Copyright ©1990,American SocietyforMicrobiology
Isolation and
Characterization
of Herpes Simplex
Virus Mutants
Containing Engineered Mutations
at the
DNA
Polymerase Locus
ALICE I. MARCY, DORNE R. YAGER, ANDDONALD M. COEN*
DepartmentofBiological Chemistry and Molecular Pharmacology, HarvardMedicalSchool,
Boston,Massachusetts02115
Received 1 November 1989/Accepted 25 January 1990
We have derived Vero cell lines containing the herpes simplex virus DNA polymerase (pol) gene that complementtemperature-sensitivepol mutants. These cell lines were used to recover virusescontaining new mutations at the pol locus. Twospontaneouslyarising host-range mutants, 6C4 and 7E4, wereisolated. These mutants did not growefficientlyon Verocells or synthesize late polypeptides but formed plaques onacell line containing the polgene (DP6 cells). Whereas mutant 6C4specifieda
wild-type-size
Polprotein, wedetectednofull-length Polprotein in 7E4-infected cell extracts.Complementationstudiesdemonstratedthat 6C4 and 7E4 contain different mutations and indicated that6C4 is in a complementationgroupdifferent from that ofpol
temperature-sensitivemutant tsC7 or tsD9.A mutant inwhich 2.2kilobases ofpolsequences were replaced
with theEscherichiacolilacZgene under the control of the herpes simplex virus thymidine kinase promoter was constructed. This mutant formed blue plaques on DP6 cells in the presence of
5-bromo-4-chloro-3-indolyl-o-D-galactoside. Using this virus in marker rescue experiments, weengineeredthree mutantscontainingdeletions
in thepolcoding region which grew efficiently on DP6 cells but not on Vero cells and whichdiffered in their
synthesis of Pol polypeptides. The lacZ insertion virus was also used to introduce a deletion in the region
upstreamof thepol longopen reading frame, which removes a short open reading frame that could encode a
10-amino-acid peptide. This mutant grew to similar titers on Vero and DP6 cells, indicating that these
sequencesarenotessentialforgrowthof the virus intissue culture.
Theherpessimplexvirus(HSV)DNApolymerase (Pol) is
essential for viral DNAreplication (1, 35, 40), serves as a
targetfor antiviral drugs(9),andprovidesanexcellentmodel
for studies ofeucaryoticreplicativeDNApolymerase inpart
because it is amenable to both biochemical and genetic
analysis. Understanding Pol functionswill begreatly
facili-tated bymutants defining protein domainsthatcontributeto
catalysis, interactions with other proteins, stability, and
intracellularlocalization.
Such a combined approach has already contributed to
effortsto correlatePol structure with itsfunctions. Genetic
mapping and sequencing of pol mutants exhibiting altered
sensitivities to nucleoside and
PPi
analogs and aphidicolinindicate that amino acids lyingindiscrete regions between
residues 597 and 961 contributetosubstratebinding (17, 18,
27, 31, 32, 45). Furtherinsights intoPolfunctionhave been
provided by temperature-sensitive (ts)
pol
mutants (6, 10).Four pol ts mutants map within a 4-kilobase region that
specifiesasingle majormRNA and longopenreading frame
(18,23, 48). Genetic analysisofthese mutants hasidentified
three complementation groups (groups 1-3, 1-4, and 1-14),
with complex overlapping patterns of complementation,
consistentwith the enzymehaving multiple functions (6, 23,
39, 46). The prototypic mutant in complementation group
1-4, tsD9, exhibits altered drug sensitivity and contains a mutation in a domain thought to participate in substrate
recognition(17, 18). Mutantsincomplementation group 1-3
specifythermolabile Pol activity in vivo (1) and in vitro (37)
and map to sequences that include the left end of the gene
(10, 23), suggestingthat theN-terminalportion of the protein
includes regions contributing to stability. The limits of
sequencestowhichcertain of these tsmutations havebeen
mappedalsoinclude a short openreadingframe which could
*Correspondingauthor.
encodea10-amino-acidpeptide (18). Thisshortopenreading
frame lies 75 basepairs (bp) downstream ofthepol
transcrip-tional start site and ends 93 bp upstream ofthe long open
reading frame (18,48). Itsrole isunknown.
The studies described herewereinitiatedtofacilitateour
understanding of Pol functions by increasing thenumberand
type ofpol mutants available for biochemical and
genetic
analysis andtoexamine theimportance ofthe10-amino-acid
upstream openreading frame for viral growth. Wewishedto be able to introduce site-specific mutations in thepol gene
into the HSV genome to allow mutant Pol
proteins
to beanalyzed in the context of a viral infection. Since we
expectedto recovervirusescontaining lethal pol mutations,
we isolated a cell line able to support the growth ofthese
viruses. As these mutants would not necessarily have a
selectable phenotype, we also needed todevelop a system
for screening pol mutants. We demonstrate the use of a
complementing
cell lineand apolmutantthatproducesblueplaquestoprovideasystemfortheefficientrecoveryof pol
mutants anddescribe the isolation and
preliminary
charac-terization of six newpol mutants, one of which lacks the
10-amino-acid openreadingframe.
MATERIALSANDMETHODS
Cellsand viruses. Verocells weregrown andmaintainedas
described previously (46). Nero
cells
are a cloned line ofG418-resistant
cells
derivedbytransformingVerocells withpSV2neo (42). HSV type 1 mutants tsC4, tsC7, and tsD9
werederived fromwild-typestrain KOS and havemutations
conferring temperature sensitivity at the pol locus (10).
MutanthrR3, kindlyprovided bySandraWeller,has alacZ insert in the ICP6gene (20).
Plasmids.The
plasmids
usedinthisstudyaredepicted
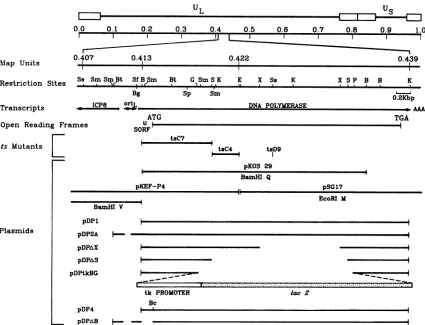
inFig. 1. Plasmid pDP1 contains the 153 bpupstream ofthe
2208
on November 10, 2019 by guest
http://jvi.asm.org/
UL
MIap Units Restriction Sites
Transcripts
Open Reading Fra
ts Mutants
Plasmids
EJ
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
0.407 0.413 0.422 0.439
-I
III
Ss Sm Sm Bt Sf B Sm Bt G SmSK E X Ss K X SP B B K
Bg Sp Sm
NL2Kbp
ICPB DNA POLYMERASE AAA
ATG
imes UF
SORF
pKEF-P4
TGA
tsC7
tsC4 tsD9
i I
pKOS 29
BamH Q
pSGI7 EcoRl M BamHI V
pDP1
pDP2A
pDPaX
pDP&S
pDPtkcBG
pDP4
tk PROMOTER lac Z
Bc
pDPtB
-FIG. 1. Maplocation ofHSV pol. The locations ofpoltranscript, open reading frames, and relevant restrictionsites and themapped limits oftsmutationsareshown. The bottomportion of the figure representsstructuresofplasmidsused in thisstudy. Restrictionenzymesare
abbreviatedasfollows:B, BamHI; Bc,BcII; Bg, Bg!I; Bt, BstII; E, EcoRI; G, BgIII; K, KpnI; P, PstI; S,SaII; Sf, SfiI; Ss, SstI; Sm, SmaI; andX, XhoI. UL, Unique long sequence;Us,uniqueshortsequence;SORF,shortupstream openreadingframe.
firstATGof the HSV polopenreading frame,theentire pol
openreadingframe, and156bp downstream ofthe
termina-tion TGA codon (HSVmapcoordinates 0.413to0.439). The
plasmid was constructed by treating pKOS29 (10) with
BamHI, filling in the overhanging ends by using Klenow
fragment, ligating on HindlIl linkers, and digesting with
HindIII and PstI. The
3.2-kbp
HindIII-PstI fragment wasthen ligated to HindIII-PstI-digested pUC19 DNA (50) to
give risetoplasmid p5'DP. Plasmid pSG17 (19)wastreated
with
KpnI
and then with T4 DNA polymerase to removeoverhanging ends, ligatedtoXbaIlinkers,anddigested with
PstI and XbaI. The 0.8-kbp PstI-XbaI fragment from this
plasmid was recovered and ligated to PstI-XbaI-digested
p5'DPtocreatepDP1. pDP1stillhasthe
original
BamHIsiteat 0.413 map units adjacent to the unique HindIII site.
PlasmidpDP2A,which containsthepol openreadingframe
andpromoter sequences, wasmade asfollows. The 363-bp
BstII-BamHI fragment containing thepolpromoter and 51
bp of5'untranslated sequences wasremoved frompKEFP4
(47), treated with Klenowfragment, and ligated to
HindIIl
linkers. The resulting fragment was digested with HindIII
andligatedtoHindIII-treated
pDP1
to createpDP2A. Viralsequences in thisplasmiddiffer from theviralgenomebythe presence ofthe HindIII site and a deletion starting 95 bp
upstream ofthe mRNA start site and extending upstream
intooriLfor62bp (present in pKEFP4; 47, 48).Thisdeletion
does not remove the binding sites for
transcription
factorsSpland TFIID upstreamofthepolmRNAcapsite
(48),
andthepolpromoterinpDP2A retainsactivity, as
judged
by itsability to expresspolin transfected cellsupon HSV
super-infection.
PlasmidpDPtkBGwas madeby first
inserting
the800-bpBamHI-BglII fragment containingtheHSVthymidine kinase
(tk)promoterfrom
plasmid pKG3.6
(26) intoBglII-digested
pDPl.Theresulting
plasmid
wasdigested withSail,
treatedwith Klenow fragment, ligated to
BglII
linkers, and thendigested with
BgIH.
ThisDNAwasthenligated
to a3.0-kbp
BamHI fragment containingthecodingsequencesfor
Esch-erichia coli
3-galactosidase
from pDP500, kindlyprovided
by Dennis Panicali(34). Plasmids
pDPAS
andpDPAX
weremadebytreating pDP1with
Sail
orXhoI, respectively,
andligase.
Site-specific mutagenesiswasused to introduce aunique
BclI site (TGATCA)11bpupstreamofthepolopen
reading
frame in
pDP1
by a procedure developed byTaylor
et al.(43). Mutagenesis was
performed by using
theoligonucleo-tide
5'-TCGGGTGIGAICAACCGC-3'
and a636-bp
HindIII-SphI
fragment
of pDP1 cloned into thebacterio-phage vector
M13mpl9.
Theoligonucleotide
hybridized
tothenoncoding pol strand, and the altered basesare
under-lined. Reagents for this
procedure
wereprovided
in a com-mercial kit(AmershamCorp.).
Clonescontaining
the muta-tion were identifieddirectly by
DNAsequencing,
using
previouslydescribed
procedures
(18). Double-strandedrep--- -
JF--I.
L-I
on November 10, 2019 by guest
http://jvi.asm.org/
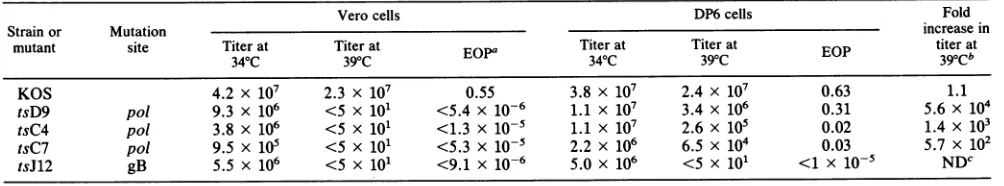
[image:2.612.90.515.74.399.2]TABLE 1. Efficiency of plating of HSV ts mutants onVero and DP6 cells
Verocells DP6cells Fold
Strain or Mutation increasein
mutant site Titerat Titer at Titer at Titerat titer at
340C 390C EOPa 34 C 39 C EOP 39oCb
KOS 4.2 x 107 2.3 x 107 0.55 3.8 x 107 2.4 x 107 0.63 1.1
tsD9 pol 9.3 x 106 <5 x 101 <5.4 x 10-6 1.1 X 107 3.4 x 106 0.31 5.6 x 104
tsC4 pol 3.8 x 106 <5 x 101 <1.3 X 10-5 1.1 X 107 2.6 x 105 0.02 1.4 x 103
tsC7 po/ 9.5 X 105 <5 X 101 <5.3 x 10-5 2.2 x 106 6.5 x 104 0.03 5.7 x 102
tsJ12 gB 5.5 x 106 <5 x 101 <9.1 x 10-6 5.0 x 106 <5 x 101 <1 X 10-5 NDC
a EOP,Efficiency ofplating(determined as titerat39°C/titerat34°C).
bEfficiency of plating on DP6cells/efficiency of platingonVerocells.
cND, Cannot bedetermined.
licative-form phage DNA was prepared, and the mutated
HindIII-SphI fragment was removed and ligated with
HindIII-SphI-digested pDP1. The presence of the new
re-striction site in the resulting clone, pDP4, was verified by
digesting plasmid DNA derived fromDam- cells (GM119,
kindly provided by Jacqueline Miller) with BclI. Plasmid
pDPAB was made by ligating the 363-bp HindIII-BamHI
fragment frompDP2A toHindIII-BclI-digested pDP4.
Transformationof Verocells. Verocellsweretransformed with pSV2neo (kindly provided by Neal DeLuca) and a
10-fold molar excess of pDP2A by a modification of the
procedure described byDeLuca et al. (14). After
transfor-mation,thecells were incubated for2daysandthenplated
to adensityof 5 x 103/cm2in thepresenceof750
jig
ofG418per ml(GIBCO Laboratories). Individual colonies appeared
after2to 3 weeksand wereamplifiedand assayedfortheir
ability tosupport the growthofpol mutant tsC7 or tsD9 at
the nonpermissive temperature (39°C). One cell line, DP6,
exhibited high complementing activity and was used for
further study.
Complementationanalysis. Complementation assays were
performedbyconductingmixedinfections under
nonpermis-sive conditions (growth at39°C on Vero cells) and assayed
under permissive conditions (growth at 34WC on DP6cells)
by procedures described previously (40, 46). The
comple-mentation indexwascalculated as (plaques for mixed A+ B
infection)/(plaques for A infection + plaques for B
infec-tion),where A and B aretwo mutants. An index of
.10
wastaken to beindicativeofcomplementation(46).
Marker transfer andrescue. AnHSV
expressing
,-galac-tosidase was recovered by cotransfecting DP6 cells with
infectious KOS DNA and pDPkBG by methods described
previously (7). Progeny from the transfection were plated
onto DP6 cells and stained after 3 days with 2 ml of
phosphate-buffered saline containing 600 to 750 ,ug of
5-bromo-4-chloro-3-indolyl-,-D-galactoside(X-Gal) per ml. At
1 or 2 days after staining, blue plaques were picked and screenedfor the ability togrow on DP6 cells and not on Vero cells. One suchvirus, HP66, was selected for further study. Marker rescue was performed as described previously (7),
using infectious HP66 DNA and the plasmids indicated in
Results.
Southern blot analysis. Viral DNA wasrecovered by the procedure described by Coen et al. (11). DNA samples were
digested with restriction enzymes and electrophoresed
throughagarose gels. The DNA was transferred to a nylon
membrane asdescribedby
Southern
(41). Membranes wereUVcross-linked (8) for 5 minimmediately after being blotted and then probed with
32P-radiolabeled
DNA fragments.DNA probes were prepared by random priming of
gel-isolated restrictionfragments with[32P]dCTP(16).
Western immunoblot analysis and immunoprecipitations.
[35S]methionine-radiolabeled
or unlabeled extracts ofin-fected cells for sodium dodecyl sulfate
(SDS)-polyacryl-amidegels,immunoblots, and
immunoprecipitation
analyseswere prepared as described previously (26). For blotting,
proteinswere electrophoresedthrough
SDS-polyacrylamide
gels and then transferred to nitrocellulose as described
by
Towbinetal. (44). Thenitrocellulosewasthen reactedwith
a 1:1,000dilution ofa mixture ofpolyclonal rabbit antisera
directedagainst
P-galactosidase-polymerase
fusionproteins
derived from bacteria (PP5 and/or BGG4; 49). Bound
anti-bodiesweredetected withagoatanti-rabbit
immunoglobulin
G-alkaline phosphataseconjugate, andvisualized withnitro
blue tetrazolium and 5-chloro-4-chloro-3-indolyl phosphate
(PromegaBiotec). ICP8 wasdetected withpolyclonal rabbit
antiserum 3-83, kindlyprovided by David Knipe.Thymidine
kinase wasdetected withpolyclonal rabbit antiserum
gener-ouslyprovided by William Summers.
RESULTS
Vero cellsexpressing DNApolymerase.As afirststep in the
isolationofnewpolmutants,wefirstclonedthepolgene and
its upstream sequences into plasmid pDP2A (Fig. 1). This
plasmid contains no other known intact HSV genes and
exhibited biological activity in a transfection assay (5) in
whichaplasmid containinganHSVorigin of DNA
replica-tion was used (M. Challberg, personal communication).
Since HSVpol isessential for viral
replication,
thepropa-gation ofviruses containing lethal pol mutations
requires
acell line expressing wild-type Pol. To thisend, we
cotrans-formed Vero cells with pDP2A and pSV2neo, which
con-tains the neo geneunder the controlofthe simianvirus 40
promoter(42).G418-resistant colonies of cellswerepicked,
expanded,andassayed for
their
abilitytosupport thegrowthof polmutanttsC7 or tsD9at 39°C.
Approximately
30% ofthe G418-resistant colonies expressed detectable
comple-mentingactivity.One cellline,DP6, which hadhighlevels of
complementing activitywas chosenforfurtherstudy.
Plaque formation by wild-type KOS and various ts
mu-tants onDP6 and Vero cellsat 34 and 39°C was
compared
(Table 1). Similartiters of
wild-type
strain KOS wereob-tainedbyusingeither DP6 orVerocells at either 34 or39°C,
indicating thatexpression oftheresidentpolin DP6cellsis not detrimental for viral infection. DP6
cells
didnot comple-ment mutant tsJ12, whichcontains aglycoprotein Bmuta-tion (13). Polymerase mutantstsC4, tsC7, and tsD9did not
formplaques onVerocells atthe elevatedtemperature but
formedplaquesonDP6cells under the sameconditions. For
tsD9, essentially wild-type levels ofplaque formationwere
obtained at 39°C. The extent of
complementation
differedon November 10, 2019 by guest
http://jvi.asm.org/
TABLE 2. Plaque formation byHSVmutantsonVero andDP6cells
Titeron:
Strain or pimtto
mutant ~~polmutation
mutant Verocells DP6cells
KOS 4.2 x 107 3.8 x 107
6C4 Spontaneous host range <5 x 101 2.5 x 107
7E4 Spontaneous host range 5 x 101 1.6x 107
HP66 2.3-kbp deletion, lacZ 1.3 x 102 4.0 x 107
insertion
ASi 2.0-kbp deletion <5 x 101 9.8 x 106 AX14 1.2-kbp deletion 5.5 x 102 2.7 x 107
AX17 1.2-kbp deletion 1.2 x 102 1.1 x 107 AB10 144-bp deletion 6.0 x 106 2.7 x 106
among thepol mutants but was reproducibly the highest with tsD9 and lower with tsC4 and tsC7. DP6 cells contain
approximately30 copies of pol DNA percell,asdetermined
by Southern analysis of cellular DNA, and showed comple-menting abilitythrough 25 passages (data notshown).
Recovery of spontaneous pol host-range mutants. Having isolated a complementing cell line, our efforts were next directed toward obtaining apoldeletion mutant. For these experiments, DP6 cells were cotransfected with infectious wild-type viral DNA and pDPAS, which contains a 2-kbp deletion in thepolgene (Fig. 1). Two mutants isolated from the progeny of this transfection, 6C4 and 7E4, formed plaqueson DP6 cells but not on Vero cells (Table 2). These mutants were plaque purified andamplifiedforfurther study.
Todeterminewhether these host-range mutantviruses
con-tained the expected deletions, the BamHI restriction pat-terns of the mutant viral DNAs were compared with plasmid
DNA containing either the wild-type pol (pDP1) or the
deleted gene (pDPAS). Unexpectedly,both of the host-range mutants showed wild-type BamHIrestriction patterns when
probed with radiolabeledpolDNA (datanot shown).
Complementation analysis of spontaneous mutants. Since
the spontaneous mutants could becomplemented by a cell
line whose only viral sequences are the pol gene, we examined their ability to complement the growth ofpol ts mutants at the nonpermissive temperature (39°C) in Vero cells. Vero cells werecoinfectedwith thehost-rangemutants and either tsC7or tsD9 at 39°C. The progeny of this infection were assayed on DP6 cells at 34°C, and the results are shown in Table 3. As previously reported (39), tsD9 and tsC7
complementedone another for growth at thenonpermissive
temperature. 6C4 complemented both of thepolts mutants
and 7E4, while 7E4complemented neither pol ts mutant.
Polypeptides synthesized in spontaneous mutant-infected
cells. To determine whether the mutations in 6C4 and 7E4
resulted in the failure of the mutants to synthesize late
polypeptides,Nero and DP6 cells infected with either of the
mutants or the wild-type virus were labeled with
[35S]methionine
from 2 to 6 h postinfection. In Nero cells, [image:4.612.58.299.105.223.2]these twomutantsfailed tosynthesizedetectable amounts of
TABLE 3. Complementation ofpol host-range and ts mutants
Complementation indexwith mutant: Mutant
tsC7 6C4 7E4
tsD9 11 11 0.5
tsC7 66 1.0
6C4 110
true latepolypeptides such as ICP1,2 (Fig. 2A, top
arrow-head) and synthesized reduced amounts of other late
poly-peptides, relative to KOS. This pattern resembled that of
Nero cells infected by KOS in the presence of 200 ,ug of phosphonoacetic acid per ml, an inhibitor of viral DNA
synthesis.In contrast,in DP6cells,the two mutants
synthe-sized polypeptides that wereindistinguishable from those of the wild-type virus (results not shown). Thus thepol
muta-tions inthese two mutants led toadecreasein thesynthesis
oflate polypeptides, presumably due to the failure to syn-thesize viral DNA. Indeed, dot blot analysis (results not
shown) indicated that the two mutants were profoundly
impaired in the synthesis of viral DNAin Nero cells. Toexamine the Pol proteins specified by these host-range mutants, we probed blots containing proteins from KOS-,
6C4-, and 7E4-infected Nero cells with antiserum PP5, which is directed against a fusion protein consisting of ,B-galactosidase and theC-terminal 261 amino acids of Pol (49).
6C4 synthesizedanimmunoreactiveprotein the same size as
wild-type Pol, while no detectable equivalent was specified
by 7E4 (Fig. 2B). Similar results were observed when PP5
and BGG4 antisera were used to immunoprecipitate [355] methionine-labeled proteins from KOS-, 6C4-, and 7E4-infected Nero cell extracts(data not shown). 6C4- and 7E4-infected Nero cell extracts contained levels of ICP8 similar to those found in KOS-infected cells (Fig. 2B); thus the absence offull-length Pol polypeptide in 7E4-infected cell extracts was notdue to inefficient infection.
Recovery of apol insertion mutant expressing
j8-galactosi-dase.The resultsof the effortstoobtain apol deletion mutant suggested that animproved screeningprocedurewas needed to enhance the efficiency of detecting recombinant viruses. Wechose to use the E. colilacZ gene in a manner similarto that used byPanicali et al. (34) and Chakrabarti et al. (4) for vaccinia virusand by Weller andcolleaguesfor HSV (3, 20, 21). With this approach, a virus that forms blue plaques in the presence ofX-Galis used as ascreening marker in order
torecover recombinants that form white plaques due to the
replacement oflacZwithsequences froma plasmid contain-ing knownpol mutations.
Plasmid pDPtkBG (Fig. 1) was used in a marker transfer
experiment with wild-type infectious DNA to obtain viral recombinants that grewonlyonDP6 cells because of thepol
deletion-lacZ insertion and formed blue plaques in the presence of X-Gal due to the expression oflacZ. One such mutant, HP66, which grew efficiently on DP6 cells and poorly on Vero cells (Table 2) waschosen forfurther study.
The position of the lacZ gene was confirmed by Southern
analysis of wild-type KOS and HP66 viral DNAs. HP66 contained a5.4-kilobaseBamHIfragmentthathybridized to
both thelacZ gene andpol(BamHI Q-fragment) probes, as expected (Fig. 3). Mutant hrR3, which contains lacZ in a
3.0-kilobase BamHI fragment in the HSV ICP6 gene (20),
was included in this analysis to confirm thespecificity of the
lacZprobe.
The ability of HP66 to grow in DP6cells and not onNero cells demonstrated that the lethal mutation inHP66 is in the
pol gene. This was verified by marker rescue experiments
using wild-type pol sequences (pDP1). Transfections of
HP66infectious viral DNA with pDP1 gave rise to
approx-imately 100-fold more recombinants that grew on Vero cells than transfections with no plasmid DNA (data not shown).
The presence of
P-galactosidase
in HP66-infected cells wasconfirmed by Western blot analysis of cells infected with KOS or HP66, using a serum specific fora
3-galactosidase-polymerase fusionprotein (49).HP66specifiednodetectableon November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.58.298.673.736.2]-Y
0
v2
(0~
COz m
Y cr I
CLr (fl 14 41-0 0 C) W6
o oe to N
Y, Y
-.4
_ .& .
n_
_ )! _NCD
5.4kb- u
3 3kb- *4. *
*.
Probe: BamO
cn CO Q
- SL
3.Okb-
*.
..:
':::::,o
.,.
lacZ
FIG. 3. Southern blotanalysisof HP66 viral DNA. Viral DNAs from wild-type KOS andmutantshrR3and HP66weredigested with BamHI, electrophoresed through 1%agarosegels, and transferred tonylon. Membraneswereprobed with either 32P-radiolabeled pol DNA(BamHIQfragment, Fig. 1)orlacZDNA.Fragment sizes(in kilobases)areindicated.
r
DP6 T Nero, I Y 2 I E
FIG. 2. Analysis of 6C4- and7E4-infected-cell polypeptides. (A) [35S]methionine-radiolabeled proteins from Nero cells mock infected
orinfected with KOS, KOS in the presence ofphosphonoacetic
acid,or6C4 and 7E4wereelectrophoresed through
SDS-polyacryl-amidegels and exposedto film. Arrowheads indicate examples of
lateproteinspresentinKOS-infected cells and absenceorreduced
inmutant-infected cells. (B) Proteins fromNerocells infected with wild-typestrain KOS andmutants6C4 and7E4wereelctrophoresed
throughSDS-polyacrylamidegels,transferredtonitrocellulose, and reacted withantiserum (oa) specific for either Pol (left)orICP8 (right). Pol inNerocells andspecifiedaprotein that is thesamesize as ,-galactosidase in both Nero and DP6 cells (Fig. 4).
Figure 4 also shows that uninfected DP6 cells make no
detectable Polprotein.
FIG. 4. Western blot analysis of HP66-infected-cell proteins.
Protein lysates from mock-, wild-type strain KOS-, and mutant HP66-infected Nero and DP6 cells were electrophoresed through
polyacrylamide gels and transferred to nitrocellulose. The
mem-braneswereprobedwithanantiserumspecificfora
Pol-,-galactosi-dase fusion protein (49). The leftmost lane contains a set of
molecularweight standards, ofwhich P-galactosidase isa compo-nent.
A <
0-
Pol-_ -- -IGP8
pol AICP8
-Pol -9-gal
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.93.268.80.591.2] [image:5.612.362.511.82.336.2] [image:5.612.370.507.439.657.2]m
x
---X Qa
T-(-><
Cao
U)
'-00
Cl) c0Q
1> C)O O
< C] y >< xoI'll 0
<: < Y- 2
205-
Pol-
116-a..
97- d
Ut
- 3.3 kb
65--3.3kb
q0,..NPp46m
E1: -2.1 kb -1.3kb
[image:6.612.74.287.78.383.2]
45-A PoI Tk
FIG. 5. Southern blot analysisofdeletion mutantviral DNAs. ViralDNAs from AS1, AX14, and AX17 andplasmidDNAsfrom pDP1,pDPtkBG, pDPRS, andpDPAX weredigested withBamHI
andelectrophoresed through 1%agarosegels. TheDNAfragments
were transferredto nylon andprobed with radiolabeled polDNA (BamHI Q fragment, Fig. 1). Fragment sizes (in kilobases) are
indicated.
Recovery ofpol host-range deletionmutants.WeusedHP66 to obtainpol deletion mutants. Infectious HP66 DNA was
cotransfected withpDPAS orpDPAX (Fig. 1), theprogeny
from the transfections were plated onto DP6 cells and
stained with X-Gal, and the white plaques were picked.
Small stocks of virus werederived from each plaque pick,
andthe viral DNAwasrecovered andanalyzed by Southern blotting. Approximately 80%of the white plaquesfrom the
pDPAS and pDPAX transfections contained the expected
2.0- and 1.2-kilobase pol deletions, examples of which are
provided (Fig. 5). Two mutants that contained the XhoI deletion(AX14andAX17)andonemutantthat contained the
SalI deletion (AS1) were plaque purified, amplified and
selected, and analyzed further. All three mutants grew
efficientlyonDP6cellsandinefficientlyonVero cells (Table
2). Stocks ofAX14, AX17, and HP66 contained low levels
(approximately 1 in 105viruses) of virusthathadwild-type growth characteristics. Presumably, thesewild-typeviruses
arose by recombination between the pol gene resident in
DP6cellsandeach deletionmutant.
We expected mutants containing the XhoI deletion to specifyatruncatedpolypeptideboth because of the deletion andbecause the deletion results inaframeshift. This
possi-bilitywas examined byimmunoprecipitating Pol from [355]
methionine-radiolabeled extracts of mock-, KOS-, AX14-,
and AX17-infected cells (Fig. 6). The predicted molecular
FIG. 6. Immunoprecipitation analysis of Pol proteins Synthe-sized by pol deletion mutants. Antisera specific for Pol(left) and thymidine kinase(right)were used toimmunoprecipitate 35S-radio-labeledproteins frommock-,AX14-, and AX17-infectedVerocells. The immunoprecipitated proteinswere separated ineithera7.5% (left) or a10%(right)SDS-polyacrylamide gel. Positions of molec-ularweight markers (inthousands) are indicated for the gel
contain-ing immunoprecipitated Pol.
weight of the truncated Pol specified by these deletion
mutantsisapproximately 65,000 (65Kpolypeptide),due to a
termination codon inthe newreading frame after the XhoI
site. Virus AX14 specifies a 96K Pol-related polypeptide. This sizeproteinisconsistent withanadditionalmutationin
AX14 which causes the reading frame to be brought back into register, allowing expression ofa largerprotein. Virus
AX17 specifiedno protein detectable with the Pol antisera
butproduced wild-type levels ofthymidine kinase, indicat-ing that the cells were efficiently infected. Analysis of
proteins in AS1-infected cells indicated that this mutant
specifies atruncatedPol consistent with the deletion muta-tion (49).
Recovery of a pol deletion mutant lacking upstream
se-quences. Since the marker transfer techniquewith the lacZ insertion mutant was useful in obtainingpol recombinants
containing mutations in the majorpol open reading frame,
we employed the same procedure to obtain a mutation
outside of theregion substituted inHP66. Previous
experi-ments torecover HSV tkrecombinants had indicated that
sequencesasfar as1,000bp awayfrom the selected marker could be transferred into the virus(2,11, 26).To examinethe
role in HSV infections of the short upstream open reading
frame (Fig. 1, SORF) which encodes a decapeptide, we
wishedtoobtainamutantlacking these sequences. Plasmid pDPAB (Fig. 1) contains twodeletions; the first
deletionisapproximately 62bpwithinoriL. Such deletions are commonwhen
oriL-containing
sequencesare cloned inbacterialvectors(38,47, 48). The second deletionremoves
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.319.555.83.357.2]BstII Sfil LL O
CI
LL °Cf)iU'C0 wa'
-'
-it
.SmaI
U-0C:
C OU 0 LLoY
m
Y a
1
-4.2
-3.6
-3.0
-1.7 -1.5 _ _
~~~~-
1.31.4
16-d-.79
_R ~~-.6586
.11~~~ -"l .566
-.45
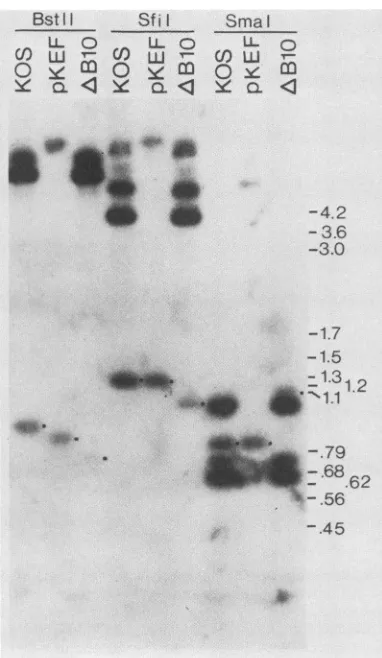
FIG. 7. Restriction analysis of
AB10
viral DNA. Viral DNA samples from wild-type strain KOS, mutant AB10, and plasmid pKEF weredigested withBstII,Sfil, orSmaI.The fragmentswere separated in a 1.5% agarose gel, transferred to nylon, and then probed with 32P-radiolabeled BamHI Q-fragment DNA (Fig. 1). Dots adjacentto bands highlight fragments discussed in the text. Positions of molecular sizemark~ers
(inkilobases) electrophoresed in parallel are indicatedattheright.the144bpto theright oftheBamHI site inthe 5' leader of
pol mRNA, which includes the short open reading frame.
InfectiousHP66 DNAandpDPABDNA werecotransfected
intoDP6cells, and theprogeny wereplatedontoboth Vero
andDP6cells.ViralDNA wasprepared from whiteplaques
and compared by
Southern
analysis. BamHI digestion ofDNA from oneplaque isolate, AB10, resulted in a5.4-kbp
BamHI fragment hybridizing with radiolabeled BamHI Q
BgU
StlI
BamHl
Sm!aI t
Sma! Bu!
If
B
Ufragment (results not shown). This size is consistent both
with the predicted loss of the BamHI site separating the
3.3-kbpBamHI Q and 2.3-kbp BamHIVfragmentsandwith
thedeletion of100 to 200bp.
AB10wasplaquepurified, amplified, andanalyzed further
by restriction
digestion
andhybridization
with radiolabeledBamHI Q fragment (Fig. 7). The relevant restriction sites
andthededuced site oftheAB10deletionsareshown in
Fig.
8. The presence ofthe 144-bp deletion removingthe short
openreading frame was verifiedby the loss of the
1.2-kbp
SfiI
fragment found in KOS andpKEF-P4
DNA and itsreplacement with a fragment about 150 bp smaller, by the
loss ofa 0.87-kbpBstEII fragment in KOS andpKEF-P4
DNA and its replacement with a fragment about 150 bp
smaller, and by the loss ofa0.87-kbp BstEII fragment in
KOS and pKEF-P4 and its replacement with a
0.72-kbp
fragment in AB10. Similarly, the 0.82-kbp SmaI fragment
whichcontainstheshortopenreading framewasmissing in
AB10 and replaced by a 1.1-kbp fragment, consistent with
the predicted loss of a SmaI site, fusing the
0.82-kbp
fragment to a0.45-kbp fragment (which lies mainly outside
the
BamHI
Q fragment), and the144-bp deletion.The analysis was notconsistent with the presence ofthe
orideletion in AB10. Forexample, the BstEII fragment in
AB10 was 0.72 kbp, not the 0.66 kbp expected if the ori
deletion was present. Similar results were obtained with a
variety of otherrestriction enzymes (not shown).
Virus AB10 formed plaques on Vero cells as well as on
DP6cells(Table 2), demonstratingthatthe144-bp
fragment
containingthe smallopenreading frame is notessential for
virusgrowth in cell culture.
DISCUSSION
Inthisreport, wedescribetheisolation ofacell line that
containsthe HSVpolgeneand itsuseintherecoveryofnew
spontaneous andconstructed HSVpolmutants.
Isolation of a cell line thatcomplementspol mutants. The
DP6cell line complementsavariety ofpolmutants,
includ-ing ts and deletion mutants. Plaque formation of pol ts
mutants at 39°C is more than 500-foldgreater on DP6 cells
than on Verocells andranges as
high
as50,000-fold
greater.Muchlower levelsof
complementation
havebeenobservedwithpol mutant tsC7 in a transient assay using a
plasmid
containing the pol and UL31 genes (15). The greater
effi-ciency of
complementation by
DP6cellsmay be duetotheuse ofa stable cell line and/or thepol
plasmid
constructused. DP6cells
complemented
tsC4 andtsC7 less wellthantsD9;such
allele-specific
differences incomplementing
abil-ity by cells expressing a wild-type gene product have also
been observed with the UL5 gene (S. Weller,
personal
But! Sma! BglSf1!
m
-ICP8
orlL
pKEFAII
D
SOR?
DNA POLR
ATG
O.O5Kbp
, ABIOSO
FIG. 8. Enlarged view ofsequences removed from
AB10.
Thelocation of the pol transcript is indicatedO-),and openreading frames are shown (=). Plasmid pKEFP4 lacks the 62 bp from oriL, and virus AB10 lacks the 144 bp between the BamHI site and 11 bp upstream of theATG for thepol longopen reading frame. A bracket over the transcript designates a region predicted to form a stable secondary structure (22). SORF, Short upstream reading frame.on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.86.277.76.405.2] [image:7.612.76.552.598.690.2]communication). Regardless, DP6 cells appear well suited
for the isolation ofnewpol mutants. Moreover, because DP6
cells were constructed by using a plasmid that contains no other intact genes besides pol, the complementation of mutantsby DP6 cells effectively maps these mutations to the pol gene.
Complementation of ts and host-rangepolmutants. Previ-ous work with pol ts mutants has shown that
complementa-tion with these mutants is complex and variable and that intragenic complementation is frequently observed (6, 39, 46). We found (Table 3) that tsD9 and tsC4 complemented each other well, as previously reported (39, 46), and that theyinturn could be complemented by one of the host-range mutants (6C4) which expresses full-length Pol polypeptide (Fig. 2). On the other hand, we detected virtually no com-plementation with host-range mutant 7E4 and the ts mutants, while 7E4 complemented 6C4 well. Given the ability of 7E4
to complement 6C4, it was somewhat surprising that we
were unable to detect full-length Pol polypeptide in
7E4-infected cells. This could be due to the failure of the Pol
antisera to recognize an altered polypeptide. Alternatively,
the 7E4 complementing activity might require only low levels of either full-length or truncated Pol polypeptide.
Ourfindings of intrageniccomplementation with the
host-range mutants areconsistent with previous findings with pol
tsmutantsand may be areflection of the numerous activities
specified by pol. Purified polymerase has been associated
with3'-5'-exonuclease activity and RNase H activity (12, 28, 33; A. I. Marcy, P. D. Olivo, M. D. Challberg, and D. M.
Coen, Nucleic AcidsRes.,inpress).The maplocationof the
tsD9 mutation (complementation group 1-4) andits altered
drug sensitivity phenotypes (17, 18, 36) suggest that it identifies the polymerase activity; perhaps the
complemen-tation group 1-3 mutations and 6C4 identify the two other
functions.The presenceof multiple complementationgroups
forone protein may also imply that the protein is active at
least in part as amultimer (24, 25). Biochemical analysis of
Polindicates that the enzyme activity sediments as a
mono-mer (28, 33) but leaves open the possibility that multimers
exist invivo.
Removal of the small upstream open reading frame. The
deletion in AB10 removes sequences that could code for a
small decapeptide. Ourinterestin apotentialfunctional role
intissueculturefor this open reading framewasinitiatedby
the observation that it shares partial homology with
sub-stratesfor cAMP-dependent proteinkinase (18). Inaddition, recentwork (H. Bludau, K. Weisshart, U. K. Freese, C. W.
Knopf, and H. Zur Hausen, Abstr. 14th Int. Herpesvirus
Conf. 1989, abstr. no. 161, p. 161) indicates that in asmall
percentageof poltranscripts the short open reading frame is
splicedinto the pol open reading frame. The deletion in AB10
would also prevent formation of these spliced transcripts by removing the splice donor site. Since this virus grows as well
on Vero cells as on the DP6 cell line, which also contains sequences for this open reading frame (Table 2), we
con-clude that the peptide and the spliced transcripts are not
essential for growth in tissue culture. It remains to be
demonstrated whether this open reading frame is also
dis-pensable inanimal models of HSV infection.
The short open reading frame is downstream from the
transcriptional start site for pol (48) and we expect it to be
translatedfrom the same mRNA as pol, but it is not known whether this protein is synthesized in infected cells.
Expres-sion of this short open reading frame would imply that the pol mRNAisbicistronic. Inthat case, polexpression could be duetotranslational reinitiation, as can occur in the HSV
thymidine kinase mRNA (26), or, less likely, due to initiation at aninternal AUG codon (29, 30).
Recovery ofnewpolmutants. The selection method
pro-vided by the lacZ insertion mutants has proven to be a useful
method for obtaining viruses containing site-specific
muta-tions (3, 4, 21, 34; this report). Our results, in which we
recovered several host-range mutants that either did not
containtheexpected mutationsorcontained more than one
mutation, emphasize the importance of demonstrating that
theengineered changeis in fact theonly relevant alteration.
We expect that the deletion mutants described here and point mutants currently being isolated (J. S. Gibbs, A. I. Marcy, and D. M. Coen, unpublished results) will be critical forefforts to separate domains of Pol that contributeto its variousenzymatic activities andbiological activities, suchas
mutation rate, antiviral drug sensitivity, and intracellular localization.
ACKNOWLEDGMENTS
We thank N. DeLuca and D.Panicali forplasmids, S. Weller for mutanthrR3,D.Knipeand W. P.Summersforantisera,J.Miller for Dam-bacteria,K.Kerns andK.Hicksfor technicalassistance, and P.Schaffer foracriticalreading of the manuscript.
A.I.M. was the recipient ofa postdoctoral fellowship fromthe American Cancer Society, and D.R.Y. was the recipient of a
postdoctoral fellowship from the National Institutes of Health. This workwassupported by Public Health ServicegrantsAI19838 and BRSGS07RR05381 from the National Institutes of Health.
LITERATURE CITED
1. Aron, G.M.,D.J. M.Purifoy,and P. A.Schaffer. 1975. DNA synthesisandDNApolymeraseactivity of herpes simplex virus type 1temperature-sensitivemutants. J.Virol. 16:498-507. 2. Boni,J., and D. M. Coen. 1989. Examination ofthe roles of
transcription factorSpl-binding sites andanoctamer motifin trans induction ofthe herpes simplex virus thymidine kinase gene. J.Virol. 63:4088-4092.
3. Carmichael,E.P., and S. K.Weller.1989.Herpessimplex virus type 1 DNAsynthesis requires the product ofthe UL8 gene: isolationandcharacterization ofanICP6::lacZ insertion muta-tion.J. Virol. 63:591-599.
4. Chakrabarti,S.,K.Brechling,and B. Moss.1985. Vaccinia virus
expression vector: coexpression of ,-galactosidase provides
visualscreening of recombinant virus plaques.Mol.Cell. Biol. 5:3403-3409.
5. Challberg, M. 1986. Amethod for identifying the viralgenes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA 83:9094-9098.
6. Chartrand, P., C. Crumpacker, P. A. Schaffer, and N. M. Wilkie. 1980. Physical mapping ofpaar mutations of herpes
simplex virus type 1 andtype 2 byintertypic markerrescue.
Virology103:311-326.
7. Chiou,H.C.,S. K.Weller,and D. M.Coen.1985. Mutations in the herpes simplex virus major DNA-binding protein gene leading to altered sensitivity to DNA polymerase inhibitors. Virology 145:213-226.
8. Church, G. M., and W. Gilbert. 1984. Genomic sequencing. Proc. Natl.Acad. Sci. USA81:1991-1995.
9. Coen,D.M.1986.Generalaspectsof virusdrugresistance with special reference toherpes simplexvirus. J. Antimicrob. Che-mother. 18(Suppl. B):1-10.
10. Coen,D.M.,D.P.Aschman,P.T.Gelep,M.J.Retondo,S.K.
Weller, andP. A. Schaffer. 1984. Fine mappingandmolecular cloning ofmutations in theherpessimplex virusDNA
polymer-aselocus. J. Virol. 49:236-247.
11. Coen,D.M., S. P.Weinheimer,and S. L.McKnight. 1986. A geneticapproachtopromoterrecognitionduringtransinduction of viral geneexpression. Science 234:53-59.
12. Crute,J. J., andI. R.Lehman. 1989. Herpessimplex-1 DNA polymerase: identification ofanintrinsic 5'-3'exonucleasewith
on November 10, 2019 by guest
http://jvi.asm.org/
ribonucleaseH activity. J. Biol. Chem. 264:19266-19270. 13. DeLuca, N., S. Person, D. J. Bzik, and W. Snipes. 1984.
Location of temperature sensitive mutants in glycoprotein gB of herpes simplex virus type 1. Virology 137:382-389.
14. DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570.
15. Dorsky, D., P. Chatis, and C. Crumpacker. 1987. Functional expression of a cloned herpes simplex virus type 1 DNA polymerase gene. J. Virol. 61:1704-1707.
16. Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction fragments to high specific activ-ity. Anal. Biochem. 132:6-13.
17. Gibbs, J. S., H. C. Chiou, K. F. Bastow, Y.-C. Cheng, and D. M. Coen. 1988.Identification of amino acidsin herpes simplexvirus DNA polymerase involved in substrate and drug recognition. Proc. Natl. Acad. Sci. USA85:6672-6676.
18. Gibbs, J. S., H. C. Chiou, J. D. Hall, D. W. Mount, M. J. Retondo, S. K. Weller, and D. M. Coen. 1985. Sequence and mapping analysis of the herpes simplex virus DNA polymerase gene predict a C-terminal substrate binding domain. Proc. Natl. Acad. Sci. USA 82:7969-7973.
19. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C. Glorioso. 1981. Cloning of herpes simplex virus type 1 se-quences and representing the whole genome. J. Virol.38:50-58. 20. Goldstein, D. J., and S. K. Weller. 1988. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characteri-zation of an ICP6 lacZ insertion mutant. J. Virol.62:196-205. 21. Goldstein, D. J., and S. K. Weller. 1988. Factor(s) present in
herpes simplex virus type-1 infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reduc-tase: characterization of an ICP6 deletion mutant. Virology 166:41-51.
22. Hall, J. D., J. S. Gibbs, D. M. Coen, and D. W. Mount. 1986. Structural organization and unusual codon usage in the DNA polymerase gene from herpes simplex virus type 1. DNA 5:281-288.
23. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C. Glorioso, and M. L. Levine. 1984. Transcriptional and genetic analyses of the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J. Virol. 49:947-955.
24. Honess, R. W. 1981. Complementation between phosphonoace-tic acid-resistant and -sensitive variants of herpes simplex viruses: evidence for an oligomeric protein with restricted intracellular diffusion as the determinant of resistance and sensitivity. J. Gen. Virol. 57:297-306.
25. Honess, R. W., and D. H. Watson. 1977. Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21:584-600.
26. Irmiere, A. F., M. M. Manos, J. G.Jacobson, J. S. Gibbs, and D. M. Coen. 1989. Effect of an ambermutation in the herpes simplex virus thymidine kinase gene on polypeptide synthesis and stability. Virology 168:210-220.
27. Knopf, C. W. 1986. Nucleotidesequence ofthe DNA polymer-ase gene of herpes simplex virus type 1strainAngelotti. Nucleic Acids Res. 14:8225-8226.
28. Knopf, K. W. 1979. Properties ofherpes simplex virus DNA polymerase and characterization of itsassociated exonuclease activity. Eur. J. Biochem. 98:231-244.
29. Kozak, M. 1986. Regulation of protein synthesis in virus in-fected animal cells. Adv. Virus Res. 31:221-292.
30. Kozak, M. 1989. The scanning model fortranslation: an update. J. Cell Biol. 108:229-241.
31. Larder, B. A., S. D. Kemp, and G. Darby. 1987. Related functional domains in virus DNA polymerases. EMBO J. 6: 169-175.
32. Larder, B. A., J. J. Lisle, and G. Darby. 1986. Restoration of wild-type pathogenicity to an attenuated DNA polymerase
mutantof herpes simplextype1.J.Gen. Virol. 67:2501-2506. 33. Ostrander, M., and Y.-C. Cheng. 1980. Properties ofherpes
simplextype1andtype2 DNApolymerase. Biochim.Biophys. Acta 609:232-245.
34. Panicali, D., A. Grzelecki, and C. Huang. 1986. Vacciniavirus vectorsutilizingthe
P-galactosidase
assayforrapid selection of recombinant viruses and measurement of gene expression.Gene 47:193-199.
35. Purifoy, D. J. M., R. B. Lewis, and K. L. Powell. 1977. Identification oftheherpesvirusDNApolymerasegene.Nature (London) 269:621-623.
36. Purifoy, D. J. M., and K. L. Powell. 1977. Herpessimplexvirus DNA polymerase as the site ofphosphonoacetate sensitivity:
temperature-sensitive mutants.J. Virol. 24:470-477.
37. Purifoy, D. J. M., and K. L. Powell. 1981. Temperature-sensitive mutants in two distinct complementation groups of herpes simplex virustype1 specify thermolabileDNA
polymer-ase. J. Gen. Virol. 54:219-222.
38. Quinn, J. P., and D. J. McGeoch. 1985. DNA sequence of the regionin the genome ofherpessimplex virus type 1 containing the genes for DNA polymerase and the major DNA binding
protein. Nucleic Acids Res. 13:8143-8163.
39. Schaffer, P. A., G. M. Aron, N. Biswal, and M. Benyesh-Melnick. 1973. Temperature-sensitive mutants of herpes sim-plex virustype 1: isolation, complementation and partial char-acterization. Virology52:57-71.
40. Schaffer, P. A., V. C. Carter, and M. C. Timbury. 1978. Collaborative complementation study oftemperature-sensitive mutants of herpes simplex virus types 1 and 2. J. Virol. 27:490-504.
41. Southern, E. M. 1975. Detection of specific sequences among DNAfragments separated by gelelectrophoresis. J. Mol. Biol. 98:503-517.
42. Southern, P. J., and P. Berg. 1982.Transformation of
mamma-lian cells to antibiotic resistance with a bacterial gene under control oftheSV40 early regionpromoter.J.Mol. Appl.Genet. 1:327-341.
43. Taylor, J. W., J. Ott, and F. Eckstein. 1985. The rapid genera-tion of oligo-nucleotide directed mutagenera-tions at high frequency using phosphorothioate-modified DNA. Nucleic Acids Res. 13:8765-8785.
44. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer ofprotein from polyacrylamide gels to nitrocellulose sheets: procedureand someapplications.Proc. Natl.Acad. Sci. USA 76:4350-4354.
45. Tsurumi, T., K. Maeno, and Y. Nishiyama. 1987. A single base change within the DNA polymerase locus of herpes simplex virus type 2 can confer resistance to aphidicolin. J. Virol. 61:388-394.
46. Weller, S. K., D. P. Aschman, W. R. Sacks, D. M. Coen, and P. A. Schaffer. 1983. Genetic analysis oftemperature-sensitive mutantsof HSV-1: the combined use ofcomplementation and physical mappingfor cistron assignment. Virology 130:290-305. 47. Weller, S. K., A. Spadaro, J. E.Schaffer, A. W.Murray, A. M. Maxam, and P. A. Schaffer. 1985. Cloning, sequencing, and functionalanalysisoforiL, aherpessimplex virus type 1origin of DNA synthesis. Mol. Cell. Biol. 5:930-942.
48. Yager, D. R., and D. M. Coen. 1988. Analysis of thetranscript of the herpes simplex virus DNA polymerase gene provides evidence that polymerase expression isinefficient at the level of translation. J. Virol. 62:2007-2015.
49. Yager, D. R., A.I. Marcy, and D. M. Coen. 1990.Translational regulation of herpes simplex virus DNA polymerase. J. Virol. 64:2217-2225.
50. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide se-quences ofM13mpl8 and pUC19 vectors. Gene 33:103-119.