thomas, D. Bradley, H. Margolis, and J. P. Kim.1996. Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science271:505–508.
28. Matzura, O., and A. Wennborg.1996. RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Comput. Appl. Biosci.12:247–249.
29.Melan, M. A., and G. Sluder.1992. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells. Implications for immu-nofluorescence microscopy. J. Cell Sci.101:731–743.
30. Melvin, S. L., G. J. Dawson, R. J. Carrick, G. G. Schlauder, C. A. Heynen, and I. K. Mushahwar.2000. Biophysical characterization of GB virus C from human plasma. J. Virol. Methods71:147–157.
31. Nerurkar, V. R., P. K. Chua, P. R. Hoffman, W. M. Dashwood, C. M. Shikuma, and R. Yanagihara.1998. High prevalence of GB virus C/hepatitis G virus infection among homosexual men infected with human immunode-ficiency virus type 1: evidence for sexual transmission. J. Med. Virol.56:123–127. 32. Okamoto, H., H. Nakao, T. Inoue, M. Fukuda, J. Kishimoto, H. Iizuka, F. Tsuda, Y. Miyakawa, and M. Mayumi.1997. The entire nucleotide sequence of two GB virus C/hepatitis G virus isolates of distinct genotypes from Japan. J. Gen. Virol.78:737–745.
33. Pessoa, M. G., N. A. Terrault, J. K. J. Detmer, M. Collins, H. M. Hassoba, and T. L. Wright.1998. Quantitation of hepatitis G and C viruses in the liver: evidence that hepatitis G virus is not hepatotropic. Hepatology27:877–880. 34. Ridpath, J. F., and S. R. Bolin.1998. Differentiation of types 1a, 1b, and 2
bovine viral diarrhoea virus by PCR. Mol. Cell. Probes12:101–106. 35. Robertson, B., G. Myers, C. Howard, T. Brettin, J. Bukh, B. Gaschen, T.
Gojobori, G. Maertens, M. Mizokami, O. Nainan, S. Netesov, K. Nishioka, I. T. Shin, P. Simmonds, D. Smith, L. Stuyver, and A. Weiner.1998. Clas-sification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch. Virol.143:
2493–2503.
36. Schmidt, W. N., D. Klinzman, D. LaBrecque, D. E. Macfarlane, and J. T. Stapleton.1995. Direct detection of hepatitis C virus (HCV) RNA from whole blood, and comparison with HCV RNA in plasma and peripheral blood mononuclear cells. J. Med. Virol.47:153–160.
37. Seipp, S., M. Scheidel, W. J. Hofmann, U. Tox, L. Theilmann, T. Goeser, and B. Kallinowski.1999. Hepatotropism of GB virus C (GBV-C): GBV-C replication in human hepatocytes and cells of human hepatoma cell lines. J. Hepatol.30:570–579.
38. Shimizu, Y. K.1999. Replication of GB virus C (hepatitis G virus) in inter-feron-resistant Daudi cells. J. Virol.73:8411–8414.
39. Shindo, M., A. M. Di Bisceglie, T. Akatsuka, T. L. Fong, L. Scaglione, M. Donets, J. H. Hoofnagle, and S. M. Feinstone.1994. The physical state of the negative strand of hepatitis C virus RNA in serum of patients with chronic hepatitis C. Proc. Natl. Acad. Sci. USA91:9719–9723.
40. Simons, J. N., S. M. Desai, D. E. Schultz, S. M. Lemon, and I. K. Mushah-war.1996. Translation initiation in GB viruses A and C: evidence for internal ribosome entry and implications for genomic organization. J. Virol.70:6126– 6135.
41. Simons, J. N., T. P. Leary, G. J. Dawson, T. J. Pilot-Matias, A. S. Muerhoff, G. G. Schlauder, S. M. Desai, and I. K. Mushahwar.1995. Isolation of novel virus-like sequences associated with human hepatitis. Nat. Med.1:564–569. 42. Simons, J. N., T. J. Pilot-Matias, T. P. Leary, G. J. Dawson, G. J. Desai, S. M. Desai, G. G. Schlauder, A. S. Muerhoff, J. C. Erker, S. L. Buijk, and M. J. Chalmers.1995. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA92:3401–3405.
43. Stapleton, J. T., D. Klinzman, W. N. Schmidt, P. Wu, D. R. LaBrecque, J.-Q. Han, M. J. Perino-Phillips, R. Woolson, and B. Alden.1999. Prospective comparison of whole blood and plasma hepatitis C virus RNA detection systems: improved detection using whole blood as the source of viral RNA. J. Clin. Microbiol.37:484–489.
44. Stapleton, J. T., J. Xiang, S. George, and D. Klinzman.Evidence for delayed human immunodeficiency virus (HIV) disease progression in in HIV-GB virus C co-infected individuals.InH. S. Margolis (ed.), Viral hepatitis and liver disease, in press. International Medical Press, London, United King-dom.
45. Tacke, M., S. Schmolke, V. Schlueter, S. Sauleda, J. I. Esteban, E. Tanaka, K. Kiyosawa, H. J. Alter, U. Schmitt, G. Hess, B. Ofenloch-Haehnle, and A. M. Engel.1997. Humoral immune response to the E2 protein of hepatitis G virus is associated with long-term recovery from infection and reveals a high frequency of hepatitis G virus exposure among healthy blood donors. Hepatology26:1626–1633.
46. Tanaka, T., T. Takeuchi, K. Inoue, S. Tanaka, and M. Kohara.1997. Acute hepatitis caused by sexual or household transmission of GBV-C. J. Hepatol.
27:1110–1112.
47. Thomas, D. L., D. Vlahov, H. J. Alter, R. Marshall, J. Astemborski, and K. E. Nelson.1998. Association of antibody to GB virus C (hepatitis G virus) with viral clearance and protection from reinfection. J. Infect. Dis.177:539–542. 48. Toyoda, H., I. Takahashi, Y. Fukuda, K. Hayakawa, and J. Takamatsu.2000. Comparison of characteristics between patients with GB virus C/hepatitis G virus (GBV-C/HGV) RNA and those with GBV-C/HGV E2-antibody in patients with hemophilia. J. Med. Virol.60:34–38.
49. Wu, J. C., W. Y. Sheng, Y. H. Huang, S. J. Hwang, and S. D. Lee.1997. Prevalence and risk factor analysis of GBV-C/HGV infection in prostitutes. J. Med. Virol.52:83–85.
50. Wu¨nschmann, S., and J. T. Stapleton.1999. Fluorescence-based quantitative methods for detecting human immunodeficiency virus type 1-induced syncy-tia. J. Clin. Microbiol.38:3055–3060.
50a.Wuenschmann, S., J. D. Medh, D. Klinzmann, W. N. Schmidt, and J. T. Stapleton.Characterization of hepatitis C virus (HCV) and HCV E2 inter-actions with CD81 and the low-density lipoprotein receptor. J. Virol., in press.
51. Xiang, J., K. J. Daniels, D. R. Soll, W. N. Schmidt, D. R. LaBrecque, and J. T. Stapleton.1999. Visualization and characterization of GB virus C (hepatitis G virus) particles: evidence for a nucleocapsid. J. Viral Hepatol.
6:S16–S22.
52. Xiang, J., D. Klinzman, J. McLinden, W. N. Schmidt, D. R. LaBrecque, R. Gish, and J. T. Stapleton.1998. Characterization of hepatitis G virus (GB-C virus) particles: evidence for a nucleocapsid and expression of sequences upstream of the E1 protein. J. Virol72:2738–2744.
53. Yanagi, M., R. H. Purcell, S. U. Emerson, and J. Bukh.1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA94:8738–8743.
54. Yanagi, M., M. St. Claire, M. Shapiro, S. U. Emerson, R. H. Purcell, and J. Bukh.1998. Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology244:161–172.